

Abstract
Progesterone acting through two isoforms of the progesterone receptor (PR), PRA and PRB, regulates proliferation and differentiation in the normal mammary gland in mouse, rat and human. Progesterone and PR have also been implicated in the etiology and pathogenesis of human breast cancer. The focus of this review is on recent advances in understanding the role of the PR isoform specific functions in the normal breast and in breast cancer. Also discussed is information obtained from rodent studies and their relevance to our understanding of the role of progestins in breast cancer etiology.
Keywords: progesterone, progesterone receptor, breast, breast cancer, animal models
I. INTRODUCTION
Reproductive factors, such as an early onset of menarche, an early age at the first full term pregnancy, a late onset of menopause, and long-term menopausal hormone therapy significantly increase or decrease breast cancer risk.1–6 Although the exact mechanisms underlying these risk factors are not known, the ovarian hormones, estrogen (E) and progesterone (P), are believed to play an important role in the etiology of breast cancer. It is hypothesized that overall lifetime exposure to estrogen is a crucial factor increasing breast cancer risk due to the proliferative effects of estrogen on the breast.7 However, recent studies have shown that progestins, used in combination with estrogens in menopausal hormone replacement therapy (HRT), increase breast risk, whereas estrogen alone HRT is not associated with increased breast cancer risk.8, 9 Understanding the mechanisms of P action in the breast is particularly important since progestins are widely used not only in HRT, but also in contraceptives and for suppression of ovarian function in the treatment of certain pathological conditions.10, 11 In the breast, progestin action is conveyed by two isoforms of the progesterone receptor (PR), PRA and PRB. In the present review we will focus on current knowledge of progesterone receptor isoform structures, functions, expression, and the molecular mechanisms of PR action in normal human breast and in breast cancer. Information obtained from rodent studies and their relevance to our understanding of the role of progestins in breast cancer etiology is also discussed.
II. STRUCTURE AND REGULATION OF PROGESTERONE RECEPTOR
A. Progesterone Receptor Structure
P mediates its effects through binding to its cognate receptor, the PR, which is a member of the nuclear steroid receptor family.12 The PR exists as two isoforms, PRA and PRB, which are expressed from a single gene in both humans and rodents. Translation of PRA and PRB protein initiates at two distinct AUG signals, with PRB containing an amino terminal extension of 164 amino acids. The PR is made up of a central DNA binding domain (DBD) and a carboxy-terminal ligand binding domain. In addition, it contains multiple activation function (AF) and inhibitory elements which enhance or repress transcriptional activation by association of these domains with transcriptional coregulators.13 The unique region of PRB contains a transcription activation domain, AF3, in addition to AF1 and AF2 that are common to PRA.14 Upon ligand binding, the PR undergoes a conformational change and dimerization which leads to the recruitment of specific coregulators and general transcription factors. Ligand bound PR dimerizes to form homo- or heterodimers with 3 classes possible: A:A, A:B or B:B. The activated PR complex binds to progestin response elements (PRE) in the promoters of target genes, resulting in the modulation of transcription of those genes. PRA and PRB are expressed in the normal mammary glands of humans, rats, and mice and in human breast cancers.
There is also a third, less studied progesterone receptor isoform, PRC.15 PRC is an N-terminally truncated 60 kDa protein that contains sequences for hormone binding, dimerization, and nuclear localization, but lacks the first zinc finger of the DBD.15 Wei et al. have reported that in T47D breast cancer cells PRC interacts with PRB and can increase the transcriptional activity of both PRA and PRB.16, 17 In contrast, Condon et al. have reported an inhibitory role for PRC in the uterus.18 They propose that PRC expression in the uterus inhibits PRB transactivation leading to a loss of uterine quiescence and the onset of labor. The expression and role of PRC in the normal mammary gland and in breast cancer remains to be determined.
In the human 19 and rat 20 PR genes, a promoter specific for PRA and another specific for PRB have been identified. However, it is not known if there are two promoters in the mouse as well. Most of what is known about the PR promoter has been learned through examination of the human PR gene in vitro in cell lines. E upregulates expression of PR in the MCF-7 breast cancer cell line.21, 22 However neither the PRA nor PRB promoters contain a canonical estrogen response element (ERE).19 Examination of the PR promoter has identified two Sp1 sites in the −80/−34 region,23 a +90 AP-1 site,24 and an ERE half site with two adjacent Sp1 sites from +571 to +595,25, 26 that confer estrogen receptor (ER)-mediated E responsiveness to the PR gene. Interestingly, an AP-1 site at +745 decreases E-mediated transcription of the PR gene, in contrast to the +90 AP-1 site that increases transcription of PR.27 These elements of the PR promoter are fairly well conserved across species, but given the extensive use of the mouse as a model for PR expression and P action, future studies to address the regulation of the mouse PR promoter are warranted.
B. Regulation of Progesterone Receptor Expression
There is limited information on regulation of the progesterone receptor in the normal human breast. In the postmenopausal breast, PR expression is increased by HRT with E relative to non-HRT users (Fig. 1).5 However, the regulation of PR isoforms was not examined in that study. Studies in breast cancer cell lines examining the regulation of PR isoform expression by hormones have produced conflicting results. Preferential stimulation of PRB by E and downregulation of both PR isoforms by treatment with P has been shown in T47D breast cancer cells.28 However, a later report has shown that E can stimulate both PRA and PRB in T47D cells, and additionally that E preferentially upregulates PRA in MCF-7 cells, while PRB is preferentially upregulated by E in ZR-75-1 cells.29 From these studies one can conclude that E upregulates PR expression in breast cancer cells; however, how E regulates PR isoforms expression in the normal breast or in vivo in breast cancer is not well understood.
Figure 1.
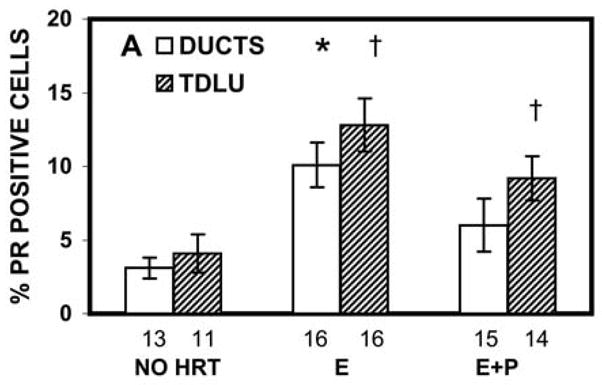
Effects of HRT on PR level in breast tissue of postmenopausal women. The number under each bar represents the number of individuals for whom ducts or TDLU could be analyzed. *, P = 0.002–0.04 that the percentage of PR-positive cells in the ducts of the E-alone group was significantly greater than in the ducts of the no-HRT or E+P groups; †, P = 0.0007 and 0.04, respectively, that the percentages of PR-positive cells in the TDLU of E-alone and E+P groups were significantly greater than in the TDLU of the no-HRT group. (Courtesy Hofseth et al.5 Copyright 1999, The Endocrine Society)
Regulation of PR isoform expression has been studied in vivo in adult wildtype, genetically unaltered mice.30 These studies examined PRA and PRB regulation in adult ovariectomized Balb/c mice treated for 3, 5, or 10 days with vehicle control, E, P, or E+P. This study showed that PRA level is increased by E and decreased by P, and that the percentage of PRA positive cells is reduced upon prolonged treatment with P or E+P. In contrast, expression of PRB is induced by P and this induction is enhanced in E+P-treated mice. Increased PRB levels coincide with the formation of alveolar structures in response to P or E+P treatment.
III. EXPRESSION OF PR ISOFORMS IN MAMMARY TISSUES
A. Detection of Progesterone Receptor Expression
PR expression has routinely been measured by biochemical methods, real-time RT-PCR, and by immunohistochemical methods. The most sensitive method of PR isoform detection is real time RT-PCR analysis. A critical issue in such experiments is correct primer design. The 5′ untranslated region (UTR) of PRA mRNA overlaps with the PRB reading frame; therefore, only the 5′ UTR of PRB mRNA should be used for the design of PRB-specific primer(s). Namely, the sequence upstream of the PRB translation start site in the human PR gene (NM_000926) – nucleotide 1455; in the mouse PR gene (NM_008829) – nucleotide 636; and in the rat PR gene (U06637) – nucleotide 2015 should be used to design PRB-specific primer(s). To design primers specific for both PRA and PRB, the sequences between PRB and PRA translation start sites should be used, instead of the sequences located in the DBD. Primers located in the DBD may amplify PRC in addition to PRB and PRA mRNAs. For quantitative analysis, it is additionally important that there be similar amplification efficiency of the primers used to detect PRB vs. primers used to detect total PR (PRA+PRB) transcripts.
