

Abstract
The NF-κB pathway is a ubiquitous stress response that activates the NF-κB family of transcription factors. Antigen receptors, receptors of the innate immune system, and certain intracellular stressors are potent activators of this pathway. The transcriptional program that is activated is both antiapoptotic and highly proinflammatory. Indeed, any compromise in engagement of the pathway results in immunodeficiency, whereas constitutive activation generates a sustained inflammatory response that may promote malignancy. As such, NF-κB activation is under tight regulation by a number of post-translational modifications, including phosphorylation and ubiquitination. This article attempts to synthesize our current knowledge regarding the regulation of NF-κB signaling by ubiquitination, specifically highlighting the biochemical basis for both positive and negative feedback loops that function in unison to generate coordinated signals that are essential for the viability of metazoan animals.
Dysregulated NF-κB pathways can be fatal. Feedback loops involving ubiquitination of components such as NEMO, RIP, and NIK ensure that their activity is precisely controlled.
INTRODUCTION TO NF-κB SIGNALING
The NF-κB family of transcription factors includes critical regulators of proinflammatory and antiapoptotic gene transcription programs. As such, they play essential homoeostatic roles in the development and orchestration of the host immune response. NF-κB-mediated transcription is the endpoint of a complex series of reactions that are initiated by a vast array of stimuli, ranging from cellular stress to the engagement of receptors that mediate innate and adaptive immunity. In this article, we review the unique aspects of the pathways that culminate in NF-κB activation and also highlight the common components that are shared by these diverse signaling cascades (see Table 1).
Table 1.
Classes of signaling components in NF-κB transduction pathways
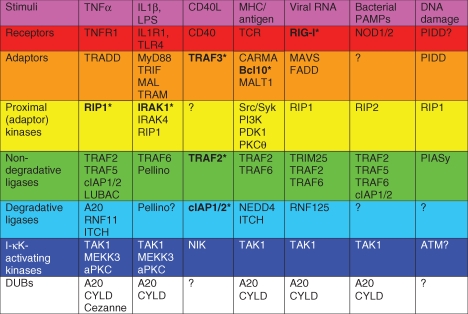 |
Representative stimuli and the cognate receptors (where known) are listed for each signaling pathway. Degradative ligases assemble polyubiquitin chains that promote substrate degradation, whereas nondegradative ligases polymerize polyubiquitin chains that promote assembly of proximal signaling complexes and subsequent activation of NF-κB signaling. PIASy, though a SUMO ligase, can therefore be classified as a nondegradative ligase in the DNA damage pathway since NEMO SUMOylation does not promote NEMO degradation and is a proximal event that is required for NF-κB activation. See text for details.
*Indicates these proteins are targets of ubiquitin editing.
Each NF-κB-activating stimulus converges on the activation of one of two kinase complexes termed inhibitors of -κB (I-κB) kinase (I-κK) complexes. The heterotrimeric I-κK complexes are composed of a regulatory subunit I-κK-γ, or NF-κB essential modulator (NEMO), and two kinases I-κK-α or I-κK-β. Canonical NF-κB signaling pathways assemble proximal signaling complexes on polyubiquitin chain scaffolds; assembly of these complexes promotes the activation of I-κK-β, which in turn phosphorylates I-κB inhibitory proteins. These inhibitors mask the nuclear localization signals (NLS) within NF-κB transcription factors, thereby retaining them in the cytosol. Phosphorylation of I-κB by I-κK-β permits the recognition of I-κB by the SCFβTrCP ubiquitin ligase complex, which subsequently targets I-κB for proteasomal degradation. Thus, heterodimeric NF-κB transcription factors such as p50/p65 are released from I-κB inhibition and enter the nucleus to activate transcription of NF-κB target genes (Hoffmann and Baltimore 2006). Alternatively, stimuli that activate noncanonical signaling pathways promote stabilization of NF-κB-inducing kinase (NIK), a labile proximal kinase. When sufficient levels accumulate, NIK phosphorylates and activates I-κK-α within the I-κK complex, which in turn phosphorylates p100, a precursor NF-κB subunit. Phosphorylated p100 is also ubiquitinated by the SCFβTrCP ubiquitin ligase complex and is subsequently processed by the proteaseome to p52, which is a transcriptionally competent NF-kB subunit in conjunction with RelB (Vallabhapurapu and Karin 2009). Thus, ubiquitination is a critical regulatory mechanism at multiple steps within NF-κB signaling cascades. We therefore provide a brief review of the ubiquitin/proteasome system.
THE UBIQUITIN/PROTEASOME SYSTEM (UPS)
Ubiquitin is a 76-amino-acid protein that when covalently linked to target proteins may alter their half-life, localization, or function. An enzymatic cascade composed of three types of proteins—the E1 ubiquitin-activating enzyme, the E2 ubiquitin-conjugating enzyme, and the E3 ubiquitin ligase—mediates the conjugation of ubiquitin to substrate proteins (Fig. 1). The E1 enzyme hydrolyses ATP and transfers a thio-ubiquitin intermediate to the active site cysteine of one of over 30 E2 enzymes. The E2 enzyme “charged” with a thio-ubiquitin may transfer the ubiquitin to a HECT domain of an E3 ligase, or instead, may bind to a RING domain (or a related variant domain) of an E3 ligase, which number in the hundreds. Importantly, the E3 also binds the substrate and orients the reactants for facile ligation of the ubiquitin carboxyl terminus to the ϵ-amino group or the amino terminus of a substrate lysine (Lys) residue (Schwartz and Ciechanover 2009).
Figure 1.

Enzymes and reactions of the ubiquitin/proteasome system.
Multiple rounds of ubiquitination, with ubiquitin itself serving as a substrate, generates polyubiquitin chains. Ubiquitin has seven Lys residues, Lys-6, Lys-11, Lys-27, Lys-29, Lys-33, Lys-48, and Lys-63, and any one of these can participate in polyubiquitin chain formation. The amino terminus of ubiquitin may also be conjugated to another ubiquitin carboxyl terminus in which case linear polyubiquitin chains are generated. Whereas the HECT domain-containing E3 ligases are intrinsically responsible for dictating ubiquitin linkage specificity, it is the E2 associated with RING-type ligases that determines the type of polyubiquitin chain formed. For example, the E2 UBC13, in cooperation with the E2 variant UEV1 and TRAF RING domains, promotes the formation of Lys-63-linked ubiquitin chains. However, the E2 UBC5, together with the RING-containing protein ROC1/RBX1 of the SCFβTrCP ligase, promotes Lys-48-linked polyubiquitination (Vallabhapurapu and Karin 2009). Embedded in the topology of the various ubiquitin chains is information that dictates signaling outcome. For example, Lys-48- and Lys-11-linked polyubiquitination usually, but not always, targets substrates for proteosomal degradation (Fig. 1). In contrast, Lys-63-linked polyubiquitin chains function as scaffolds to assemble signaling complexes and thereby participate in diverse cellular processes ranging from DNA repair to activation of NF-κB signaling (Fig. 1) (Pickart and Fushman 2004; Ikeda and Dikic 2008). Distinct ubiquitin-binding domains (UBDs) are the receptors that bind different ubiquitin chain conformations and dictate the fate of ubiquitinated substrates. For example, the ubiquitin-associated (UBA) domains of proteasome subunits Rpn13/ARM1 and Rpn10/S5a preferentially bind Lys-48-linked chains, thereby recruiting substrates for degradation. Alternatively, the UBAN (UBD in ABIN and NEMO) motif of NEMO binds linear and Lys-63-linked polyubiquitin chains, thereby shuttling NEMO and the associated I-κK proteins, to activated signaling complexes (Ikeda and Dikic 2008).
