

Abstract
Traumatic brain injury (TBI) is a major cause of neurological disability across all ages, but the elderly are particularly vulnerable and have a worse prognosis than younger individuals. To advance the understanding of long-term pathogenesis induced by TBI in the elderly, aged mice (21–24 months) were given a controlled cortical impact (CCI) injury to the sensorimotor cortex, and their brains were analyzed by MRI and histopathology at 1 and 2 months after CCI injury, a post-acute period. A T2 hypointensity was observed in the ipsilateral thalamus but not in the contralateral thalamus or in the thalamus of sham operated, control mice. The hypointensity was colocalized with increased histochemical staining of iron, a paramagnetic substance that causes a shortening of the T2 relaxation time. Since iron catalyzes reactions that lead to toxic free radicals, the deposition of iron in the thalamus raises the possibility that it promotes pathogenesis following TBI. Astrocyte gliosis and microgliosis were also observed in the ipsilateral thalamus in the post-acute period. The ipsilateral internal capsule displayed a trend for a T2 hypointensity, however, unlike the thalamus it did not have an increase of iron or GFAP staining, but it did have evidence of microgliosis. In summary, areas of T2 hypointensity were revealed in both the thalamus and internal capsule during the post-acute period following CCI injury, but the underlying pathology appeared to be distinct between these regions.
Keywords: T2 hypointensity, iron, traumatic brain injury, GFAP, Iba1
Traumatic brain injury (TBI) is a leading cause of neurological disability across all age categories. The elderly, whose numbers are expected to double in the United States from 2000 to 2030 [17], are more likely to experience TBI, and they have a worse prognosis, than younger individuals [9, 28]. There is a lack of effective interventions following TBI, and advancing the understanding of neuropathological changes that are triggered by TBI has the potential to reveal new therapeutic targets. During the post-acute period following TBI, e.g., when patients have left the hospital and are undergoing outpatient therapy, ongoing pathogenesis is possibly of critical importance to long-term patient outcome [4] and yet has received very little study in animal models. A potentially important treatment target is iron due to its ability to act as a catalyst of redox reactions that lead to the production of toxic free radicals. CNS levels of iron increase with age as well as in a variety of neurological diseases [35], and in multiple sclerosis there is evidence that increases in iron are associated with brain atrophy or cognitive impairment [3, 5]. However, the role of iron in the pathogenesis of TBI is poorly understood.
The objectives of this study were to determine whether abnormal iron deposits develop following TBI in aged subjects, and to determine whether these deposits are associated with ongoing inflammatory processes. In order to investigate responses in aged subjects, 21–24 month old male C57BL/6 mice, obtained from the National Institute on Aging colonies, were utilized. These mice were given a controlled cortical impact (CCI) injury to the sensorimotor cortex, or a sham operation as described previously [25, 26]. In brief, a moderate CCI injury was induced in the sensorimotor cortex (AP = 0, ML = +2.0 from bregma) using a flat-faced, 3 mm diameter rounded tip with a strike velocity of 1.5 m/s, a 1 mm depth of impact and a contact time of 85 msec.
At a post-acute period (1 or 2 months), mice received MRI as previously described [25, 26]. Briefly, animals were scanned with a 9.4T Varian INOVA horizontal MRI scanner with a 31cm room temperature bore (Varian Inc., Palo Alto, CA) using a 400mT/m gradient coil set. Given the small size of mouse brains, approximately 10mm × 16mm × 6mm, and the desire for high resolution, an inductively coupled surface coil was used. Anatomically coronal images were acquired with TR=2500ms, TE= 45ms, 0.5mm slice thinkness, 16 × 16 mm field of view. Analysis was performed with NIH Image J to measure mean intensities in 0.244 mm2 regions of interest in the thalamus and internal capsule ispilateral to the injury and also at the corresponding contralateral locations. No intensity normalization nor homogeneity correction techniques were applied.