Biochemical methods to analyze PR isoform expression, such as immunoblot analysis and immunoprecipitation, may provide important information about PR isoform molecular sizes and post-translational modifications. They can provide quantitative analysis of expression levels when used for homogeneous cell cultures, such as isolated primary mammary epithelial cell cultures or breast cancer cell lines. However, when used to quantify PR levels in protein extracts of whole mammary gland, an important confounding factor is the unknown contribution of stromal proteins to the total protein in extracts. This is particularly relevant in mammary tissues that exhibit changes in overall epithelial content, such as the pubertal gland versus adult virgin gland versus pregnant mammary gland. A further limitation to biochemical methods is sensitivity of detection. For example, in the mouse whole mammary gland samples the dilution of PR present in mammary epithelial cells by the stromal cell component that lacks PR often results in PR levels below the limit of detection.31, 32
Immunohistochemistry with antibodies that are specific for PRA or PRB is a suitable method to determine the cell type-specific expression, intracellular distribution, and colocalization of PR isoforms within the same cells. In some cases, interpretation of studies of PR isoform expression has been confounded by lack of information about the specificity of the antibody used to detect only PRA, only PRB or both PRA and PRB. The study by Mote et al. in human breast tissue and cells has shown that of 11 antibodies generated against human PR, 10 detect both PRA and PRB and 1 detects only PRB by immunoblot analysis.33 However, in immunohistochemical analysis 8 of the antibodies tested have detected only PRA, and 2 detect both PRA and PRB. Only one of the antibodies tested has been specific for PRB. Prior to that study it has often been assumed that antibodies that detect both PRA and PRB by immunoblot also detect both isoforms by immunohistochemistry. Since a number of the commercially available anti-PR antibodies detect only PRA or both PRA and PRB, the interpretation of immunohistochemical analyses which draw conclusions about specific PR isoform expression must be viewed in this context.
B. Cell-Type Specific Expression of PR Isoforms
The mammary gland is a composite organ. It consists of epithelial and stromal components. The epithelium, in turn, consists of two distinct cell types: luminal epithelial cells that line the ducts and lobules, and myoepithelial cells that form the contractile network surrounding the luminal epithelium. The stroma is also heterogeneous; it consists of adipocytes, fibroblasts, blood and lymphatic vessels, nerve bundles and wandering cells of the immune system. Immunohistochemical studies in human breast 34, mouse 35 and rat mammary gland 36 showed that PR expression is confined to the luminal epithelium. PR isoforms are expressed unevenly in different cell types within the mammary gland. Analysis of PR isoform specific expression in the normal human premenopausal breast has revealed that both PRA and PRB are expressed in luminal cells and colocalized in the same cells.34 In the mouse, both PRA and PRB are expressed in the luminal epithelium 35. In the rat, PRA expression is confined to luminal epithelium, whereas PRB is expressed in both luminal and myoepithelial cells.37 The function of PRB in myoepithelial cells is not known. We speculate that P may induce proliferation and/or differentiation of the myoepithelial cells in the rat. Whether PRB is expressed in human myoepithelial cells or plays a role in myoepithelial cell function is unknown.
PR isoforms have not been detected in human or mouse mammary stroma by immunohistochemistry.34, 35 However, further investigation of mouse and rat stroma indicate that PRB may be expressed in lymph nodes and that PRA appears to be expressed in blood vessels and nerve fibers in the rat mammary gland (Kariagina & Haslam, unpublished observations). The specific cell types that express PR and the role of P action in the stromal component of the gland during normal development and/or in mammary cancer remains to be determined.
IV. MECHANISMS OF PROGESTERONE RECEPTOR ACTION
A. Genomic Actions of PR
Genomic actions of PR occur in the nucleus. In this case, PR actions are mediated by direct binding of the PR DBD to progesterone response elements (PRE) and result in modulation of gene transcription.38 Much of what is known about mechanisms of PR action comes from in vitro studies in cell lines. In recent studies by Roemer et al.,39 a less conserved carboxyl-terminal extension (CTE) within a highly conserved DBD of human PR has been described. This CTE domain may bind to DNA outside the consensus PRE and stabilize the PR-DNA interaction. Such additional interaction may be particularly important for regulation of genes that have a weak half-PRE or a PRE that deviates from the canonical sequence.
PR may be unevenly distributed inside the nucleus in the form of distinct foci. It has been shown that PR foci colocalize with transcriptional co-activators p300 and steroid receptor coactivator (SRC) SRC-1 and are localized to regions of active euchromatin.40 In the normal human breast, focal distribution of PR is predominant in the luteal phase of menstrual cycle, when P level is increased.40 In T47D breast cancer cells engineered to express only PRA or only PRB, both isoforms show predominantly even distribution throughout the nucleus in the absence of progestins, and form distinct nuclear foci after hormone treatment.40 In primary breast cancer, it has been shown that the focus formation occurs with similar frequency in cancers from pre- and post-menopausal women regardless of circulating P levels, indicating that the ligand–dependence of PR to form nuclear foci, typical for the normal breast, is lost in cancer.40
B. Regulators of PR Transciptional Activity
The transcriptional activity of PR isoforms depends on their interactions with several families of auxiliary proteins such as molecular chaperons, coactivators, and corepressors. Recent studies have shown that the co-chaperone immunophillin, FKBP52, is critical for PRA mediated gene transcription in the mouse uterus.41 The loss of FKBP52 in the mouse results in a partial reduction in the extent of mammary gland sidebranching and alveolar formation in response to E+P treatment.41 Interestingly, the loss of FKBP52 does not alter the ability of PRA or PRB to bind the ligand. These data suggest that in vivo, FKBP52 may be involved in events downstream of ligand binding. Another novel co-chaperone protein GCUNC-45 has been shown to directly bind both PRA and PRB in T47D human breast cancer cells.42 In a cell-free system, addition of GCUNC-45 in 2-fold molar excess over Hsp90 results in almost complete reduction of hormone binding by PR. However, the subsequent addition of co-chaperone FKBP52 reduces binding of GCUNC-45 to the chaperone-PR complex and FKBP52 increases PR hormone binding in a dose-dependent manner. These studies support the concept that even when PR levels are unaltered the functional outcome of PR signaling may be modulated by auxiliary proteins and that further studies of steroid receptor interactions with auxiliary proteins are warranted.
SRCs bind the ligand-binding domain of steroid receptors and enhance the transcriptional activities of steroid receptors. Several studies have established that SRCs may modulate the functional activity of steroid receptors. Studies in bigenic mice expressing indicator of PR activity and carrying the genetically engineered disruption of either SRC-1 or SRC-3 have shown that the absence of SRC-1 does not impair PR responses to E+P treatment in mammary epithelium, whereas the absence of SRC-3 abrogates the PR responses after E+P treatment in mammary gland.43 In another study, the genetically engineered loss of SRC-2 exclusively in cells expressing PR leads to impaired sidebranching and alveolar formation after E+P treatment.44 Together, these results demonstrate that SRC proteins modulate the physiological function of PR in mammary tissue in vivo. In the human breast, SRC-2 is expressed in a distinct punctate nuclear pattern similar to the pattern of PR expression.44 However the role of SRC-2 in the human breast is currently unknown. It remains to be determined if different SRCs act in an isoform specific manner.
The transcriptional activity of PR can also be modulated by proteins originally discovered as regulatory proteins in other cellular signaling pathways. For example, protein inhibitors of activated STAT (signal transducers and activators of transcription) (PIAS), PIAS1 or PIAS3, which are the inhibitors of STAT1 or STAT3 respectively, may substantially inhibit PR-induced gene transcription. In the T47D breast cancer cell line, PIAS3, similar to PIAS1,45 induces sumoylation of PR and significantly inhibited PRB-dependent gene-transcription in hormone-dependent manner. 46 It has been shown that PIAS3 inhibits the DNA-binding ability of PRB and alters the subcellular distribution of PRB protein. Whether similar regulatory mechanisms operate in the normal human breast to regulate PR transcription is currently unknown.
The transcriptional activity of PR fluctuates throughout the cell cycle with the highest transcriptional activity occurring during S phase.47, 48 Cyclin A/cyclin dependent kinase 2 (cdk2) complexes have been shown to function as coactivators of PR-dependent transcription. In HeLa cells, cyclin A increases transcriptional activity of PRA and PRB independent of their phosphorylation status.49 In the S phase, the cyclin A/cdk2 complex is recruited to PR bound to DNA and stimulates the additional recruitment of coactivator SRC-1.47 In the G1 phase when cyclin A is absent, PR transcriptional activity and the recruitment of SRC-1 are diminished. It will be of interest to determine if PR transcriptional activity is also increased during S phase in the normal human breast and in breast cancer.