Ubiquitination can be reversed by proteases termed deubiquitinases (DUBs) (Fig. 1). The human genome encodes approximately 100 DUBs that fall into five families: Four are papain-like cysteine proteases (ubiquitin carboxy-terminal hydrolase, UCH; ubiquitin specific protease, USP; ovarian tumor domain, OTU; and Machado-Joseph disease proteases, MJD) and the fifth is a metalloprotease (JAMM). In addition to protease domains, DUBs often contain protein interaction motifs, including UBDs, that direct their recruitment to particular signaling complexes and promote preferential binding to specific poly-Ub chain linkages (Reyes-Turcu and Wilkinson 2009). DUBs are critical regulators of NF-κB signaling pathways. For example, genetic ablation of the OTU DUB A20 causes rampant inflammation in multiple organ systems because of unchecked NF-κB activity, resulting in perinatal lethality (Lee et al. 2000). Furthermore, mutation or deletion of TNFAIP3, which encodes A20, is associated with deregulation of NF-κB signaling in a variety of lymphomas (Compagno et al. 2009; Kato et al. 2009; Novak et al. 2009; Schmitz et al. 2009) and TNFAIP3 polymorphisms are associated with the autoimmune disorder systemic lupus erythematosus (Musone et al. 2008). The USP DUB CYLD is also a tumor suppressor that negatively regulates NF-κB signaling by deubiquitinating a number of critical pathway components. Somatic mutations are linked to familial cylindromatosis and loss of CYLD expression has been implicated in a variety of malignancies including colonic, hepatocellular, and renal carcinomas, as well as multiple myeloma (Sun 2009). Thus, cycles of ubiquitination and deubiquitination, each mediated by distinct enzymes, control cellular processes that are essential for the maintenance of cellular homeostasis.
UPS REGULATION OF NF-κB SIGNALING: GENERAL CONCEPTS
Translating Polyubiquitination into I-κK Activation
Having introduced the NF-κB transcription factors and the UPS, we can now address some general concepts in the signaling pathways that activate NF-κB. The first is how the polymerization of nondegradative ubiquitin chains promotes I-κK-β activation. There are several pieces of evidence suggesting that polyubiquitination plays an essential role in the activation of I-κK-β. First, NEMO is both a ubiquitin binding protein and a target of ubiquitination in classical NF-κB signaling pathways. Genetic ablation studies have shown that NEMO is essential for activation of I-κK-β (Makris et al. 2000; Rudolph et al. 2000). Indeed, mutations in NEMO that disrupt ubiquitin binding cause anhidrotic ectodermal dysplasia and immunodeficiency caused by improper activation of NF-κB signaling (Chen and Sun 2009). Finally, as indicated previously, mutation or deletion of the DUBs A20 and CYLD deregulate NF-κB signaling, thereby promoting unchecked inflammation and tumorigenesis.
Several models of how ubiquitination might promote I-κK-β activation have been proposed. These include: (1) conformational changes in NEMO induced by polyubiquitin binding that promote I-κK-β activation, (2) induced proximity of I-κK-β proteins within the I-κK complex that activates trans-phosphorylation, and (3) induced proximity of I-κK kinases, such as TAK1 or MEKK3, with the I-κK complex. Structural studies have indicated that NEMO binds Lys-63- and linear polyubiquitin chains, which is critical for NF-κB activation in vivo (Rahighi et al. 2009; Lo et al. 2009; Ivins et al. 2009). Furthermore, it was suggested that NEMO association with polyubiquitin could induce sufficient conformational changes to activate associated I-κK-β (Rahighi et al. 2009). The role of NEMO polyubiqutination in I-κK-β activation is less certain. Several polyubiquitination sites have been mapped on NEMO but only Lys-392, the equivalent of human Lys-399, has been tested in a murine knock-in model. These mice were more resistant to lipopolysaccharide (LPS)-induced endotoxic shock but had no defect in T-cell receptor (TCR)-induced proliferation, both of which are mediated by NF-κB signaling (Ni et al. 2008). This finding could suggest that ubiquitination of Lys-392 on NEMO is not important for TCR-induced NF-κB activation. However, it is also possible that alternative sites on NEMO may be sufficiently ubiquitinated following TCR activation if Lys-392 is not available for modification. Thus, NEMO ubiquitination on specific residues may be more important in certain NF-κB signaling pathways than in others. Knockout studies have established a role for MEKK3 and TAK1 as I-κK kinases in certain pathways. MEKK3 is thought to associate with RIP1 in the TNFR1 pathway and activate I-κK-β that is recruited to proximal signaling complexes via NEMO (Vallabhapurapu and Karin 2009). Similarly, the TAB2 and -3 regulatory proteins within the TAK1/TAB complex have UBDs that facilitate binding to Lys-63 ubiquitinated substrates. It is thought that corecruitment of TAK1/TAB and I-κK complexes to polyubiquitinated proteins within proximal signaling complexes may also allow TAK1 to phosphorylate I-κK-β via induced proximity (Chiu et al. 2009).
Thus, there is ample evidence for each of the three models to explain how polyubiquitination of proteins that participate in classical NF-κB signaling pathways could promote I-κK activation. In theory, none of the three models are mutually exclusive and all could contribute to I-κK activation. For example, polyubiquitin chains on one NEMO subunit could bind the UBD of another NEMO subunit to promote the assembly of a larger I-κK complex. This could promote low-level I-κK activity via trans-phosphorylation, and subsequent recruitment of this oligomeric I-κK complex to activated signaling complexes via polyubiquitin/UBD interactions could further enhance I-κK activity by induced proximity with I-κK kinases such as TAK1. Such a scenario is all the more plausible given that NEMO can be modified by and can associate with both Lys-63-linked and linear polyubiquitin chains: One type of chain could promote assembly of oligomeric I-κK complexes, whereas the other type of chain could promote the recruitment of I-κK complexes to activated signaling networks.
Inactivation of NF-κB Signaling
Down-regulation of NF-κB signaling is as important as NF-κB activation. Unchecked NF-κB activity may promote rampant inflammation or tumorigenesis, as revealed by mutations or deletions of the DUBs CYLD and A20. In addition to DUBs, a number of inhibitory proteins down-regulate NF-κB signaling, including RIP3, MyD88s, IRAK-M, SARM, and I-κB. RIP3, MyD88s, and SARM competitively block the recruitment of key signaling proteins to activated complexes (Barton and Medzhitov 2004; O'Neill and Bowie 2007); IRAK-M blocks the dissociation of IRAK1 and -4 from MyD88, which is a critical step to activate NF-κB downstream of IL1R1 and TLR (O'Neill and Bowie 2007); and I-κB masks the NLS on NF-κB hetrerodimers to block nuclear translocation (Hoffmann and Baltimore 2006). DUBs may also collaborate with ubiquitin ligases to inactivate and degrade critical mediators of NF-κB signaling. This form of regulation is termed ubiquitin editing and usually regulates the ubiquitination status of essential positive regulators of NF-κB signaling complexes. First, Lys-63 polyubiquitin chains are depolymerized by DUBs to disassemble the platform on which signaling complexes are organized. Then, Lys-48 polyubiquitin chains are polymerized on the same protein to target it for proteasomal degradation (Wertz et al. 2004; Heyninck and Beyaert 2005; Newton et al. 2008). However, it is important to note that ubiquitin editing need not be restricted to regulation of Lys-48 and Lys-63-linked polyubiquitination or even to ubiquitination in general. In fact, ubiquitin editing can be broadly conceptualized as the concerted removal of any modification that promotes signaling complex assembly, such as Lys-63-linked polyubiquitination, linear ubiquitin chains, or even ligation with the ubiquitin-like protein SUMO, combined with modifications that promote substrate degradation, such as Lys-11 or Lys-48 polyubiquitination. Table 1 includes a summary of proteins regulated by ubiquitin editing in NF-κB signaling pathways.