Animals were sacrificed and tissue was processed according to Onyszchuk et al. [26]. To localize iron deposition within CNS structures, iron histochemical staining was performed as described previously [19] except that the dehydration, xylenes and proteinase K steps were eliminated from the procedure. Astrocytes and microglia were stained by immunohistochemistry against glial fibrillary acidic protein (GFAP) and ionized calcium-binding adapter molecule 1 (Iba1), respectively [29]. Image analysis was performed on coronal sections from levels that corresponded to the locations of the T2 hypointensity (see Table 1). For each marker, the analyzed sections were stained at the same time, and thus, were processed with identical staining conditions. The NIH ImageJ program was used to measure staining density, rather than counting cell numbers, as abnormal iron deposits were not restricted to cell bodies. Image analysis of staining density was also utilized for immunohistochemical staining as microglia undergo hypertrophy and astrocytes undergo pronounced upregulation of GFAP expression as well as hypertrophy following CCI [23], and we have found that staining density is a consistent measure of glial activation following CCI [26]. Statistical analyses were performed using the Wilcoxon two sample test, the Wilcoxon matched-pairs signed-ranks test, and the Student-t distribution. All procedures involving animals were approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
Table 1.
Relative Staining in Aged Mice with CCI Injury or Sham Operation
| Measure | Structure | Group | n | Contra:Ipsilateral1 | p value2 |
|---|---|---|---|---|---|
| T2 hypointensities3 | Thalamus | Sham | 3 | 1.014 ± 0.020 s.d. | 0.024 |
| CCI injured4 | 6 | 1.355 ± 0.134 s.d. | |||
| Internal Capsule | Sham | 3 | 1.116 ± 0.078 s.d. | 0.095 | |
| CCI injured 4 | 6 | 1.318 ± 0.190 s.d. | |||
| Iron histochemistry5 | Thalamus | Sham | 7 | 1.000 ± 0.024 s.d. | 0.001 |
| CCI injured 4 | 7 | 1.564 ± 0.142 s.d. | |||
| Internal Capsule | Sham | 7 | 0.960 ± 0.078 s.d. | 0.710 | |
| CCI injured | 7 | 0.945 ± 0.092 s.d. | |||
| Iba16 | Thalamus | Sham | 8 | 1.006 ± 0.007 s.d. | 0.015 |
| CCI injured 4 | 7 | 1.087 ± 0.056 s.d. | |||
| Internal Capsule | Sham | 8 | 0.995 ± 0.022 s.d. | 0.035 | |
| CCI injured 4 | 7 | 1.096 ± 0.045 s.d. | |||
| GFAP6 | Thalamus | Sham | 8 | 1.006 ± 0.023 s.d. | 0.035 |
| CCI injured 4 | 7 | 1.067 ± 0.033 s.d. | |||
| Internal Capsule | Sham | 8 | 1.010 ± 0.052 s.d. | 0.752 | |
| CCI injured | 7 | 1.057 ± 0.057 s.d. |
A ratio greater than 1 indicates that the ipsilateral structure was darker than the contralateral structure which is due to a greater hypointensity (MRI) or darker staining (iron histochemistry, Iba1, and GFAP) in the former structure.
Two-tailed Wilcoxon two sample test, comparing CCI injured with sham ratios.
Mean intensities measured from T2-weighted MR images in 0.244 mm2 square regions at center of ipsilateral hypointensity and at corresponding contralateral location.
Ipsilateral structure was significantly darker than the contralateral structure. The Wilcoxon matched-pairs signed-ranks test, significance p ≤ 0.05.
Mean brightness intensities of two areas (210 μm diameter circles) with maximal staining in the ipsilateral structure and two corresponding areas in the contralateral structure.
Mean brightness intensities of two areas (150 μm × 150 μm squares) with maximal staining in the ipsilateral structure and two corresponding areas in the contralateral structure.