C. PR Phosphorylation
The transcriptional activity of PR can be modulated by its phosphorylation status.50 There are thirteen constitutive and ligand-dependent serine phosphorylation sites identified in human PR and six sites are located only in PRB, implying that transcriptional activity of PRB may be regulated differently by cellular kinase pathways.51 Phosphorylation on Serine 294 is ligand-dependent. In progestin treated T47D cells expressing only PRB, Ser294 phosphorylation by mitogen-activated protein kinase (MAPK) stimulates proteasome-dependent PR degradation.52 Paradoxically, such downregulation of PR protein coincides with the highest PR transcriptional activity. The authors propose that transcriptional activity of PR is tightly coupled with receptor turnover, which is regulated by ligand-dependent phosphorylation on Ser294. Another phosphorylation site, Serine 400, may be constitutive or ligand-induced. In HeLa cells expressing only PRB, Ser400 phosphorylation by cdk2 is not required for the increase of ligand-dependent transcriptional activity, but significantly increases ligand-independent PRB-mediated transcription.53
Treatment of T47D cells with the synthetic progestin R5020 leads to rapid activation of Erk through interaction with unliganded estrogen receptor and leads to the subsequent phosphorylation of PR on serine 294.54 Additionally, Msk1 becomes activated and a complex of these three proteins (Erk, PR, and Msk1) is recruited to PREs and leads to phosphorylation of histone H3 and nucleosome remodeling. Thus, P induction of target genes may be modulated by rapid activation of kinase pathways.54 Another study in T47D cells has shown that PRB, but not PRA, is also able to induce robust and sustained activation of Erk1/2 that increases cyclin D1 protein levels.55 This process occurs through PRB-induced transcriptional upregulation of Wnt-1 leading to matrix metalloprotease-mediated activation of epidermal growth factor receptor (EGFR) and persistent activation of c-Src and Erk1/2. Sustained activation of this pathway results in increased progestin-stimulated anchorage-independent growth, and suggests a physiological link between growth factor and PR genomic signaling.
D. Non-Genomic Actions of PR
The existence of nongenomic pathways activated by PR has recently been described. PR can also interact with cytokine and growth factor signaling pathways at multiple levels to influence signaling cascades that play important roles in mammary cell proliferation and differentiation. In T47D cells expressing only PRB, epidermal growth factor (EGF) and P act synergistically on promoters that drive the cell-growth regulatory genes c-fos and p21, neither of which contains a PRE.56 This synergy is believed to be mediated through MAPK activation. Human PR has also been shown to mediate rapid activation of the Src/Ras/Raf/MAPK signaling pathway through a Pro-Xaa-Xaa-Pro-Xaa-Arg motif located in the N-terminal domain of both PRA and PRB.57 This activity is distinct from PR transcriptional activity and is not dependent upon the DBD of PR. Both PRA and PRB can bind to Src, but only PRB produces strong stimulation of Src kinase activity and downstream effectors.
Another signaling pathway influenced by P and PR involves STATs.56 STAT5 is important for normal lobuloalveolar development and lactational function of the mammary gland.58 Nuclear localization, DNA binding and regulation of target genes by STAT5 requires phosphorylation that is induced by growth factors and cytokines, such as prolactin, via Janus kinases. Studies in T47D cells expressing only PRB show that P acts to upregulate STAT5 mRNA and that P/PRB action is also implicated in STAT5 nuclear localization. This is believed to occur through a direct interaction of STAT5 and PR, and it is believed that STAT5 is translocated to the nucleus as a companion with PR.56
V. FUNCTIONS OF PR ISOFORMS
A. PR Target Genes
Much of the information about PR isoform specific effects on gene regulation that may be relevant to breast cancer has been generated from in vitro studies of human breast cancer cell lines. Differential gene regulation by PRA and PRB has been demonstrated by expression profiling of progestin-treated T47D breast cancer cell lines, engineered to express only PRA or only PRB.59, 60 In the presence of ligand, PRB has greater transcriptional activity than PRA. Of 94 genes that are P-regulated, 59 are uniquely upregulated by PRB, 4 uniquely by PRA and 19 by both isoforms. Twelve genes have been found to be downregulated by P; 6 are regulated by both isoforms, 6 uniquely by PRB, and none by PRA. In the absence of ligand, PR also can be transcriptionally active.61 It has been shown that 54 genes are regulated by unliganded PR; 11 are upregulated by PRB, 33 upregulated by PRA and 10 upregulated by both isoforms. In contrast to liganded PR, PRA is the more active isoform in the unliganded state. A number of genes regulated by both unliganded PRA and PRB are associated with tumor size, nodal metastasis and cell aggressiveness. Additionally, genes regulated by unliganded PRA are associated with an invasive, poorly differentiated and aggressive tumor phenotype.
Among the genes regulated by PRB only, there are several genes essential for steroid biosynthesis and general cellular metabolism (11-β-hydroxysteroid dehydrogenase 2, glucose-6-phosphatase), cell adhesion (integrin α6) and several transcription factors (C/EBPβ, STAT5a).59 In particular, STAT5a is essential for normal lobuloalveolar development and lactational function of the mammary gland.58 Conversely, PRA alone mediates induction of genes, such as docking protein HEF1, anti-apoptotic gene Bcl-XL, and orphan receptor estrogen-related receptor α (ERRα).59 Interestingly, ERRα has been shown to stimulate the transcription of ERα-dependent genes in BT-474 mammary carcinoma cells expressing high levels of ErbB2, suggesting that ERRα may contribute to the poor responsiveness to anti-estrogen therapy in ERα positive ErbB2 overexpressing tumors.62 It is possible that the mechanisms underlying the poor prognosis associated with PRA overexpression in breast cancers additionally involve the up-regulation of Bcl-XL and ERRα genes.
In the normal adult premenopausal breast, PR isoforms are localized within the same cell and expressed in 1:1 ratio, whereas in breast cancer this ratio is often shifted toward PRA predominance.34, 63, 64 To mimic the PRA:PRB ratio in the normal breast and in breast cancer Clarke’s group has engineered T47D cells expressing near equimolar PRA:PRB ratio and cells expressing 5-fold excess of PRA over PRB.63 When both PRA and PRB are expressed in the same cell, the ratio between these isoforms may be critical in determining the profile of P-responsive genes. This study shows that the shift of PRA:PRB ratio from 1.8 to 5.1 does not change the global profile of gene expression in T47D cells 6 hours after P treatment. However, at 48 hours after P treatment many genes have been differentially activated in the cells with low vs. high PRA:PRB ratio. Of 601 P-regulated genes, 54 genes are up- or down-regulated only in the cells with high PRA:PRB ratio and 28 genes respond to P treatment only in the cells with low PRA:PRB ratio. Thus, the majority of the genes (519) are regulated in a similar fashion regardless the PRA:PRB ratio. Interestingly, among the genes regulated by P in the cells with the high PRA:PRB ratio, there are genes involved in cellular metabolism and cell adhesion. Further experiments have shown that P treatment decreases adhesion and affects the shape of T47D cells in a PRA-dependent manner. These results suggest that PRA may contribute to the aggressiveness of breast cancer cells by decreasing the cell-to-cell and cell-to-matrix adhesion.
In additional studies, expression profiling in PRA+PRB positive T47D cells has been carried out with P and five clinically relevant synthetic progestins: 3-ketodesogestrel, levonorgestrel, medroxyprogesterone acetate, norethindrone acetate, and timegestone.65 These synthetic progestins show a high degree of similarity in their transcriptional responses and each progestin regulates between 77 and 91% of the genes regulated by P. Thus, P and synthetic progestins have a similar and fairly specific mode of action.