UPS REGULATION OF NF-κB SIGNALING: SPECIFIC PATHWAYS
Introduction
As mentioned previously, an array of stimuli may activate host receptors to initiate the upstream signaling events that culminate in the activation of NF-κB transcription factors. Although each signaling cascade is activated by unique stimuli and contains highly specialized components, the pathways share some functionally common elements, including: (1) adaptors that link the activated receptor to downstream effector proteins, (2) proximal kinases that propagate signaling, sometimes in a kinase-independent manner by serving as adaptor proteins, (3) nondegradative ligases, (4) degradative E3 ligases, (5) distal I-κK kinases, and (6) DUBs. These components, and the pathways that they participate in, are listed in Table 1.
TNFR1
Tumor necrosis factor-α (TNF-α) was first described in 1975 as a factor that caused tumor necrosis in a murine sarcoma model (Carswell et al. 1975). Since then, TNF-α has been characterized as a cytokine that activates a variety of cellular responses including NF-κB signaling, as depicted in Figure 2. TNFR1 is trimerized on TNF-α binding, thereby promoting recruitment of the adaptor protein TNF receptor associated protein with a death domain (TRADD). TRADD assembles at least two distinct signaling complexes that initiate opposing signaling pathways: Complex 1-mediated activation of antiapoptotic and proinflammatory mediators, including NF-κB, or complex 2-mediated activation of apoptosis. Activation of apoptosis by complex 2 has been extensively reviewed elsewhere (Varfolomeev and Vucic 2008); in this article, we focus on NF-κB activation by complex 1.
Figure 2.

TNFR1 and TLR4 signaling pathways. Lys-48-linked ubiquitin chains are shown in red, and Lys-63-linked ubiquitin chains are shown in green. See text for additional details.
In addition to TRADD, complex 1 includes receptor interacting protein-1 (RIP1), cellular inhibitor of apoptosis protein-1 (cIAP1), cIAP2, and TNF receptor associated factor-2 (TRAF2). RIP1 is a kinase that is essential for TNF-α-mediated NF-κB signaling in a kinase-independent manner (Ting et al. 1996; Kelliher et al. 1998). On TNF stimulation, TNFR1-associated RIP1 is rapidly modified by Lys-63-linked polyubiquitin chains. These Lys-63-linked chains create a scaffold to recruit the I-κK complex via NEMO. Mutations in NEMO that abolish binding to Lys-63-linked polyubiquitin chains also disrupt association of NEMO with RIP in TNF-α-stimulated cells, block the recruitment of I-κK to TNFR1, and inhibit I-κK activation (Ea et al. 2006; Wu et al. 2006). The UBDs in the TAB2/3 regulatory proteins of the TAK1 kinase complex also bind Lys-63-linked polyubiquitin chains, thus TAB2/3 is recruited to polyubiquitinated RIP on TNF-α stimulation (Kanayama et al. 2004). The subsequent activation of the TAK1 complex promotes I-κK activation (Kovalenko and Wallach 2006). Lys-377 is likely the primary residue that is ubiquitinated on RIP and appears to be important for NF-κB activation, given that a Lys-377 mutation to arginine attenuates RIP ubiquitination, prevents the recruitment of TAK1 and I-κK complexes to TNFR1, and inhibits I-κK activation (Ea et al. 2006). TNFR1-stimulated signaling pathways may also activate MEKK3 and aPKC that in turn phosphorylate the I-κK complex.
Given the importance of RIP modification with Lys-63 polyubiquitin chains in propagating TNFR1 signaling, the definitive identification of RIP E3 ligases is of significant interest. The initial candidate was TRAF2, a RING domain E3 that is essential for TNF-α-stimulated and NF-κB signaling (Yeh et al. 1997; Tada et al. 2001), possibly in collaboration with the heterodimeric E2 UBC13/UEV1 (Shi and Kehrl 2003). Evidence for TRAF2-mediated RIP ubiquitination is based on TRAF2 RNAi (Wertz et al. 2004) and gene ablation (Lee et al. 2004) experiments, which decreased RIP ubiquitination in the absence of TRAF2. However, these data could simply reflect that TRAF2 is an adaptor that recruits another ligase to RIP1. Thus, formal proof demonstrating direct ubiquitination of RIP1 by TRAF2 is still needed. TRAF5 is another RING domain E3 that is thought to participate in TNFR1-induced NF-κB activation because TRAF2 and -5 are functionally redundant in promoting NF-κB activation (Tada et al. 2001). However, formal proof of TRAF5 ligase activity is missing, and TRAF5 recruitment to complex 1 has not been reported.
The cIAP1 and cIAP2 (cIAP1/2) proteins are another pair of RING E3s that associate with activated TNFR1 (Shu et al. 1996; Srinivasula and Ashwell 2008). Because cIAP1/2 are recruited to TNFR1 via TRAF2, they are candidate TRAF2-associated RIP1 ligases. Indeed, cIAP1/2 promote RIP1 polyubiquitination in vivo, and in vitro experiments revealed that cIAP1/2 couple with the E2 UBCH5 to catalyze Lys-63-linked RIP polyubiquitination. Furthermore, deletion of both cIAP1 and -2 revealed their essential and redundant roles in TNF-α-induced NF-κB activation (Varfolomeev et al. 2008; Mahoney et al. 2008; Bertrand et al. 2008). It is therefore unclear what specific functions TRAF2 and -5 and cIAP1/2 have in RIP1 ubiquitination and NF-κB activation. It is possible that TRAF2 (and perhaps TRAF5) simply recruits cIAP1/2 to TNFR1 but has no additional role in RIP1 ubiquitination. Alternatively, TRAF2 and -5 could regulate cIAP1/2 ubiquitin ligase activity, possibly via ubiquitination, or perhaps TRAF2 and -5 ubiquitinate RIP1 on different sites and/or in different contexts than cIAP1/2 to fine-tune TNFR1-induced NF-κB activation.
The E2 UBC13 was first implicated in TNFR1 signaling when a dominant–negative version of UBC13 was reported to block TNF-α- and TRAF2-induced NF-κB activity (Deng et al. 2000). However, UBC13 genetic ablation experiments performed by two separate groups have generated conflicting results. In each set of studies, homozygous UBC13 ablation was embryonic lethal, thus necessitating study of hemizygous UBC13−/+ mice (Fukushima et al. 2007) or mice conditionally deficient in UBC13 (Yamamoto et al. 2006a). Although UBC13+/− macrophages and splenocytes displayed blunted activation of NF-κB in response to TNF-α (Fukushima et al. 2007), UBC13−/− MEFs displayed no alteration in TNF-α-induced NF-κB signaling relative to wild-type MEFs (Yamamoto et al. 2006a). These discrepancies may be attributable to other E2 enzymes or E2/ligase pairs that can substitute for UBC13 in certain cell lines. For example, it was shown in a cell-free system that UBC4/5 can promote I-κB-α phosphorylation with an unidentified ligase (Chen et al. 1996). Furthermore, TRAF2 and -5 fail to bind UBC13 in vitro (Yin et al. 2009), implicating another E2 and/or E2/ligase pair in TNFR1-induced NF-κB activation. The recent identification of a distinct E2/E3 enzyme complex that modifies NEMO with linear polyubiquitin chains and is essential for TNF-α-activated NF-κB signaling may explain some of the discrepancies revealed in the previous studies (Tokunaga et al. 2009).