Large areas of increased signal intensity on T2-weighted images (T2 hyperintensity), corresponding to the lesion, were observed in the cortex ipsilateral to the CCI and these extended into the hippocampus (Fig. 1A, B) similar to that previously described [26]. In contrast, areas of lower signal intensity (T2 hypointensity) were visible by eye in the thalamus and internal capsule of the ipsilateral hemisphere (Fig. 1A, B). Quantitative image analysis revealed that the ipsilateral thalamus in injured animals had a greater T2 hypointensity (darker) than the contralateral thalamus or sham-operated animals (Table 1). T2 hypointensity was not observed in the thalamus at acute time points, e.g., 7 days or earlier (unpublished observations). Some noticeable signal reductions in the ipsilateral internal capsule of injured animals were observed, but while these were statistically darker than those in the contralateral internal capsule they were not significantly different from sham animals (Table 1).
Fig. 1.
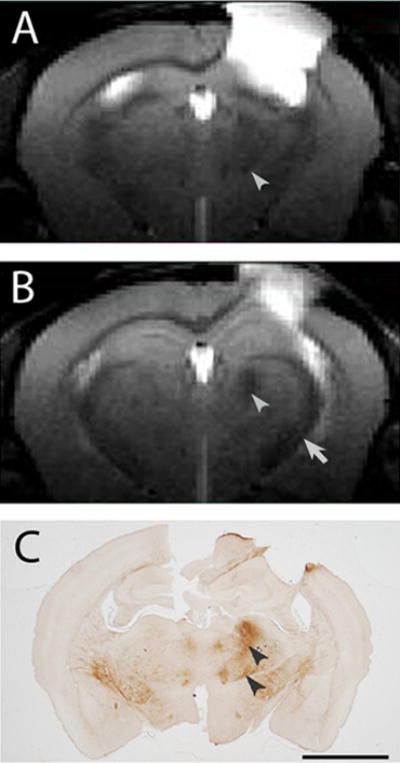
A, B) T2 weighted images, 0.5 mm thick, coronal, and centered at −1.0 mm (A) and −1.5 mm (B) relative to Bregma from an aged C57BL/6 mouse (~24 months) given a CCI injury 2 months previously. The primary injury site appears as a hyperintensity in the sensorimotor cortex. T2 hypointense areas are present in the ipsilateral thalamus (arrowheads) and ipsilateral internal capsule (arrow) relative to the contralateral structures. C) Abnormal iron deposits (arrowheads), revealed by iron histochemistry, co-localize to the T2 hypointensity in the thalamus (the same animal as for A, B). Bar = 2 mm (A–C).
Iron histochemistry revealed a high staining density in similar ipsilateral thalamic locations as the T2 hypointensity of animals that received a CCI and there was a low density of iron staining in the contralateral thalamus (Fig. 1A–C) or in sham operated, control mice, which did not have a T2 hypointensity (correlation coefficient of contralateral:ipsilateral ratio values between T2 and iron deposits = 0.694; p ≤ 0.062). There was a high density of punctate labeling in the ipsilateral thalamus but punctate deposits were not observed in the contralateral thalamus (Fig. 2A, B) nor in the thalamus of sham operated animals. At the margins of thalamic areas with punctate deposits, labeled cells could be observed with morphologies of degenerating oligodendrocytes or ameboid microglia (Fig. 2C). Generally, the density of punctate deposits was less at the margins of these areas and the punctate structures were not generally concentrated in oligodendrocytes (Fig. 2F). In the contralateral thalamus, labeling of oligodendrocytes and myelin (Fig. 2A) was similar to that seen in the thalamus of sham-operated animals (not shown). Image analysis revealed that the iron staining density in the ipsilateral thalamus was significantly greater than in the contralateral thalamus in CCI-injured animals as well as to staining in the equivalent structure in the sham-operated animals (Table 1).
Fig. 2.