B. Regulation of Proliferation by PR Isoforms
1. PR and Cyclin D1 expression
The ability to increase proliferation is one of the most important actions of P in the breast. This effect can be mediated by direct or indirect activation of the genes important for cell cycle initiation and progression. Studies from the Horwitz laboratory have demonstrated that P induces one round of proliferation in T47D cells followed by cell cycle arrest in the G1 phase.66 This cell cycle arrest is associated with induction of the cyclin-dependent kinase inhibitors (CDKI) p21 and p27. The proliferative action of P may be mediated in part by induction of cyclin D1, a protein essential for cell cycle initiation and transition through the G1 phase to the S phase.67 However, the cyclin D1 promoter lacks a canonical PRE. In T47D cells stably expressing only PRB, P treatment increases cyclin D1 mRNA transcription up to 5-fold 6 hours after treatment. This induction of cyclin D1 has been abolished by inhibitors of MAPK, c-Src, and, partially, by the inhibitor of PI3K, indicating that the increase of cyclin D1 expression results from PRB interaction with multiple cytoplasmic signaling pathways.68
Studies conducted in our laboratory have shown that in the pregnant mouse and rat mammary glands PRB expression is highly colocalized with nuclear cyclin D1 expression and with the proliferation marker, BrdU.35, 37 In the adult virgin rat mammary gland, cells co-expressing both PRA and PRB are also highly colocalized with cyclin D1. However, these cells appear to be arrested in the G1 phase of the cell cycle, due, in part, to the increased nuclear expression of CDKI p21 and p27.37 To our knowledge, no studies on colocalization of PR isoforms and cyclin D1 have been performed in the normal human breast.
2. PR and Expression of Cyclin Dependent Kinase Inhibitors
The promoters of CDKI p21 and p27 lack canonical PRE and appear to be regulated by P indirectly. In T47D cells that co-express PRA and PRB, P treatment induces the p27 promoter activity and p27 mRNA expression via tethering of liganded PR through Sp1 protein to the p27 promoter.69 Similarly, in T47D cells expressing only PRB, P activates the p21 promoter through an Sp1 site.70 Coactivator proteins CBP/p300 and TReP-132 have been shown to be critical for p21 and p27 promoter activation by PR.70, 71 Interestingly, PR directly (through PRE) stimulates mRNA expression of TReP-132 in T47D cells.71 This suggests the existence of a regulatory feedback loop that, upon P exposure, allows induction of CDKI expression by PR that results in the inhibition of proliferation.
Our studies in the rat mammary gland have shown that both p21 and p27 are highly expressed in the nuclei of the proliferatively quiescent adult virgin mammary gland and are highly colocalized in cells that co-express both PRA and PRB. In the pregnant gland, where the proliferation rate is high, both p21 and p27 are expressed at very low levels.37 Studies in human breast cancers have shown that the levels of p27 mRNA, but not p21 mRNA, are positively correlated to PR expression and are negatively correlated with the tumor grade.69 Future studies of the relationship between PR isoform expression and p21 or p27 expression may provide an important insight into mechanisms of how P regulates proliferation in the normal human breast and in breast cancer.
3. Paracrine Mechanisms of Progesterone Receptor Action
Immunohistochemical studies demonstrating the lack of colocalization of steroid receptors with markers of proliferation have been interpreted to mean that in the normal breast PR positive cells rarely proliferate.36, 72 These observations have formed the basis for the proposed paracrine mechanism of P action in mammary gland. Some candidate proteins, such as Receptor Activator of Nuclear factor Kappa B Ligand (RANKL) and members of the Wnt family, are reported to be induced in PR expressing cells and are proposed to mediate P action in PR negative cells through paracrine mechanism(s).32, 73 Studies in PRA knockout (PRAKO), PRB knockout (PRBKO), and total PR knockout (PRKO) mice suggest that PRB may mediate stimulation of RANKL expression in E+P treated mammary glands.32 The same study suggested that in PRBKO mice PRA may retain the ability to mediate Wnt-4 activation by P to sustain ductal sidebranching in the absence of PRB. Studies of RANK, the receptor for RANKL, overexpressing mice have shown sustained proliferation throughout pregnancy and day 1 of lactation compared to wildtype mice that normally exhibit reduced proliferation after day 15 of pregnancy.74 In the human, out of 16 Wnt family members expressed in normal breast, only two Wnts, Wnt-1 and Wnt-4, are overexpressed in breast carcinomas.75 RANKL expression is lost in 86% of breast cancers.76 The relationship between human PR isoform expression and the regulation of RANKL or Wnt proteins in the normal human breast has not been established.
VI. PR ISOFORM EXPRESSION AND HORMONAL REGULATION OF NORMAL MAMMARY GLAND DEVELOPMENT
A. Human
In humans, the hormonal regulation and molecular mechanisms involved in breast development have not been well studied. Much of what is assumed to be true about the development of the human breast is based on what is known about mammary gland development from animal studies. At birth the breast consists of a rudimentary ductal system. The breast remains in a dormant state until the onset of puberty. During early puberty, P is not produced. This anovulatory state continues for 1–6 years.3 With the onset of ovulatory menstrual cycles, P synthesis and secretion occurs during the latter half of the menstrual cycle (luteal phase), and mature adult breast lobuloalveolar elements are formed. Thus, the adult human breast exhibits a ducto-lobular histo-architecture and lobules are present prior to pregnancy. Lobuloalveolar development is thought to be mediated by P acting through PR. The roles of P and PR are inferred from studies in PR gene-deleted mice since these mice fail to develop a lobuloalveolar mammary gland morphology upon exposure to E and P.77 E is thought to play an important role in P action since PR expression is upregulated by E. There is agreement that DNA synthesis in breast epithelium, as measured by tritiated thymidine incorporation,78, 79, 80 or cell proliferation as measured by PCNA or Ki67 expression,5 is increased during the luteal phase of the menstrual cycle when progesterone is present (Fig. 2). The greatest proliferative activity occurs in the terminal duct lobular unit (TDLU), the site of origin of most breast cancers.5 The breast attains its maximum development during pregnancy; proliferation results in an increase in lobule numbers and size. The epithelial proliferation and lobular expansion are thought to be due in large part to elevated E and P levels produced during pregnancy. The beginning of secretory activity is characteristic of the largest and most fully differentiated lobule.81 After pregnancy and lactation, the breast undergoes a regression resulting in an increase in the number of small lobules and a concomitant decline in the number of larger lobules.81
Figure 2.
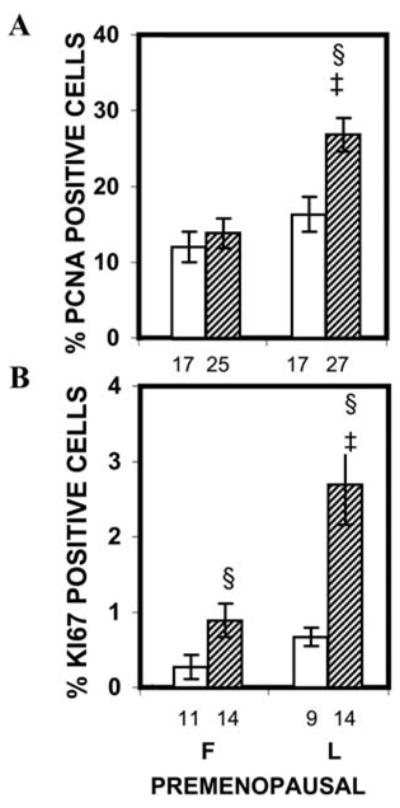
Epithelial proliferation indices in normal breast tissue of cycling premenopausal women. The number under each bar represents the number of individuals for whom ducts or TDLU could be analyzed. ‡, P < 0.05 that the percentages of PCNA-positive cells in the TDLU of the luteal-phase group were significantly greater than in the TDLU of the follicular-phase group; §, P < 0.05 that the percentages of PCNA- and Ki67-positive cells were greater in the TDLU than in the ducts of the same group. (Courtesy Hofseth et al.5 Copyright 1999, The Endocrine Society)
Progestins also promote proliferation in the postmenopausal breast 5. Analysis of proliferation indices using PCNA and Ki67 antibodies in the adjacent normal breast tissue from postmenopausal women who have undergone benign breast biopsies has been carried out. Both proliferation indices are significantly higher in the TDLU of women receiving E+P HRT compared to no HRT or E alone HRT (Fig. 3A).5 Additionally, the breast epithelial density in women taking either form of HRT is significantly greater than in non-users of HRT, and treatment with E+P HRT is associated with a significantly greater breast epithelial density than with E alone HRT (Fig. 3B–E). These data provide compelling evidence that E has a mitogenic effect in the human breast, and that E+P combination has a greater effect.5 The authors hypothesize that the increased breast cell proliferation and increased epithelial density provide a potential mechanistic basis for the increased breast cancer risk associated with E+P HRT.
Figure 3.