Genetic ablation experiments established A20 as a critical negative regulator of TNF-α-induced NF-κB signaling (Lee et al. 2000), and functional studies later revealed that A20 contains both an OTU DUB domain and a C2/C2 ZnF E3 ligase domain. As such, A20 is a dual-function ubiquitin editing enzyme for RIP1: The A20 DUB domain first depolymerizes Lys-63-linked ubiquitin chains from RIP1, and the A20 E3 ligase motif then promotes the ligation of Lys-48-linked ubiquitin chains on RIP1 (Wertz et al. 2004). Several modulators of A20 ubiquitin ligase activity have also been identified. TAXBP1, a binding protein of the hTLV TAX protein, is an A20-binding protein and cooperates with A20 to attenuate TNF-α signaling by recruiting the HECT-domain ubiquitin ligase Itch (Shembade et al. 2008). The RING domain E3 RNF11 also collaborates with TAXBP1 and Itch to promote RIP1 degradation after TNF-α treatment (Shembade et al. 2009), although the precise roles for each E3 are unknown. A number of additional RIP1 DUBs have also been reported. The A20-like protein cellular zinc finger antiNF-κB (Cezanne) also has an OTU domain and promotes RIP1 deubiquitination on TNF-α stimulation (Enesa et al. 2008), and CYLD is also proposed to deubiquitinate RIP1, among other critical targets, in TNF-α-induced NF-κB signaling pathways (Sun 2009). In vivo evidence of RIP1 ubiquitin editing was recently revealed by ubiquitin linkage-specific antibodies (Newton et al. 2008), thus the interplay between the various ubiquitin modifying enzymes in orchestrating RIP1 ubiquitination and degradation will be interesting to elucidate.
IL1R1/TLR4
Interleukin-1 (IL1) receptor-1 (IL1R1) and Toll-like receptors (TLR) are transmembrane proteins that share a common intracellular Toll and IL1 receptor (TIR) domain. As such, they recruit related complexes of signaling proteins with distinct variations that fine-tune regulation and mediate signaling specificity. Like TNFR1, IL1R1-activated signaling pathways may also activate MEKK3 and aPKC that in turn activate the I-κK complex. IL1R1 is activated by the potent inflammatory cytokine IL1-β, whereas TLRs, of which there are at least 10 in humans, recognize pathogen-associated molecular patterns (PAMPs) such as LPS and viral nucleic acids. On activation, most TLRs and IL1R1 recruit the adaptor protein MyD88 either directly or via MAL (also known as TIRAP) through TIR/TIR interactions. TLR4 can also recruit TRIF via TRAM, and TLR3 directly recruits TRIF (for more extensive reviews, see O'Neill and Bowie 2007; Verstrepen et al. 2008).
Here, we focus on TLR4 as a prototypical TIR domain-containing receptor. A wealth of studies have investigated the mechanistic details leading to I-κK activation, thus the model presented here highlights general concepts (Fig. 2). LPS binding activates two primary pathways downstream of TLR4 that culminate in TAK1 activation. In one branch, TRAM and TRIF are recruited to TLR4, and TRIF recruits both TRAF6 and RIP1 to the proximal receptor signaling complex. It is thought that TRAF6 and RIP1 Lys-63-linked polyubiquitination both facilitate TAK1 activation (Schauvliege et al. 2007; Vallabhapurapu and Karin 2009). In the other branch, recruitment of MyD88 via MAL promotes the assembly of a proximal signaling complex that includes IRAK1 and -4 and TRAF6. IRAK4 phosphorylates IRAK1, which promotes dissociation of IRAK1 and bound TRAF6 from TLR4. This complex subsequently associates with the TAK1/TAB complex, perhaps via TAB2/3 binding to TRAF6 Lys-63-linked chains. Pellino RING ligases are also recruited and may ubiquitinate IRAK1 with Lys-63-linked chains. IRAK1 and Pellino proteins are ubiquitinated and targeted for proteasomal degradation, perhaps by Pellino ligases that promote Lys-48 and Lys-11 polyubiquitination. IRAK1 degradation and TAK/TAB phosphorylation may facilitate the release of TAK1/TAB complex and associated TRAF6, but the role of such translocation in NF-κB activation is unclear (Moynagh 2009). Thus, most evidence indicates that Lys-63-linked ubiquitination is critical for TAK1 and IKK activation via MyD88 and MAL even though the precise mechanisms are unknown.
Indeed, TRAF6, IRAK1, NEMO, and the Pellino proteins are all reported targets of Lys-63-linked ubiquitination, and the ubiquitination sites that are critical for propagating NF-κB signaling have been mapped on IRAK1 (Conze et al. 2008) and TRAF6 (Lamothe et al. 2007). Nevertheless, the E2 and E3 enzymes responsible for activating NF-κB signaling downstream of IL1R1 and TLR are not completely characterized. For example, conditional UBC13−/− B cells, bone marrow-derived macrophages, and MEFs have no defects in NF-κB signaling after stimulation with IL1-β or TLR agonists including LPS, CpG DNA, or bacterial lipopeptide (Yamamoto et al. 2006a). These results suggest that UBC13 in collaboration with TRAF ligases may not be responsible for catalyzing Lys-63 polyubiquitination, and/or that Lys-63 polyubiquitination is not important for IL1R1 or TLR signaling pathways. In contrast, UBC13+/− macrophages and splenocytes showed decreased NF-κB activity in response to LPS (Fukushima et al. 2007). Adding to the complexity of interpreting these studies, it was also reported that LPS treatment promoted less TRAF6 ubiquitination in UBC13+/− spleen lysates and splenocytes relative to wild-type controls (Fukushima et al. 2007). However, it is not clear whether the lysates were denatured before TRAF6 immunoprecipitation to dissociate noncovalently bound proteins; if not, the polyubiquitination detected in TRAF6 immunoprecipitates could reflect the ubiquitination status of TRAF6-associated proteins (including UBC13), rather than TRAF6 itself. Finally, the role of the TRAF6 RING in TLR- and IL1R1 signaling is unclear. One study showed that the TRAF6 RING and first ZnF were dispensable for NF-κB activation in complementation experiments with TRAF6-deficient cells (Kobayashi et al. 2001), whereas similar complementation studies (Lamothe et al. 2007; Conze et al. 2008; Walsh et al. 2008) and structure/function studies (Yin et al. 2009) indicate that the TRAF6 RING domain is essential. Similarly, structure/function studies have shown that TRAF6 interaction with UBC13 is essential for IL1-induced NF-κB activation (Yin et al. 2009). It is therefore possible that in certain conditions alternate E2s, ligases, and/or even polyubiquitin chains can propagate IL1R1- and TLR-induced NF-κB signaling, such as the LUBAC ligase complex that polymerizes linear ubiquitin chains that are essential for IL1-β-induced NF-κB signaling (Tokunaga et al. 2009).
Ubiquitin editing also appears to be an important regulatory mechanism for NF-κB activation in IL1R1 and TLR4 signaling pathways. IRAK ubiquitination and degradation on receptor activation was first reported 12 years ago (Yamin and Miller 1997), but it was only recently shown that IRAK is ubiquitinated with Lys-63 polyubiquitination before degradation (Ordureau et al. 2008). Furthermore, ubiquitin-linkage-specific antibodies revealed that IRAK1 is a target of ubiquitin editing after IL1-β stimulation (Newton et al. 2008). A20 (Boone et al. 2004) and CYLD (Sun 2009) have been shown to remove Lys-63 polyubiquitin chains from TRAF6 and thereby regulate TLR4 and IL1R1 signaling, but the enzymes responsible for IRAK1 ubiquitin editing are largely unknown. Certain Pellino proteins are reported to synthesize Lys-11, Lys-48, and Lys-63 polyubiquitin chains and are therefore attractive candidates for IRAK ligases in addition to TRAF6 (Moynagh 2009), and Pellino genetic ablation studies will likely further elucidate the physiological targets.