A) Oligodendrocytes and myelin are labeled by iron histochemical staining in the contralateral thalamus relative to the CCI injury. B) A high density of punctate iron deposits is present in the ipsilateral thalamus. C) Degenerating oligodendrocytes and ameboid microglial cells, located at the periphery of areas with a high density of punctate labeling, are stained by iron histochemistry. D) Iron histochemistry reveals labeled reactive microglia in the subcortical white matter and in the overlying cortex. E) Reactive microglial cells in the cortex labeled by iron histochemistry. F) A high power, Normarski field illustrating that most of the punctate labeling is not present within stained oligodendrocytes. G, H) Iba1 immunohistochemistry reveals microglia in the contralateral thalamus (G) and a pronounced microgliosis in the ipsilateral thalamus (H). I, J) GFAP immunohistochemistry reveals astrocytes in the contralateral thalamus (I) and astrocyte gliosis in the ipsilateral thalamus (J). Bars = 40 μm (A–C), 80 and 40 μm (D, E), 10 μm (F), 50 μm (G, H), and 50 μm (I, J).
In general, the histochemical staining of iron in the ipsilateral internal capsule was similar to that in the contralateral internal capsule and to that in sham-operated animals as determined by visual inspection and image analysis (Table 1). However, pathological iron deposits were occasionally observed in other CNS regions, such as within reactive microglia in the subcortical white matter or cortex (Fig. 2D, E).
In the post-acute period following CCI injury, both Iba1 and GFAP immunohistochemical staining were significantly greater in the ipsilateral thalamus of injured mice than in the contralateral thalamus of injured animals or in the thalamus of sham-operated animals (Fig. 2G–J; Table 1). In injured animals, Iba1 immunohistochemical staining was significantly greater in the ipsilateral internal capsule than in the contralateral internal capsule and sham-operated animals (Table 1). GFAP immunohistochemical staining in the internal capsule was more variable than for Iba1, with no statistical differences between the ipsilateral and contralateral internal capsule in CCI-injured animals or between CCI-injured and sham animals (Table 1).
Examination of thionin stained sections from the level that corresponded to the T2 hypointensity revealed a paucity of neurons in the ipsilateral thalamus of the CCI injured mice compared to the contralateral thalamus (Fig. 3) or to the thalamus of sham operated animals.
Fig. 3.
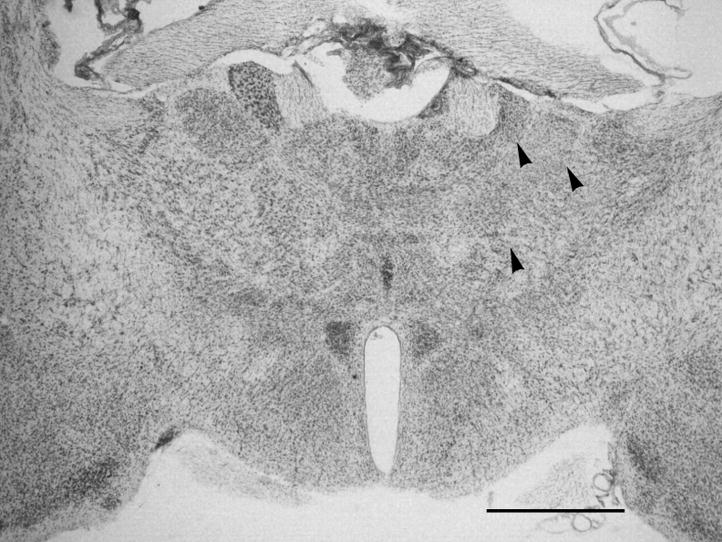
A thionin stained section from an aged mouse at 60 days following CCI at the level corresponding to T2 weighted images as well as to the level of sections used for iron histochemistry and GFAP and Iba1 immunohistochemical staining. Note the lower density of neurons in ipsilateral thalamic nuclei (arrowheads) compared to the contralateral thalamus. Bar = 1 mm.