Effects of HRT on epithelial proliferation indices and breast epithelial density in postmenopausal women. A. Epithelial proliferation indices in normal breast tissue of postmenopausal women. The number under each bar represents the number of individuals for whom ducts or TDLU could be analyzed. *, P = 0.002-0.0001 that the percentages of PCNA- and Ki67-positive cells in the TDLU of the E + P group were significantly greater than in TDLU of no-HRT or E-alone groups; †, P = 0.007-0.002 that the percentages of PCNA-positive cells in the TDLU or ducts of the E group or ducts of the E + P group were significantly greater than in the TDLU or ducts of the no-HRT group; §, P < 0.05 that the percentages of PCNA- and Ki67-positive cells were greater in the TDLU than in the ducts of the same group. B–D. Photomicrographs of breast tissue from postmenopausal women; (B) no HRT, (C) E alone HRT, (D) E+P HRT. TDLUs and ducts are indicated by arrows. Magnificantion, ×40. E. Effects of HRT on breast epithelial density in postmenopausal women. The number under each bar represents the number of individuals for whom epithelial density was determined. *, P = 0.001-0.01 that the percentages of epithelial area in the E-alone HRT or E + P HRT groups were significantly greater than that of the no-HRT group. †, P = 0.02 that the percentage of epithelial area in the E + P group was significantly greater than that of the E-alone group. (Courtesy Hofseth et al.5 Copyright 1999, The Endocrine Society)
There is limited information on PR isoform expression during normal human breast development. In the normal adult premenopausal breast approximately 12% of mammary epithelial cells are PR positive, and this percentage does not change throughout the menstrual cycle.82 A single study of PR isoform expression in the human breast has analyzed adult premenopausal cycling women, ranging from 21 to 50 years of age.34 Based on immunohistochemical analysis, PRA and PRB are expressed at a ratio of 1:1 and there is uniform colocalization of PRA with PRB in the same cells. The proportion of PR positive cells ranges from 10–20%, with marked variation throughout a section, and with PR positivity in individual ducts or lobules ranging from 0–90%. The significance of this variation is not known.
In the adult human breast, PR positive cells generally do not proliferate, suggesting that PR mediates proliferation via an indirect mechanism at this stage.36, 72 Analyses of colocalization of PR isoforms with proliferation markers in the human breast have not been isoform specific. However, PRA and PRB are co-expressed in the same cell in the human breast 34, therefore it can be inferred that cells containing both PRA and PRB promote proliferation through an indirect mechanism. However, it should be noted that proliferation occurs in the adult human breast in only 2% of epithelial cells.72 In order to fully understand the relationship between PR isoform expression and proliferation, it will be necessary to study stages of mammary gland development that display higher levels of proliferation, such as puberty and pregnancy.
B. Mouse
The mouse is the most extensively studied and best understood model of P action in the normal mammary gland. The virgin, non-pregnant mouse mammary gland exhibits a predominantly ductal histo-architecture. Postnatal growth and development of the mouse mammary gland is initiated at the onset of ovarian cycling at puberty, coincident with the production of ovarian hormones (review 83). Pubertal development of the gland proceeds via ductal elongation accompanied by secondary and tertiary branching of the ductal tree. Based upon studies of ovariectomized pubertal mice treated with E or progestin, E plays a dominant role in ductal elongation.84 In contrast, acute treatment with E+P has only a minimal additional effect on proliferation and does not produce sidebranching or alveologenesis, suggesting that the pubertal mammary gland is less sensitive to P than to E.84 Maturation of the gland is achieved when the ducts grow to the limit of the mammary stromal fat pad, at which time the gland becomes proliferatively quiescent.
In adulthood, maturation of the mammary gland is accompanied by the acquisition of responsiveness to P. In adult ovariectomized mice, acute treatment and longer-term treatment with E produces a transient proliferative effect, whereas treatment with E plus P significantly increases proliferation resulting in sidebranching and the start of alveologenesis.30, 84 Elevated levels of E and P during pregnancy lead to the reinitiation of proliferation that leads to extensive ductal sidebranching, alveologenesis and the organization of alveoli into lobules. Upon parturition, maximal secretory activity takes place during lactation. After weaning, the gland undergoes extensive involution and regression to a pre-pregnancy-like state, but it contains a higher proportion of regressed alveoli compared to the nulliparous gland.
Immunohistochemical analysis of the mouse PR has shown that PRA and PRB expression are temporally and spatially separated during mammary gland development in the Balb/c strain.35 Similar results have been observed in the C57Bl/6 mouse strain (Drolet & Haslam, unpublished observations). PRA is the predominant isoform expressed in pubertal and adult virgin mammary glands. The percentage of PRA positive cells decreases in the mature gland and decreases further at mid-pregnancy. PRB is the major isoform expressed during pregnancy, and is mainly expressed in alveolar epithelial cells. During pregnancy, the majority of PR positive cells contain only PRB and colocalization of PRA and PRB occurs in only a small proportion of epithelial cells. Neither PRA nor PRB protein expression is detected during lactation. In the postlactational, fully regressed gland, PRA expression is permanently reduced to the level seen during pregnancy, whereas PRB expression is maintained in a small percentage of remaining alveolar cells.
To determine how PRA expression is related to proliferation during puberty and pregnancy, we analyzed PRA and PRB colocalization with the proliferation marker, BrdU.35 In the pubertal, virgin gland PRA is the predominant isoform and is infrequently colocalized with BrdU. Therefore, the majority of PRA expressing cells do not proliferate in the pubertal mammary gland. During mid-pregnancy PRB colocalizes extensively with BrdU and proliferating cells do not express PRA. Thus, PRB expressing cells proliferate and differentiate during pregnancy, but similar to puberty, PRA expressing cells do not proliferate.
C. Rat
In contrast to the predominantly ductal histo-architecture of the virgin, non-pregnant mouse mammary gland, the virgin rat mammary gland has a ducto-lobular histo-architecture similar to that of the human breast. The major unit of the human breast is the terminal duct lobular unit (TDLU) and the mammary lobule of rat mammary gland is considered to be a counterpart of the human TDLU. At puberty, with the onset of ovarian cycles, the mammary gland grows to form a highly branched ductal tree with extensive newly forming lobules similar to that reported for the human breast.85 During pregnancy, proliferation leads to increased numbers of alveolar cells resulting in increased lobule size. After pregnancy and lactation, the mammary gland undergoes involution; however, the size and numbers of lobules remains greater than in age-matched virgin animals.85 In contrast to the mouse, less is known about the specific roles of E and P in duct and lobule development in the rat.
Immunohistochemical analyses of PRA and PRB expression during rat mammary gland development similar to that described above for the mouse have recently been performed.37 Several aspects of PRA and PRB protein expression differ from those of the mouse, while others are remarkably similar. In contrast to the mouse, in the rat both PRA and PRB are highly expressed at puberty, sexual maturity and are frequently co-localized within the same cell.37 Also in contrast to the mouse, PRB expression in the rat mammary gland remains relatively constant across all developmental states except lactation. Similar to the mouse, PRA expression decreases significantly from puberty to sexual maturity reaching a nadir during pregnancy and is permanently decreased after lactational involution.37
Recent studies in the rat have analyzed how PR isoform expression is related to proliferation during puberty and pregnancy by colocalization studies of PRA and PRB with BrdU.37 Overall, PR isoform expression and colocalization with proliferation are remarkably similar to the mouse. PRA infrequently colocalizes with BrdU in the pubertal, virgin gland. The lack of colocalization is similar to previous findings in the young virgin rat showing that proliferating cells do not express PR.36 In contrast to the mouse, PRB is frequently colocalized with proliferation in end buds of the pubertal rat. Similar to the mouse, during pregnancy there is also extensive colocalization of PRB with BrdU. Also, most PRA expressing cells do not proliferate in the pregnant mammary gland.
D. Comparison of human, mouse, and rat PR isoform expression and normal mammary gland development
The studies described above reveal interesting differences in human, rat and mouse PR isoform expression patterns in mammary gland. PR isoform expression in the rat most closely resembles the pattern reported for the human with regard to extensive co-expression of PRA and PRB in the same cells. In contrast, in the mouse, PRA is predominantly expressed in the non-pregnant state and PRB is expressed most highly during pregnancy.35 Furthermore, colocalization of PRA with PRB occurs in only a small percentage of cells.35 This suggests that in the mouse, heterodimer formation between PRA and PRB does not play a major role in P action in the mammary gland.
One possible interpretation of the difference in PR isoform expression between mouse vs. rat and human may be the predominance of a ductal organization of mammary epithelium in the adult, non-pregnant mouse (Fig 4A). This is particularly true in Balb/c strain mice used in our study. The pubertal and adult mouse contains a simple ductal system with little sidebranching. In contrast, in the pubertal and adult human and rat there is concomitant development of lobules and ducts (Fig 4B, C). Studies of PRB gene-deleted mice have shown that PRB expression is required for alveologenesis and lobule formation.32 Therefore, PRB expression may be a defining characteristic of mammary lobule formation and/or maintenance and may explain why PRB positive cells are more abundant in rat mammary glands across development whereas PRB is only highly expressed in the mouse mammary gland during pregnancy (Fig 5B). In this regard, the maintenance of sidebranching and some alveolar structures in the mouse mammary gland after pregnancy (Fig 4A) may also be due to the low level of PRB expression after pregnancy (Fig 5B).