Noncanonical NF-κB Signaling: CD40
Most TNFR family members can activate both canonical and noncanonical NF-κB signaling pathways. A subset of receptors including CD40 and BAFF-R on B cells, LTB-R on stromal cells, TWEAK-R on endothelial cells, and RANK on osteoclasts primarily activate the noncanonical pathway, making these receptors key systems for study (Hacker and Karin 2006). Here, we focus on CD40 as a model system of NF-κB activation by noncanonical signaling pathways (Fig. 3).
Figure 3.

CD40 signaling pathways. Lys-48-linked ubiquitin chains are shown in red, and Lys-63-linked ubiquitin chains are shown in green. See text for additional details.
A key feature of noncanonical NF-κB signaling is the proteasomal processing of p100 to p52. Generation of p52 allows the active NF-κB heterodimer to enter the nucleus and promote transcription. Processing of p100 is initiated by IκK-α-mediated phosphorylation, which is activated by NIK. NIK is normally maintained at low levels in the cytosol as a result of efficient degradation by the ubiquitin/proteasome system (Vallabhapurapu and Karin 2009). Thus, the identification and mechanistic characterization of the ligase(s) that promote NIK degradation has been the subject of intense research.
Several clues to the identity and function of NIK ligases have been provided by studies using knockout mice, RNAi, and small molecule antagonists of signaling components of noncanonical NF-κB signaling pathways. In unstimulated cells, NIK is constitutively bound to TRAF2 and -3, which are nonredundant negative regulators: Deficiency in TRAF2 or -3 promotes constitutive NIK activation, and crossing TRAF2 or -3 deficient mice with NIK−/− mice resolves the pathology (Wallach and Kovalenko 2008). Furthermore, inhibition of cIAP1 or -2 expression using RNAi or small molecule antagonists also activates NIK (Wu et al. 2007). Because TRAF2 and -3 are thought to catalyze nondegradative Lys-63 polyubiquitination, it was proposed that TRAF2 and -3 participate in NIK degradation by serving as a molecular link to cIAP1/2, which could assemble degradative polyubiquitin chains on NIK. Additional evidence to support this hypothesis includes degradation of endogenous cIAP1/2 on activation with ligands that activate noncanonical NF-κB signaling (CD40, BAFF, and TWEAK) and genetic deletion of cIAP1/2 in multiple myeloma cells, which is correlated with NIK stabilization, p100 processing, and chronic NF-κB activation (Varfolomeev and Vucic 2008).
Additional studies have investigated the coordinated activities of TRAF2 and -3 with cIAP1/2 in NIK degradation in response to CD40 activation. These studies suggested that on receptor ligation, TRAF3 serves as an adaptor to recruit NIK, as well as TRAF2 and associated cIAP1/2, to receptors. TRAF2 then modifies cIAP1/2 with Lys-63-linked polyubiquitination, thereby enhancing the ability of cIAP1/2 to catalyze Lys-48-linked polyubiquitination of TRAF3. The resultant degradation of TRAF3 dissociates NIK from the cIAPs, thereby permitting NIK accumulation, p100 processing, and NF-κB activiation (Wallach and Kovalenko 2008). Collectively, these studies point to cIAP1/2, and possibly to TRAF2 and -3, as targets of ubiquitin editing: On receptor activation, these proteins are initially modified with Lys-63-linked chains (Wallach and Kovalenko 2008; Varfolomeev and Vucic 2008), and are subsequently modified with degradative polyubiquitin linkages that target them for proteasomal destruction (Wu et al. 2007; Wallach and Kovalenko 2008). Notably, in the case of noncanonical NF-κB signaling, ubiquitin editing promotes NF-κB activity, as opposed to canonical NF-κB signaling in which ubiquitin editing attenuates NF-κB activity.
These reports may explain why TRAF2 and -3 have nonredundant functions in CD40 signaling but, as with any new information in complex systems, the findings prompt further investigation. For example, the role of the TRAF3 RING domain is unclear. Complementation experiments of TRAF3 null MEFs suggested that a TRAF3 RING mutant is unable to destabilize endogenous NIK or inhibit endogenous p100 processing, implicating the importance of the TRAF3 RING domain in regulating noncanonical NF-κB signaling (He et al. 2007). Conversely, experiments in HEK 293T cells with RING-deleted TRAF3 (Vallabhapurapu et al. 2008) and in TRAF2−/− or TRAF3−/− MEFs reconstituted with TRAF2/TRAF3 chimeras suggest that TRAF3 serves as an adaptor only, and the TRAF3 RING is not required to regulate p100 processing (Zarnegar et al. 2008). Additionally, how TRAF2 and -3 generate polyubiquitin chains is uncertain. Structure/function studies suggest that only TRAF6, but not TRAF2 or -3 RING domains, interact with UBC13. Thus, the E2 enzymes that cooperate with TRAF2 and -3 are unknown (Yin et al. 2009). This could explain why conditional ablation of UBC13 in B cells does not alter p100 processing in response to anti-CD40 or BAFF stimulation (Yamamoto et al. 2006a), and suggests that another ligase and/or E2 that catalyze Lys-63 polyubiquitination participate in noncanonical NF-κB signaling. Finally, it is unclear whether NIK requires additional regulation for full kinase activity, or whether simple accumulation of critical protein levels is sufficient for activation (Wallach and Kovalenko 2008).
Antigen Receptors: TCR
Antigen receptors are activated on ligation with MHC-bound antigenic peptides from pathogens that are presented on the surface of antigen-presenting cells. Receptor ligation activates a prototypical signaling cascade that is common to an array of receptors, including angiotensin II- or lysophosphatidic acid-activated G-protein coupled receptors as well as a number of receptors containing immunoreceptor tyrosine-based activation motifs (ITAMs). ITAM-containing receptors include TCR, BCR, the natural killer (NK) cell receptor NKG2D, and the osteoclast receptor OSCAR (Thome 2008). In each case, receptor activation initiates a phosphorylation cascade that promotes the assembly of a complex that is composed of CARMA1, Bcl10, and MALT1 (the CBM signalosome), which ultimately activates I-κK to promote NF-κB transcription.
Here, we focus on TCR as a prototype of antigen receptor signaling (Fig. 4). TCR ligation with a peptide/MHC complex activates TCR-associated Src/Syc family kinases. A subsequent phosphorylation cascade that includes PI3K and PDK1 culminates in PKC-Θ phosphorylation. Activated PKC-Θ then phosphorylates CARMA1, resulting in membrane recruitment and assembly of the CBM signalosome. CBM assembly promotes polymerization of Lys-63-linked ubiquitin chains and subsequent activation of TAK1 to stimulate NF-κB-mediated transcription (Bhoj and Chen 2009).
Figure 4.

TCR signaling pathways. Lys-48-linked ubiquitin chains are shown in red, and Lys-63-linked ubiquitin chains are shown in green. See text for additional details.