In a prior study, thalamic T2 hypointensity observed in rats following CCI or fluid percussion injury was associated with a reduction in neuronal cell numbers and an increase in astrocyte and microglial immunohistochemical staining [24]. In the present study, we confirm the T2, neuronal and glial findings and make several extensions. Foremost, we show that the distribution of iron deposits in the thalamus corresponds anatomically with the T2 hypointensity in aged mice receiving CCI. Iron deposits and T2 hypointensity were also observed in the ipsilateral thalamus of adult 6 month old mice receiving CCI (not shown), but the results were comparable to those for aged mice receiving CCI indicating that age was not a determinant in the development of the pathological features. Increases in iron levels are thought to account for a T2 hypointensity in other neurological disorders [6, 30, 36], and it is likely that iron deposits are a significant contributor to the T2 hypointensity in the thalamus following TBI. Increased iron staining could be due to a variety of sources: hemorrhage, increased iron uptake from the blood or CSF, or neuronal or oligodendrocyte death leading to the release of tightly bound iron from sites where it is normally utilized (e.g., mitochondria). Hemorrhage is a questionable source of the iron deposits in the thalamus during the post-acute CCI injury since hemorrhage is readily observed in other structures, e.g., cortex and internal capsule in the fluid percussion injury model [15], but not the thalamus. Increased iron uptake into the brain has been suggested for other neurological disorders [18], and it is a plausible source of elevated iron levels in the thalamus following TBI. Elevated uptake would be expected to give rise to a concentration of staining around vessels [34], but this was not the case in the mice receiving a CCI injury in this study. A more likely explanation is the release of tightly bound iron from sites where it is normally utilized. The punctate deposits are suggestive of a degenerative process. This is consistent with thalamic neurons undergoing degeneration following CCI [26] or fluid percussion injury [15], and in the present study there was a paucity of neurons in the ipsilateral thalamus of the CCI injured mice compared to contralateral thalamus or sham control animals at the level that corresponded to the T2 hypointensity. Other models of cortical injury also have shown retrograde degeneration of neurons in the thalamus in mice and rats [1, 22]. Thalamic neurons rapidly die off by 1 week following cortical injury with only one-third of normal levels of neurons remaining [1, 22]. By 1 and 2 months after injury the number of remaining neurons in the rat is 28% and 20%, respectively, and this loss slowly progresses so that only 17% of the neurons are still present by 24 weeks [1]. It is plausible that an accumulation of abnormal iron deposits in the thalamus contributes to the progressive loss of neurons at these later time points and this could counteract recovery efforts especially in aged subjects where motor improvement is slower than for younger subjects.
Although much of the stainable iron in the normal brain appears predominantly in oligodendrocytes [31], and degenerating oligodendrocytes and myelin likely contribute to the abnormal punctuate staining, the large increase of iron deposition that is stained in the thalamus following CCI is likely due, at least in part, to the release of iron from degenerating neurons, which are the prevalent cell type within nuclei of the thalamus. Iron bound to heme is generally not thought to be stained by the Perls iron histochemical procedure [21], although some iron associated with heme is apparently accessible to this staining method since red blood cells [11] and degenerating mitochondria in Gomori-positive astrocytes [33] can be stained. Heme, which is utilized by many enzymes, is broken down by heme oxygenase yielding carbon monoxide, biliverdin, and free iron. The expression of heme oxygenase is elevated in the thalamus following TBI [32]. Thus, as thalamic neurons undergo degeneration [26] iron bound to heme can be released and transition from an suboptimal stainable state (i.e., bound within heme) to a readily stainable one (i.e., loosely associated with other molecules), which could account for the increase in stained iron in the present study. In addition, the freed iron would have greater access to protons and water and thus have greater ability to affect the T2 relaxation signal leading to a hypointensity. Also, free or loosely bound iron is more likely to catalyze redox reactions that produce toxic free radicals that can promote pathology [13], and reactive oxygen species appear to be involved in the pathogenesis of TBI [16]. Thus, iron chelation treatment might have value to lessen the secondary injury of TBI. The iron chelator deferoxamine was shown to improve spatial memory performance following TBI, but not some pathological measures of disease [20]. Dextran-coupled deferoxamine has a longer plasma half-life than deferoxamine and its administration was found to allow for an improved grip test compared to the administration of deferoxamine or dextran alone following head injury in mice [27]. However, deferiprone, an iron chelator that crosses the blood-brain barrier [12] and ameliorates iron-catalyzed oxidative damage effects in other experimental conditions [2, 8, 10, 14], has the potential to lessen pathology within the CNS following TBI to a greater degree than deferoxamine or dextran-deferoxamine.