Figure 4.

Mammary gland morphology during development in the mouse, rat and human. A. Mammary gland whole mounts were prepared from pubertal, adult, mid-pregnant, and involuted Balb/c mice (scale bar, 1 mm). B. Mammary gland whole mounts were prepared from pubertal, adult, mid-pregnant, and involuted Sprague-Dawley rats (scale bar, 1 mm). C. Mammary gland whole mounts from the pubertal, adult, and pregnant human breast. Magnificantion, ×2.5. (Courtesy of Russo&Russo.86 Copyright 2004, Springer-Verlag) Note the presence of lobuloalveolar structures (indicated by arrows) in the pubertal rat and human mammary gland, whereas these structures are not present in the mouse until pregnancy. The presence of end buds in the pubertal mouse and rat is indicated by arrowheads.
Figure 5.
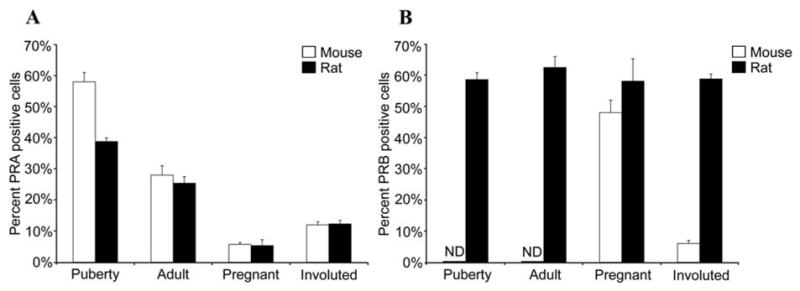
Immunohistochemical detection of PRA and PRB in the mouse and rat mammary gland. Immunohistochemistry was performed on sections from pubertal, adult, pregnant, and involuted mammary glands from mice and rats using antibody specific for (A) PRA or (B) for PRB and the percent PRA or PRB positive cells was determined. PRB was not detected (ND) in the pubertal and adult mouse mammary gland.
Notably, puberty and pregnancy exhibit similarities in the pattern of PR isoform expression and colocalization with proliferation in the mouse and rat. In both species, there is the highest PRA expression at puberty, the lowest PRA expression at pregnancy, and high PRB expression during pregnancy (Fig 5A). Additionally, both the mouse and rat display permanent decreases in PRA expression following pregnancy and involution (Fig 5A). In the mouse and in the rat, PRA generally does not colocalize with proliferation markers, whereas PRB does colocalize with proliferation markers (Fig 6A, B). The percentage of proliferating cells and low colocalization of PRA with proliferation is similar in the pubertal and pregnant glands of the rat and mouse (Fig 6A). In contrast to PRA, in both the mouse and the rat proliferation is extensively colocalized with PRB expression (Fig 6B).
Figure 6.
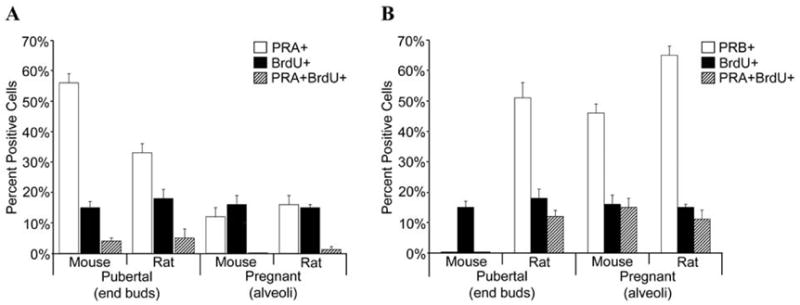
Colocalization of PRA and PRB with the proliferation marker BrdU in the mouse and rat mammary gland. Dual immunofluorescence detection of (A) PRA and BrdU or (B) PRB and BrdU was performed on mammary gland sections from pubertal and pregnant mice and rats. The percent PRA positive (PRA+), BrdU positive (BrdU+) and PRA+BrdU positive (PRA+BrdU+) and the percent PRB positive (PRB+), BrdU positive (BrdU+) and PRB+BrdU positive (PRB+BrdU+) luminal epithelial cells were determined in pubertal end buds and pregnant alveoli.
As stated earlier, our knowledge of PR isoform expression in the human comes from a single study of the adult premenopausal human breast (age range 20 to 50 years).34 Thus, analysis of human breast tissue at puberty and pregnancy could be very informative about possible changes in PR isoform expression during the course of human breast development. Defining the differences and similarities among human, rat and mouse PR isoform expression and colocalization with proliferation can provide important insights into the functions of PR isoforms in the normal human breast and in breast cancer. Additionally, such studies underscore the value of different animal models for advancing our understanding of PR isoform-specific functions in mammary gland. In this regard, the dominance of PRA expression in the virgin mouse mammary gland provides a unique opportunity to investigate the functional role of PRA in normal development.
VII. PR ISOFOM EXPRESSION AND BREAST CANCER
A. Human Breast Cancer
1. PR Isoform Expression in Primary Human Breast Cancer
The equimolar ratio of PRA:PRB expression, typical for the normal breast, may be significantly altered in breast cancer. A study measuring PRA:PRB ratios by immunoblot has shown that in PR-positive breast tumors, a significant proportion of tumors expresses very low levels of PRB and consequently had a high PRA:PRB ratio. An altered PRA:PRB ratio based upon immunoblot analysis is also reported in tumors from node positive patients. Patients with a high PRA: PRB ratio, often caused by PRA levels higher than normal, are 2.76 times more likely to relapse than patients with lower ratios.87
An immunohistochemical analysis of PRA and PRB expression has been carried out on human breast proliferative disease without atypia (PDWA), atypical ductal hyperplasia (ADH), ductal carcinoma in situ (DCIS), and invasive breast lesions.34 In both normal breast and PDWA, PRA and PRB are coexpressed in the same cells in comparable amounts. There is a significant increase in heterogeneous expression of PR isoforms in ADH and DCIS and a significant increase in predominant expression of PRA or PRB with progression to malignancy. Predominance of PRA is especially evident in DCIS and invasive breast lesions. The authors conclude that alteration in the PRA:PRB ratio is an early event in the development of breast cancer and that altered PR isoform expression may play a role in the etiology of breast cancer and affect tumor growth. Another possible interpretation is that tumors that have a high ratio of PRA:PRB originate from normal cells that have a high ratio of PRA:PRB. Clearly, more information about the pattern and ratio of PRA: PRB expression in the normal breast is needed for a better understanding of the significance of altered PRA:PRB ratios in breast cancers.
Mutations of the tumor suppressor genes, BRCA1 and BRCA2, are associated with a significantly increased risk of breast cancer. In the human breast, there are several reports on altered PR expression in mammary tissue from BRCA1 or BRCA2 carriers. According to King et al.,88 in carriers of BRCA1 but not BRCA2 mutation, PR expression is significantly higher in normal breast tissue adjacent to the tumor than in sporadic breast cancer cases. However, the authors did not discriminate between PR isoforms in this study. In a study of non-cancerous, prophylactic mastectomy-derived breast tissues of both BRCA1 and BRCA2 mutation carriers, Mote et al. report higher expression of the PRA isoform and loss of PRB.89 ER expression is no different in BRCA mutation carriers compared to non-carriers. Is the ratio of PRA:PRB in normal mammary gland related to future breast cancer risk? To answer this question, an analysis of PR isoform expression at different developmental stages of the human breast is needed. The rat mammary gland has a high degree of similarity to the human breast with regard to histo-architecture, and similar to the human breast, a high percentage of cells co-express PRA and PRB. However, the ratio of PRA:PRB changes significantly with the developmental state of the gland such that the ratio of PRA:PRB is close to 1 at puberty and then progressively decreases during adulthood, pregnancy and post partum.37 In the rat, the ratio of PRA:PRB has functional significance for the proliferative, morphological and differentiative changes occurring at different stages of development (i.e. puberty vs. pregnancy.) Whether or not an altered PRA:PRB ratio in human breast cancer is a consequence of neoplastic transformation or reflects the developmental state when breast cancer was initiated remains to be determined.