TRAF2 and -6 are candidate ligases that may polymerize Lys-63-linked Ub chains downstream of activated TCRs. Although TRAF2 or -6 knockout animals have no deficiency in TCR-induced NF-κB activation, RNAi of both TRAF2 and -6 almost completely abolished I-κK activation downstream of TCR. Because MALT1 has TRAF2 and -6 binding motifs, it was proposed that TRAF oligomerization by assembly of the CBM complex activates TRAF E3 activity and subsequent generation of Lys-63 polyubiquitin chains (Sun et al. 2004). It will be important to confirm these findings with TRAF2/6 double knockout mice. Given that TRAF proteins have been shown in other pathways to recruit additional ligases that catalyze Lys-63-linked polyUb chains, such as cIAP1/2 and Pellinos, additional studies with TRAF2 and -6 mutants that are deficient in ligase recruitment motifs will also be required to determine the relative contribution of each ligase in TCR-induced NF-κB activation. Indeed, UBC13-deficient thymocytes are severely deficient in TAK1 activation, but IκB-α phosphorylation and degradation are only modestly affected (Yamamoto et al. 2006b). These findings suggest that other upstream ligases that collaborate with alternate E2s and that activate additional I-κK-activating kinases could be involved in TCR-mediated NF-κB activation.
TRAF6, MALT1, NEMO, and Bcl10 have all been proposed as targets for Lys-63 polyubiquitination, and primary ubiquitination sites have been mapped in most cases, revealing their importance in propagating NF-κB signaling (Sun et al. 2004; Zhou et al. 2004; Oeckinghaus et al. 2007; Wu and Ashwell 2008). Interestingly, Bcl10 is also destabilized following TCR activation. It has been shown that Bcl10 is ubiquitinated and targeted for lysosomal degradation by NEDD4 and Itch, ligases that are important for regulation of immune responses (Scharschmidt et al. 2004). Thus, Bcl10 is another example of a critical signaling component that is regulated by ubiquitin editing: Bcl10 is rapidly modified by Lys-63 polyubiquitination to facilitate TCR activation, and is subsequently tagged with degradative polyubiquitin modifications to down-regulate Bcl10 and thereby attenuate TCR signaling.
Recent reports have also implicated Bcl10, as well as A20, as substrates of the caspase-like activity of MALT1. In both cases, MALT1 proteolytic activity is not required for TCR-induced NF-κB activation, in agreement with previous reports showing that substitution of the putative MALT1 catalytic Cys residue only modestly reduces NF-κB activity (Uren et al. 2000) (Lucas et al. 2001). Rather, MALT1-mediated proteolysis seems to be important for fine-tuning TCR-induced NF-κB activation. Bcl10 cleavage promotes T-cell adhesion to fibronectin following TCR activation, which is important for T-cell stimulation, migration, and extravasation (Rebeaud et al. 2008). Unlike most cells in which A20 is induced by NF-κB as a negative-feedback mechanism to prevent protracted NF-κB signaling, A20 is constitutively expressed in lymphoid cells. Thus, MALT1-induced A20 cleavage is proposed to inactivate A20 and thereby release the constitutive brake on TCR-induced NF-κB signaling. These studies provide an additional mechanism for regulation of TCR-induced NF-κB signaling and motivate the search for additional caspase substrates. This is especially significant given the genetic (Hacker and Karin 2006) and biochemical (Sun et al. 2008) evidence that caspase-8 is critically required for NF-κB activation in response to TCR ligation, yet the mechanistic details of how caspase-8 achieves these effects remain unclear.
NF-κB Activation by Intracellular Stimuli: NOD2, RIG-I, and DNA Damage
Although the previous examples of NF-κB-activating signaling pathways are all initiated by extracellular ligands that activate transmembrane receptors, a number of stimuli may also initiate NF-κB signaling from within the cytosol or nucleus. These include microbe-derived PAMPs that bind to nulceotide-binding domain leucine-rich repeat (NLR) proteins or RIG-I-like receptors (RLR), as well as DNA damage (Fig. 5). Other noxious stimuli, such as oxidation and endoplasmic reticulum stress, may also activate NF-κB signaling. However, the signaling components are less well characterized, thus these pathways will not be discussed here and the reader is instead referred to several excellent reviews (Brzoska and Szumiel 2009; Zhang and Kaufman 2008).
Figure 5.

NF-κB activation by intracellular receptors. Lys-48-linked ubiquitin chains are shown in red, Lys-63-linked ubiquitin chains are shown in green, SUMOylation is indicated by blue circles, and monoubiquitination is indicated by green circles. See text for additional details.
The NLR proteins are defined by a tripartite domain organization that includes an amino-terminal protein/protein interaction domain (CARD, pyrin, or BIR), a central nucleotide-binding oligomerization domain (NOD) that promotes activation-induced oligomerization, and a carboxy-terminal leucine-rich repeat (LRR) domain that is important for detection of PAMPs. Here, we discuss NOD1 and -2, the most extensively studied members of the NLR family (Shaw et al. 2008). NOD1 is widely expressed, whereas NOD2 expression is limited to intestinal Paneth cells, dendritic cells, and monocytes and macrophages. Both are activated by distinct components of bacterial cell walls that induce homo-oligomerization. These activation-induced conformational changes promote the recruitment of RIP2, followed by RIP2 ubiquitination with Lys-63-linked chains. Several ligases have been proposed for RIP2. Some reports suggest that TRAF2 and -5 promote RIP2 polyubiquitination in the NOD1 signaling pathway, whereas TRAF6 may be important for polyubiquitination of targets downstream of NOD2 (Vallabhapurapu and Karin 2009). However, another group showed that, like RIP1 in the TNFR signaling pathway, cIAP1 and -2 promote RIP2 Lys-63 polyubiqutination in the NOD1/2 pathways. Interestingly, cIAP1/2 did not seem to be redundant as in the TNFR1 signaling pathway, because ablation of either cIAP1 or cIAP2 attenuated NF-κB signaling (Bertrand et al. 2009). Lys-63 polyubiquitinated RIP2 then recruits TAK1 via associated TAB proteins, leading to I-κK activation (Reardon and Mak 2009). A20 ablation results in exaggerated NOD2 signaling, suggesting that A20 also edits polyubiquitination of NOD2 signaling components. Indeed, RIP2 polyubiquitination is markedly enhanced in A20 null cells, and A20 deubiquitinates RIP2 in vitro. Notably, mutations in A20 and in NOD2 are associated with Crohn's disease (Hitotsumatsu et al. 2008). NEMO is also ubiquitinated with Lys-63-linked chains following NOD2 activation, thereby activating I-κK. Interestingly, the NOD2 mutations that are found in Crohn's disease inhibit NEMO ubiquitination and NF-κB signaling, underscoring the importance of proper regulation of ubiquitination in NLR signaling pathways (Chen et al. 2009).
RLR family members include RIG-I, MDA5, and LGP2, all of which bind cytosolic viral RNA via helicase domains. Here, we focus on RIG-I as the prototypical RLR. Binding of viral RNA promotes association of the CARD motif of RIG-I with the CARD of MAVS, a mitochondrial-localized adaptor protein. The RIG-I/MAVS interaction is also facilitated by TRIM25, which polyubiquitinates RIG-I with Lys-63-linked chains. More specifically, a RIG-I Lys-172-Arg point mutant that cannot be ubiquitinated by TRIM25 neither interacts with MAVS nor activates NF-κB. MAVS also contains TRAF3 binding sites, which are important for IRF3 activation, and TRAF2 and -6 binding sites, which may promote IKK activation (Kawai and Akira 2008). FADD and RIP1 may also participate in RIG-I-mediated NF-κB activation downstream of MAVS (Hacker and Karin 2006). RIG-I activity is down-regulated by ubiquitin editing: CYLD depolymerizes Lys-63-linked polyubiquitin chains on RIG-I (Sun 2009) and the ligase RNF125 targets RIG-I and MAVS for proteasomal degradation (Chiu et al. 2009).