Unlike in the thalamus, iron staining was not increased in the ipsilateral internal capsule compared to the contralateral internal capsule of CCI injured animals, despite the appearance of greater T2 hypointensity in the ipsilateral internal capsule compared to the contralateral internal capsule. However, there was evidence of an increased microglial response in the ipsilateral internal capsule compared to the contralateral internal capsule. These data suggest that distinct pathological processes are at work between these different structures and they indicate that neuronal loss is a key determinant of the deposition of iron in a CNS region, e.g., thalamus, during the post-acute period.
In summary, CCI injury to the sensorimotor cortex led to the accumulation of abnormal iron deposits and increased astrocyte and microglial staining in the thalamus that corresponded spatially with a thalamic T2 hypointensity. These changes, observed at one and two months post-injury, are suggestive of ongoing, post-acute pathological processes. The continued presence of activated astrocytes and reactive microglia well after the primary injury has taken place, in areas removed from the site of primary injury, implicates chronic neuroinflammation in the long-term damage process, consistent with previous animal [29] and human [7] studies. Chronic neuroinflammation may act as a contributor to the elevated iron deposits and/or as a response to the elevated iron levels. Since iron is a catalyst for redox reactions that can lead to the production of free radicals that induce tissue damage, further studies are warranted to examine the role of abnormal iron deposits and chronic inflammation on the pathology that develops from TBI.
Acknowledgments
We thank Dr. Yong-Yue He for assistance with the CCI procedures. The study was supported in part by the Steve Palermo Endowment and grants from the National Institutes of Health (R21 AG026482, R01 NS039123, P20 RR016475 and P30 NICHD HD 02528). The Hoglund Brain Imaging Center is supported by C76 HF00201.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Agarwala S, Kalil RE. Axotomy-induced neuronal death and reactive astrogliosis in the lateral geniculate nucleus following a lesion of the visual cortex in the rat. J Comp Neurol. 1998;392:252–263. doi: 10.1002/(sici)1096-9861(19980309)392:2<252::aid-cne7>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 2.Barnabe N, Zastre JA, Venkataram S, Hasinoff BB. Deferiprone protects against doxorubicin-induced myocyte cytotoxicity. Free Radic Biol Med. 2002;33:266–275. doi: 10.1016/s0891-5849(02)00873-0. [DOI] [PubMed] [Google Scholar]
- 3.Bermel RA, Puli SR, Rudick RA, Weinstock-Guttman B, Fisher E, Munschauer FE, 3rd, Bakshi R. Prediction of longitudinal brain atrophy in multiple sclerosis by gray matter magnetic resonance imaging T2 hypointensity. Arch Neurol. 2005;62:1371–1376. doi: 10.1001/archneur.62.9.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bramlett HM, Dietrich WD. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125–141. doi: 10.1016/S0079-6123(06)61009-1. [DOI] [PubMed] [Google Scholar]
- 5.Brass SD, Benedict RH, Weinstock-Guttman B, Munschauer F, Bakshi R. Cognitive impairment is associated with subcortical magnetic resonance imaging grey matter T2 hypointensity in multiple sclerosis. Mult Scler. 2006;12:437–444. doi: 10.1191/135248506ms1301oa. [DOI] [PubMed] [Google Scholar]