Recently, a functional relationship between PR and expression of BRCA1 has been observed in vitro in T47D and MCF-7 breast cancer cells.90 Exogenous BRCA1 inhibits the activity of endogenous and transiently transfected PRA and PRB in human breast cancer cells. Conversely, small interfering RNA knockdown of endogenous BRCA1 enhances progestin stimulated activity of PR. These results indicate a potential inhibitory effect of BRCA1 on progestin action that may be lost in carriers of BRCA1 or BRCA2 mutations, and raise the possibility of increased progestin activity in these individuals. If the mitogenic activity of progestins plays a role in breast cancer risk and progestin activity is increased as a result of BRCA1 and BRCA2 mutations, it is conceivable that this may be one mechanism by which BRCA1 and 2 mutations increase breast cancer risk.
2. PR Isoform Expression and Function In Vivo in Human tumor Xenograft Studies
The effect of PR isoform expression on tumor cell behavior has also been investigated in vivo in human breast tumor cells xenografted into nude mice. T47D breast cancer cells that express PR independent of E have been engineered to express only PRA, only PRB, no PR, or both PRA and PRB.91 Each of the cell lines has been grown into solid tumors in ovariectomized nude mice treated with exogenous E. PRA expressing cells grow into tumors that were only half the size of PRB expressing tumors. Both types of tumors can grow in the absence of ligand (P). Tamoxifen, an estrogen receptor antagonist, inhibits the growth of PRA expressing tumors while PRB expressing tumors are not affected. In contrast to the differences in growth behavior in vivo, there is no significant difference in growth rate of PRA and PRB expressing cells in cell culture. Thus, in vivo models to study hormone-dependent growth parameters are additionally important. One interesting aspect of this study is that PRA expressing tumors did not display the aggressive growth behavior associated with PRA dominance in cell culture 63 or associated with PRA dominance in primary tumors.87 The authors suggest that PRA tumors may represent a class of small sized human breast cancers that despite their small size have a high metastatic potential.
Solid tumors derived from T47D breast cancer cells expressing PRA or PRB have also been investigated for their response to P or MPA.92 In ovariectomized nude mice, P or MPA alone does not support tumor growth, and the addition of either hormone does not alter E-dependent growth of the tumors. However, treatment of either PRA or PRB expressing tumors, but not PR negative tumors, with either P or MPA leads to increased expression of myoepithelial cell keratins 5 and 6 in a subpopulation of tumor cells. It is thought that during normal development a sub-population of luminal cells can give rise to myoepithelial cells.93 Sartorius et al. suggest that progestins may lead to differentiation of tumors characterized by a transition from luminal to myoepithelial phenotype.92 In this regard, expression of myoepithelial cytokeratins has been linked to poorly differentiated, grade 3 cancers, and a shorter overall disease free survival.94 This raises the possibility that exposure of tumors to progestins may have a detrimental effect on tumor phenotype.
B. Mammary Cancers in the Mouse
Mouse models have provided additional insight into the role of PR in the etiology of mammary cancer. Balb/c mice have been inoculated with MPA-induced but progestin-independent mammary carcinoma cell lines, 59-2-HI or 32-2-HI and allowed to grow tumors up to 50 mm2 in size. These mice were subsequently treated with synthetic antisense oligonucleotide against both PRA and PRB isoforms or with the PR antagonists RU-486 or ZK 299 for 10 days.95 Both treatment with antisense oligonucleotide against PR and treatments with PR antagonists reduce the tumor size and inhibit the proliferation of progestin-independent tumors. These results indicate that P signaling is essential for the maintenance of these tumors despite the fact that tumor growth is not progestin-dependent. P has been shown to upregulate genes, such as Bcl-XL, that inhibit apoptosis.59 Thus, it is possible that the anti-apoptotic activity of P could promote the maintenance of tumors without promoting growth.
Progesterone receptor knockout mice (PRKO), lacking both PRA and PRB, have been examined for abnormalities of mammary gland development and susceptibility to mammary tumor development.77, 96 PRKO mice are unable to develop ductal sidebranching and when treated with DMBA showed reduced mammary tumor incidence compared to wildtype controls.96 These results further suggest that PR and P responsiveness may be relevant to susceptibility to mammary tumorigenesis. Whether there is a differential susceptibility to DMBA-induced mammary cancer in PRA gene-deleted vs. PRB gene-deleted mice is currently unknown.
Interesting results have been obtained in studies of PRA overexpressing transgenic mice.97 These mice develop aberrant mammary epithelial structures that also exhibit distinct alterations in gene expression and growth potential associated with transformation. In the abnormal structures there is a decrease in p21 expression, an increase in cyclin D1 expression, an increase in cell proliferation, and a decrease in ER alpha expression. In the same mice, in mammary ducts with normal morphology there is a decrease in p21 expression only. These results suggest that abnormally high expression of PRA is associated with changes in morphology and gene expression associated with neoplasia. Parallel studies in PRB overexpressing transgenic mice revealed no apparent tumor-associated abnormalities when compared with wildtype mice.97
A functional relationship between PR and expression of BRCA1 has been described in the mouse mammary gland.90 Targeted deletion of BRCA1 exon11 causes increased mammary growth in pubertal mice, indicative of altered hormone sensitivity. P treatment of either intact or ovariectomized BRCA1 deficient mice produces an exaggerated P-induced growth response compared to wildtype mice. This study suggests that BRCA1 regulates the proliferative activity of P. It is not known if this effect is mediated by PRA and/or PRB. Based on the finding that PRB expression is lost in the normal breast tissue of women carrying BRCA1 and BRCA2 mutations, it is of interest to know if PRA or PRB isoform expression is altered in the BRCA1 exon 11 deleted mice.
The importance of P signaling for the tumor development in BRCA mutation carriers has been analyzed in double transgenic mice carrying deletion of BRCA1 exon 11 and inactivation of p53 gene, BRCA1f11/f11p53f5&6/f5&6Crec mice.98 The adult virgin BRCA1f11/f11p53f5&6/f5&6Crec mice show increased ductal sidebranching, precocious alveologenesis, and increased PRA expression compared to age-matched wildtype mice. Mammary tumors in BRCA1f11/f11p53f5&6/f5&6Crec mice first appear at 5 months of age, and by the 8 months of age all mice develop tumors. When mice have been treated with PR antagonist RU-486, no palpable tumors are detected by 12 months of age. Cell culture studies of epithelial cells isolated from BRCA1f11/f11p53f5&6/f5&6Crec mice and human BRCA1 depleted MCF10A cells, have shown that BRCA1 decreases the stability of PR protein through stimulation of PR polyubiquitination.98 These results indicate that in BRCA1 mutation carriers P signaling may be enhanced due to the increased stability of PR proteins. The observation that exogenous BRCA1 decreases the activity of PR in breast cancer cells 90 indicates a potential inhibitory effect of wildtype BRCA1 on progestin action in the normal mammary gland. These findings illustrate another level of complexity in the regulation of P action and raise the possibility that there may be additional PR regulatory factors that have not yet been identified.
VIII. PROGESTOGENIC ACTIVITY OF ENDOCRINE DISRUPTORS
Endocrine disruptors (ED) have become a pressing health concern in the modern world. While many studies have been conducted to investigate the detrimental effects attributed to the estrogenic activity of several EDs. However, it has recently been shown that some disruptors, in fact, have separate, independent progestogenic activity. Jung et al. 99 have shown that EDs, such as 4-tert-octylphenol, nonylphenol, and bisphenol A, have progestogenic activity in addition to estrogenic activity. In the mouse uterus, expression of calcium-binding protein, calbindin-D9k is predominantly regulated by P. The abovementioned EDs stimulate the expression of calbindin-D9k, and this stimulation is strongly inhibited by the PR antagonist RU-486. These data suggest that potential EDs should be tested not only for estrogenic but also for progestogenic activity and raises the possibility that EDs may have progestogenic activity in the mammary gland. Development of mammary gland markers of progestin-specific activity in animal models may facilitate screening for progestogenic activity of EDs. An additional important question to be answered is whether EDs have tissue-specific and/or PR isoform specific effects.
IX. SUMMARY AND CONCLUSION
Epidemiological studies have identified breast cancer risk factors related to reproductive history and exposure to exogenous hormones. Two key hormones associated with these risk factors are E and P. P has been shown to increase proliferation indices in the breast epithelium in premenopausal women during the luteal phase of the menstrual cycle and in postmenopausal women receiving combined E+P HRT, whereas HRT with E alone does not increase breast cancer risk. Therefore, progestins are implicated in breast cancer etiology. While the major focus has been studying the role of E in breast cancer etiology, less is known about the role of progestins.