DNA damage is a potent activator of NF-κB activity, but many of the pathway components and mechanistic details are still being elucidated. Indeed, the “receptor” for DNA damage is unknown, although the leucine-rich repeats of PIDD are thought to play a role in detecting DNA damage. Modification of nuclear NEMO by PIASy with the ubiquitin-like protein SUMO also appears to be an early event in transducing DNA damage signals. NEMO SUMOylation also requires RIP and PIDD, which associate via their respective death domains, and complex with NEMO on DNA damage. SUMOylated NEMO accumulates in the nucleus and is subsequently phosphorylated by ATM, a protein kinase that is activated by DNA damage. NEMO is then monoubiquitinated by unknown enzymes on the same residues that were SUMOylated and is exported with ATM to the cytosol, where it associates with I-κK. Both NEMO monoubiquitination and ATM are required for I-κK activation, but the mechanistic details are unknown (Janssens and Tschopp 2006; Brzoska and Szumiel 2009).
CONCLUSIONS AND FUTURE PERSPECTIVES
The study of NF-κB signaling has opened up a Pandora's box, highlighting the interplay between phosphorylation and the various forms of ubiquitination that work in a concerted manner to both activate and extinguish a signaling cascade of central importance to the livelihood of metazoan animals. As we learn from the cornucopia of information that characterization of this pathway has yielded and will continue to yield, it will be important to bear in mind a number of questions. To begin, are mechanisms like ubiquitin editing more generally applicable to other signaling pathways? With the advent of ubiquitin chain-specific antibodies and improved mass spectroscopic techniques, this pressing question should be answerable. Secondly, what determines whether ubiquitin ligases such as cIAP 1/2 synthesize Lys-48- or Lys-63-linked chains? We suspect that linkage specificity is determined by the nature of the E2 enzyme, but how is docking of various E2s to the same ligase regulated? Thirdly, what are the respective roles of the RING domains in TRAFs and cIAP1/2, and why are these ligases often recruited simultaneously to proximal signaling complexes? Are the TRAFs bona fide ubiquitin ligases and if so, do they exclusively synthesize Lys-63 polyubiquitin chains? Can TRAFs polymerize Lys-63-linked chains in conjunction with E2 enzymes other than UBC13? Is it possible that the purpose of simultaneously recruiting multiple ubiquitin ligases in proximal signaling complexes is that each type of ligase predominantly polymerizes one type of ubiquitin chain? Addressing these questions by knocking down components in transformed cells or by overexpression studies may be misleading because of ineffective knockdown and the propensity of the TRAFs to promiscuously oligomerize on overexpression. Definitive answers to these important questions in a physiological context will likely require the generation of RING-mutant knock-in mice and careful analysis of any compromise in ubiquitin chain generation and in NF-κB signaling. Finally, what regulates the negative regulators like A20 and CYLD? Both DUBs may be modulated by phosphorylation (Reyes-Turcu et al. 2009; Sun 2009), but is this their only mode of regulation? Whatever the answers, the field promises to teach us much about how NF-κB, and cellular signaling in general, is regulated. Most importantly, much of what we learn is likely to have therapeutic benefits.
ACKNOWLEDGMENTS
The authors would like to thank Eugene Varfolomeev, Domagoj Vucic, and Nobuhiko Kayagaki for critical reading of the manuscript, and Allison Bruce for graphics assistance. Our apologies to our colleagues whose important contributions are not cited due to space constraints.
Footnotes
Editors: Louis M. Staudt and Michael Karin
Additional Perspectives on NF-κB available at www.cshperspectives.org
REFERENCES
- Barton GM, Medzhitov R 2004. Toll signaling: RIPping off the TNF pathway. Nat Immunol 5:472–474 [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M 2009. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30:789–801 [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30:689–700 [DOI] [PubMed] [Google Scholar]
- Bhoj VG, Chen ZJ 2009. Ubiquitylation in innate and adaptive immunity. Nature 458:430–437 [DOI] [PubMed] [Google Scholar]
- Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, et al. 2004. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 5:1052–1060 [DOI] [PubMed] [Google Scholar]
- Brzoska K, Szumiel I 2009. Signalling loops and linear pathways: NF-κB activation in response to genotoxic stress. Mutagenesis 24:1–8 [DOI] [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B 1975. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci 72:3666–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ 2009. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33:275–286 [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T 1996. Site-specific phosphorylation of IκBα by a novel ubiquitination-dependent protein kinase activity. Cell 84:853–862 [DOI] [PubMed] [Google Scholar]
- Chen G, Shaw MH, Kim YG, Nunez G 2009. NOD-like receptors: Role in innate immunity and inflammatory disease. Ann Rev Pathol 4:365–398 [DOI] [PubMed] [Google Scholar]
- Chiu YH, Zhao M, Chen ZJ 2009. Ubiquitin in NF-κB signaling. Chem Rev 109:1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. 2009. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 459:717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD 2008. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-κB activation. Mol Cell Biol 28:3538–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ 2000. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361 [DOI] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ 2006. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22:245–257 [DOI] [PubMed] [Google Scholar]
- Enesa K, Zakkar M, Chaudhury H, Luong LA, Rawlinson L, Mason JC, Haskard DO, Dean JL, Evans PC 2008. NF-κ B suppression by the deubiquitinating enzyme cezanne: A novel negative feedback loop in pro-inflammatory signaling. J Biol Chem 283:7036–7045 [DOI] [PubMed] [Google Scholar]
- Fukushima T, Matsuzawa S, Kress CL, Bruey JM, Krajewska M, Lefebvre S, Zapata JM, Ronai Z, Reed JC 2007. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci 104:6371–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker H, Karin M 2006. Regulation and function of IKK and IKK-related kinases. Sci STKE 2006:re13. [DOI] [PubMed] [Google Scholar]
- He JQ, Saha SK, Kang JR, Zarnegar B, Cheng G 2007. Specificity of TRAF3 in its negative regulation of the noncanonical NF-κ B pathway. J Biol Chem 282:3688–3694 [DOI] [PubMed] [Google Scholar]
- Heyninck K, Beyaert R 2005. A20 inhibits NF-κB activation by dual ubiquitin-editing functions. Trends Biochem Sci 30:1–4 [DOI] [PubMed] [Google Scholar]
- Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, Lee BL, Shiffin N, Advincula R, Malynn BA, et al. 2008. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 28:381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D 2006. Circuitry of nuclear factor κB signaling. Immunol Rev 210:171–186 [DOI] [PubMed] [Google Scholar]
- Ikeda F, Dikic I 2008. Atypical ubiquitin chains: New molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO reports 9:536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivins FJ, Montgomery MG, Smith SJ, Morris-Davies AC, Taylor IA, Rittinger K 2009. NEMO oligomerisation and its ubiquitin-binding properties. Biochem J 421:243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Tschopp J 2006. Signals from within: The DNA-damage-induced NF-κB response. Cell Death Differentiation 13:773–784 [DOI] [PubMed] [Google Scholar]
- Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ 2004. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 15:535–548 [DOI] [PubMed] [Google Scholar]
- Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J, Asakura Y, et al. 2009. Frequent inactivation of A20 in B-cell lymphomas. Nature 459:712–716 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S 2008. Toll-like receptor and RIG-I-like receptor signaling. Ann New York Acad Sci 1143:1–20 [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P 1998. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8:297–303 [DOI] [PubMed] [Google Scholar]
- Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. 2001. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO 20:1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A, Wallach D 2006. If the prophet does not come to the mountain: Dynamics of signaling complexes in NF-κB activation. Mol Cell 22:433–436 [DOI] [PubMed] [Google Scholar]
- Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG 2007. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I κ B kinase activation. J Biol Chem 282:4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A 2000. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 289:2350–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Shank J, Cusson N, Kelliher MA 2004. The kinase activity of Rip1 is not required for tumor necrosis factor-α-induced IκB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem 279:33185–33191 [DOI] [PubMed] [Google Scholar]
- Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, Eliezer D, Wu H 2009. Structural basis for recognition of diubiquitins by NEMO. Mol Cell 33:602–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, Yamaoka S, Seto M, Nunez G 2001. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-κB signaling pathway. J Biol Chem 276:19012–19019 [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG 2008. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc Natl Acad Sci 105:11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M 2000. Female mice heterozygous for IKK γ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell 5:969–979 [DOI] [PubMed] [Google Scholar]
- Moynagh PN 2009. The Pellino family: IRAK E3 ligases with emerging roles in innate immune signalling. Trends Immunol 30:33–42 [DOI] [PubMed] [Google Scholar]
- Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, Shifrin N, Petri MA, Kamboh MI, Manzi S, et al. 2008. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Gen 40:1062–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, et al. 2008. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134:668–678 [DOI] [PubMed] [Google Scholar]
- Ni CY, Wu ZH, Florence WC, Parekh VV, Arrate MP, Pierce S, Schweitzer B, Van Kaer L, Joyce S, Miyamoto S, Ballard DW, Oltz EM 2008. Cutting edge: K63-linked polyubiquitination of NEMO modulates TLR signaling and inflammation in vivo. J Immunol 180:7107–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak U, Rinaldi A, Kwee I, Nandula SV, Rancoita PM, Compagno M, Cerri M, Rossi D, Murty VV, Zucca E, et al. 2009. The NF-κB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 113:4918–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364 [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Wegener E, Welteke V, Ferch U, Arslan SC, Ruland J, Scheidereit C, Krappmann D 2007. Malt1 ubiquitination triggers NF-κB signaling upon T-cell activation. EMBO J 26:4634–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Smith H, Windheim M, Peggie M, Carrick E, Morrice N, Cohen P 2008. The IRAK-catalysed activation of the E3 ligase function of Pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem J 409:43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Fushman D 2004. Polyubiquitin chains: Polymeric protein signals. Curr Opin Chem Biol 8:610–616 [DOI] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. 2009. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136:1098–1109 [DOI] [PubMed] [Google Scholar]
- Reardon C, Mak TW 2009. cIAP Proteins: Keystones in NOD Receptor Signal Transduction. Immunity 30:755–756 [DOI] [PubMed] [Google Scholar]
- Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, Gaide O, Guzzardi M, Iancu EM, Rufer N, et al. 2008. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol 9:272–281 [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Wilkinson KD 2009. Polyubiquitin binding and disassembly by deubiquitinating enzymes. Chem Rev 109:1495–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, Wilkinson KD 2009. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Ann Rev Biochem 78:363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW 2000. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Develop 14:854–862 [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt E, Wegener E, Heissmeyer V, Rao A, Krappmann D 2004. Degradation of Bcl10 induced by T-cell activation negatively regulates NF-κ B signaling. Mol Cell Biol 24:3860–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauvliege R, Janssens S, Beyaert R 2007. Pellino proteins: Novel players in TLR and IL-1R signalling. J Cell Mol Med 11:453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, Klapper W, Vater I, Giefing M, Gesk S, et al. 2009. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med 206:981–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AL, Ciechanover A 2009. Targeting proteins for destruction by the ubiquitin system: Implications for human pathobiology. Ann Rev Pharmacol Toxicol 49:73–96 [DOI] [PubMed] [Google Scholar]
- Shaw MH, Reimer T, Kim YG, Nunez G 2008. NOD-like receptors (NLRs): Bona fide intracellular microbial sensors. Current Opinion Immunol 20:377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, Harhaj EW 2008. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol 9:254–262 [DOI] [PubMed] [Google Scholar]
- Shembade N, Parvatiyar K, Harhaj NS, Harhaj EW 2009. The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-κB signalling. EMBO J 28:513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Kehrl JH 2003. Tumor necrosis factor (TNF)-induced germinal center kinase-related (GCKR) and stress-activated protein kinase (SAPK) activation depends upon the E2/E3 complex Ubc13-Uev1A/TNF receptor-associated factor 2 (TRAF2). J Biol Chem 278:15429–15434 [DOI] [PubMed] [Google Scholar]
- Shu HB, Takeuchi M, Goeddel DV 1996. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci 93:13973–13978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ashwell JD 2008. IAPs: What's in a name? Mol Cell 30:123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC 2009. CYLD: A tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death Differentiation doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, Kong L, Xu L, Hilliard B, Hu S, et al. 2008. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell 133:415–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ 2004. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 14:289–301 [DOI] [PubMed] [Google Scholar]
- Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, et al. 2001. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κ B activation and protection from cell death. J Biol Chem 276:36530–36534 [DOI] [PubMed] [Google Scholar]
- Thome M 2008. Multifunctional roles for MALT1 in T-cell activation. Nat Rev Immunol 8:495–500 [DOI] [PubMed] [Google Scholar]
- Ting AT, Pimentel-Muinos FX, Seed B 1996. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J 15:6189–6196 [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, et al. 2009. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol 11:123–132 [DOI] [PubMed] [Google Scholar]
- Uren AG, O'Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM 2000. Identification of paracaspases and metacaspases: Two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 6:961–967 [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M 2009. Regulation and function of NF-κB transcription factors in the immune system. Ann Rev Immunol 27:693–733 [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, Vignali DA, Bergsagel PL, Karin M 2008. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat immunol 9:1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E, Vucic D 2008. (Un)expected roles of c-IAPs in apoptotic and NFκB signaling pathways. Cell Cycle 7:1511–1521 [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D 2008. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J Biol Chem 283:24295–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R 2008. TLR-4, IL-1R and TNF-R signaling to NF-κB: Variations on a common theme. Cell Mol Life Sci 65:2964–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach D, Kovalenko A 2008. Self-termination of the terminator. Nat Immunol 9:1325–1327 [DOI] [PubMed] [Google Scholar]
- Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y 2008. TRAF6 autoubiquitination-independent activation of the NFκB and MAPK pathways in response to IL-1 and RANKL. PloS one 3:e4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430:694–699 [DOI] [PubMed] [Google Scholar]
- Wu CJ, Ashwell JD 2008. NEMO recognition of ubiquitinated Bcl10 is required for T cell receptor-mediated NF-κB activation. Proc Natl Acad Sci 105:3023–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Tschopp J, Lin SC 2007. Smac mimetics and TNFα: A dangerous liaison? Cell 131:655–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD 2006. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation [corrected]. Nat Cell Biol 8:398–406 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Okamoto T, Takeda K, Sato S, Sanjo H, Uematsu S, Saitoh T, Yamamoto N, Sakurai H, Ishii KJ, et al. 2006a. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol 7:962–970 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Saitoh T, Sakurai H, Uematsu S, Kawai T, Ishii KJ, Takeuchi O, Akira S 2006b. Cutting Edge: Pivotal function of Ubc13 in thymocyte TCR signaling. J Immunol 177:7520–7524 [DOI] [PubMed] [Google Scholar]
- Yamin TT, Miller DK 1997. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem 272:21540–21547 [DOI] [PubMed] [Google Scholar]
- Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, et al. 1997. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7:715–725 [DOI] [PubMed] [Google Scholar]
- Yin Q, Lin SC, Lamothe B, Lu M, Lo YC, Hura G, Zheng L, Rich RL, Campos AD, Myszka DG, et al. 2009. E2 interaction and dimerization in the crystal structure of TRAF6. Nat Struct Mol Biol 16:658–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, et al. 2008. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 9:1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM 2004. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 427:167–171 [DOI] [PubMed] [Google Scholar]