- 6.Brass SD, Chen NK, Mulkern RV, Bakshi R. Magnetic resonance imaging of iron deposition in neurological disorders. Top Magn Reson Imaging. 2006;17:31–40. doi: 10.1097/01.rmr.0000245459.82782.e4. [DOI] [PubMed] [Google Scholar]
- 7.Brooks WM, Stidley CA, Petropoulos H, Jung RE, Weers DC, Friedman SD, Barlow MA, Sibbitt WL, Jr, Yeo RA. Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J Neurotrauma. 2000;17:629–640. doi: 10.1089/089771500415382. [DOI] [PubMed] [Google Scholar]
- 8.Chenoufi N, Hubert N, Loreal O, Morel I, Pasdeloup N, Cillard J, Brissot P, Lescoat G. Inhibition of iron toxicity in rat and human hepatocyte cultures by the hydroxypyridin-4-ones CP20 and CP94. J Hepatol. 1995;23:166–173. doi: 10.1016/0168-8278(95)80331-9. [DOI] [PubMed] [Google Scholar]
- 9.Coronado VG, Thomas KE, Sattin RW, Johnson RL. The CDC traumatic brain injury surveillance system: characteristics of persons aged 65 years and older hospitalized with a TBI. J Head Trauma Rehabil. 2005;20:215–228. doi: 10.1097/00001199-200505000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Eybl V, Kotyzova D, Kolek M, Koutensky J, Nielsen P. The influence of deferiprone (L1) and deferoxamine on iron and essential element tissue level and parameters of oxidative status in dietary iron-loaded mice. Toxicol Lett. 2002;128:169–175. doi: 10.1016/s0378-4274(01)00541-0. [DOI] [PubMed] [Google Scholar]
- 11.Forge JK, Pedchenko TV, LeVine SM. Iron deposits in the central nervous system of SJL mice with experimental allergic encephalomyelitis. Life Sci. 1998;63:2271–2284. doi: 10.1016/s0024-3205(98)00512-8. [DOI] [PubMed] [Google Scholar]
- 12.Fredenburg AM, Sethi RK, Allen DD, Yokel RA. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1′ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology. 1996;108:191–199. doi: 10.1016/0300-483x(95)03301-u. [DOI] [PubMed] [Google Scholar]
- 13.Gaasch JA, Lockman PR, Geldenhuys WJ, Allen DD, Van der Schyf CJ. Brain iron toxicity: differential responses of astrocytes, neurons, and endothelial cells. Neurochem Res. 2007;32:1196–1208. doi: 10.1007/s11064-007-9290-4. [DOI] [PubMed] [Google Scholar]
- 14.Glickstein H, El RB, Link G, Breuer W, Konijn AM, Hershko C, Nick H, Cabantchik ZI. Action of chelators in iron-loaded cardiac cells: Accessibility to intracellular labile iron and functional consequences. Blood. 2006;108:3195–3203. doi: 10.1182/blood-2006-05-020867. [DOI] [PubMed] [Google Scholar]
- 15.Graham DI, Raghupathi R, Saatman KE, Meaney D, McIntosh TK. Tissue tears in the white matter after lateral fluid percussion brain injury in the rat: relevance to human brain injury. Acta Neuropathol. 2000;99:117–124. doi: 10.1007/pl00007414. [DOI] [PubMed] [Google Scholar]
- 16.Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- 17.He SMW, Velkoff V, DeBarros K. 65+ in the United States. Current Population Reports U.S. Census. 2005:P23–209. [Google Scholar]
- 18.Ke Y, Qian ZM. Brain iron metabolism: neurobiology and neurochemistry. Prog Neurobiol. 2007;83:149–173. doi: 10.1016/j.pneurobio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 19.LeVine SM. Oligodendrocytes and myelin sheaths in normal, quaking and shiverer brains are enriched in iron. J Neurosci Res. 1991;29:413–419. doi: 10.1002/jnr.490290317. [DOI] [PubMed] [Google Scholar]
- 20.Long DA, Ghosh K, Moore AN, Dixon CE, Dash PK. Deferoxamine improves spatial memory performance following experimental brain injury in rats. Brain Res. 1996;717:109–117. doi: 10.1016/0006-8993(95)01500-0. [DOI] [PubMed] [Google Scholar]
- 21.Meguro R, Asano Y, Odagiri S, Li C, Iwatsuki H, Shoumura K. Nonheme-iron histochemistry for light and electron microscopy: a historical, theoretical and technical review. Arch Histol Cytol. 2007;70:1–19. doi: 10.1679/aohc.70.1. [DOI] [PubMed] [Google Scholar]
- 22.Muessel MJ, Klein RM, Wilson AM, Berman NE. Ablation of the chemokine monocyte chemoattractant protein-1 delays retrograde neuronal degeneration, attenuates microglial activation, and alters expression of cell death molecules. Brain Res Mol Brain Res. 2002;103:12–27. doi: 10.1016/s0169-328x(02)00158-4. [DOI] [PubMed] [Google Scholar]
- 23.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 24.Obenaus A, Robbins M, Blanco G, Galloway NR, Snissarenko E, Gillard E, Lee S, Curras-Collazo M. Multi-modal magnetic resonance imaging alterations in two rat models of mild neurotrauma. J Neurotrauma. 2007;24:1147–1160. doi: 10.1089/neu.2006.0211. [DOI] [PubMed] [Google Scholar]
- 25.Onyszchuk G, Al-Hafez B, He YY, Bilgen M, Berman NE, Brooks WM. A mouse model of sensorimotor controlled cortical impact: characterization using longitudinal magnetic resonance imaging, behavioral assessments and histology. J Neurosci Methods. 2007;160:187–196. doi: 10.1016/j.jneumeth.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onyszchuk G, He YY, Berman NE, Brooks WM. Detrimental effects of aging on outcome from traumatic brain injury: a behavioral, magnetic resonance imaging, and histological study in mice. J Neurotrauma. 2008;25:153–171. doi: 10.1089/neu.2007.0430. [DOI] [PubMed] [Google Scholar]
- 27.Panter SS, Braughler JM, Hall ED. Dextran-coupled deferoxamine improves outcome in a murine model of head injury. J Neurotrauma. 1992;9:47–53. doi: 10.1089/neu.1992.9.47. [DOI] [PubMed] [Google Scholar]
- 28.Rutland-Brown W, Langlois JA, Thomas KE, Xi YL. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehabil. 2006;21:544–548. doi: 10.1097/00001199-200611000-00009. [DOI] [PubMed] [Google Scholar]
- 29.Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol. 2008:372–380. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takeuchi Y, Yoshikawa M, Tsujino T, Kohno S, Tsukamoto N, Shiroi A, Kikuchi E, Fukui H, Miyajima H. A case of aceruloplasminaemia: abnormal serum ceruloplasmin protein without ferroxidase activity. J Neurol Neurosurg Psychiatry. 2002;72:543–545. doi: 10.1136/jnnp.72.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: The role of iron. Glia. 2008 doi: 10.1002/glia.20784. [DOI] [PubMed] [Google Scholar]
- 32.Yi JH, Hazell AS. N-acetylcysteine attenuates early induction of heme oxygenase-1 following traumatic brain injury. Brain Res. 2005;1033:13–19. doi: 10.1016/j.brainres.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 33.Young JK, McKenzie JC. GLUT2 immunoreactivity in Gomori-positive astrocytes of the hypothalamus. J Histochem Cytochem. 2004;52:1519–1524. doi: 10.1369/jhc.4A6375.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zamboni P. The big idea: iron-dependent inflammation in venous disease and proposed parallels in multiple sclerosis. J R Soc Med. 2006;99:589–593. doi: 10.1258/jrsm.99.11.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Zabad RK, Wei X, Metz LM, Hill MD, Mitchell JR. Deep grey matter “black T2” on 3 tesla magnetic resonance imaging correlates with disability in multiple sclerosis. Mult Scler. 2007;13:880–883. doi: 10.1177/1352458507076411. [DOI] [PubMed] [Google Scholar]