Mechanistic studies carried out in breast cancer cell lines in vitro or as xenografts in vivo have provided important insight into the mechanisms of P action mediated by the two isoforms of the PR, PRA and PRB. These studies have shown that PRA and PRB regulate different genes and have demonstrated a number of different mechanisms by which liganded and unliganded PR can affect tumor cell proliferation and behavior in a PR isoform specific manner. Mechanistic studies in the mouse have revealed PR isoform-specific functions during normal mammary gland development. P action through PRB is essential for lobuloalveolar development during pregnancy. Less is known about the specific functions of PRA. It is proposed that PRA mediates sidebranching and regulates proliferation in a paracrine manner during normal mammary gland development. Interestingly, overexpression of PRA in mice causes changes in gene expression and mammary morphology associated with neoplasia that are not observed with PRB overexpression. Therefore, additional studies elucidating how P signaling through PRA contributes to mammary cancer development are needed.
At present, major gaps exist in our knowledge about the mechanisms of P action in the normal human breast. In normal adult premenopausal human breast both PRA and PRB are coexpressed in the same cell. The relative contributions of PRA and PRB in normal breast cell proliferation have not been identified. At the same time, the alteration of PRA:PRB ratio occurs at early stages of breast cancer development and the predominance of PRA is often associated with more aggressive tumor phenotype. In addition, normal breast tissues from BRCA1 and BRCA2 mutation carriers analyzed for alterations in PR isoform expression showed a predominance of PRA and a lack of PRB. Studies in cell cultures and in BRCA1 gene-deleted mice have shown that wildtype BRCA1 inhibits PR function and decreases PR stability. It is possible that the loss of inhibition of PRA activity in mutation carriers may contribute to the increased risk of breast cancer.
While studies on human breast tissues are challenging, a more detailed analysis of the normal human breast is needed to advance our knowledge about P action and the role of PR isoforms in the normal breast and in breast cancer. It is anticipated that studies of the normal development of the human breast can inform us about alterations in P action and responses in breast cancer. For example, ligand-independent transcriptional activity has been identified in a breast cancer cell line. It is not known if a similar phenomenon occurs in normal mammary cells. Additionally, analysis of PRA:PRB ratio throughout the development of human breast (puberty vs. adult nulliparous vs. adult parous vs. postmenopausal) may provide insights into how expression of PR isoforms is related to breast cancer development.
While the mouse has been a useful model for the study of progestin action in the normal breast, there are significant differences in PR isoform expression between the mouse and human. In this regard, PR isoform expression in the rat mammary gland appears to be more similar to the human breast and the rat model offers promise for elucidating PR actions that more closely resemble PR actions in the human. Further study of PR isoform expression and progestin effects in rat mammary cancers, which share important features of hormone-dependence with human breast cancer, is warranted.
There have been many advances in technologies for genetic manipulation and manipulation of signaling pathways in the mammary gland in vivo and in human mammary cells in vitro, as well as advances in gene expression profiling. Using a combination of these approaches it is anticipated that studies of P action and PR isoform-specific functions in the normal mammary gland and during tumor development in animal models can inform us about changes that occur in breast cancer. Most importantly, such studies may provide novel preventive, diagnostic, and therapeutic strategies for breast cancer.
Acknowledgments
The authors thank Dr. Jose Russo for providing images of human breast tissue and Dr. Mathew Edick for critical reading of the manuscript. This publication was made possible by the Breast Cancer and the Environment Research Centers grant number U01 ES012800 from the National Institute of Environmental Health Sciences (NIEHS), and the National Cancer Institute (NCI), NIH, DHHS. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NCI, NIH. This work was also supported by Department of Defense Breast Cancer Research Program Fellowship DAMD17-03-1-0605 to M.D.A.
References
- 1.MacMahon B, Trichopoulos D, Brown J, Andersen AP, Aoki K, Cole P, deWaard F, Kauraniemi T, Morgan RW, Purde M, Ravnihar B, Stromby N, Westlund K, Woo NC. Age at menarche, probability of ovulation and breast cancer risk. Int J Cancer. 1982;29(1):13–16. doi: 10.1002/ijc.2910290104. [DOI] [PubMed] [Google Scholar]
- 2.Kvale G, Heuch I. Menstrual factors and breast cancer risk. Cancer. 1988;62(8):1625–1631. doi: 10.1002/1097-0142(19881015)62:8<1625::aid-cncr2820620828>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 3.Henderson BE, Pike MC, Casagrande JT. Breast cancer and the oestrogen window hypothesis. Lancet. 1981;2(8242):363–364. doi: 10.1016/s0140-6736(81)90675-9. [DOI] [PubMed] [Google Scholar]
- 4.Clavel-Chapelon F. Differential effects of reproductive factors on the risk of pre-and postmenopausal breast cancer. Results from a large cohort of French women. Br J Cancer. 2002;86(5):723–727. doi: 10.1038/sj.bjc.6600124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab. 1999;84(12):4559–4565. doi: 10.1210/jcem.84.12.6194. [DOI] [PubMed] [Google Scholar]
- 6.Aupperlee M, Kariagina A, Osuch J, Haslam SZ. Progestins and breast cancer. Breast Dis. 2005;24:37–57. doi: 10.3233/bd-2006-24104. [DOI] [PubMed] [Google Scholar]
- 7.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21(3):427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 8.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 9.Greiser CM, Greiser EM, Doren M. Menopausal hormone therapy and risk of breast cancer: a meta-analysis of epidemiological studies and randomized controlled trials. Hum Reprod Update. 2005;11(6):561–573. doi: 10.1093/humupd/dmi031. [DOI] [PubMed] [Google Scholar]
- 10.Kumle M, Weiderpass E, Braaten T, Persson I, Adami HO, Lund E. Use of oral contraceptives and breast cancer risk: The Norwegian-Swedish Women’s Lifestyle and Health Cohort Study. Cancer Epidemiol Biomarkers Prev. 2002;11(11):1375–1381. [PubMed] [Google Scholar]
- 11.Marchbanks PA, McDonald JA, Wilson HG, Folger SG, Mandel MG, Daling JR, Bernstein L, Malone KE, Ursin G, Strom BL, Norman SA, Wingo PA, Burkman RT, Berlin JA, Simon MS, Spirtas R, Weiss LK. Oral contraceptives and the risk of breast cancer. N Engl J Med. 2002;346(26):2025–2032. doi: 10.1056/NEJMoa013202. [DOI] [PubMed] [Google Scholar]
- 12.Graham JD, Clarke CL. Expression and transcriptional activity of progesterone receptor A and progesterone receptor B in mammalian cells. Breast Cancer Res. 2002;4(5):187–190. doi: 10.1186/bcr450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108(4):465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 14.Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, Horwitz KB. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994;54(14):3868–3877. [PubMed] [Google Scholar]
- 15.Wei LL, Gonzalez-Aller C, Wood WM, Miller LA, Horwitz KB. 5′-Heterogeneity in human progesterone receptor transcripts predicts a new amino-terminal truncated “C”-receptor and unique A-receptor messages. Mol Endocrinol. 1990;4(12):1833–1840. doi: 10.1210/mend-4-12-1833. [DOI] [PubMed] [Google Scholar]
- 16.Wei LL, Norris BM, Baker CJ. An N-terminally truncated third progesterone receptor protein, PR(C), forms heterodimers with PR(B) but interferes in PR(B)-DNA binding. J Steroid Biochem Mol Biol. 1997;62(4):287–297. doi: 10.1016/s0960-0760(97)00044-7. [DOI] [PubMed] [Google Scholar]
- 17.Wei LL, Hawkins P, Baker C, Norris B, Sheridan PL, Quinn PG. An amino-terminal truncated progesterone receptor isoform, PRC, enhances progestin-induced transcriptional activity. Mol Endocrinol. 1996;10(11):1379–1387. doi: 10.1210/mend.10.11.8923464. [DOI] [PubMed] [Google Scholar]
- 18.Condon JC, Hardy DB, Kovaric K, Mendelson CR. Upregulation of the Progesterone Receptor (PR)-C Isoform in Laboring Myometrium by Activation of NF-{kappa}B May Contribute to the Onset of Labor through Inhibition of PR Function. Mol Endocrinol. 2006;20(4):764–775. doi: 10.1210/me.2005-0242. [DOI] [PubMed] [Google Scholar]
- 19.Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, Chambon P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. Embo J. 1990;9(5):1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraus WL, Montano MM, Katzenellenbogen BS. Cloning of the rat progesterone receptor gene 5′-region and identification of two functionally distinct promoters. Mol Endocrinol. 1993;7(12):1603–1616. doi: 10.1210/mend.7.12.8145766. [DOI] [PubMed] [Google Scholar]
- 21.Nardulli AM, Greene GL, O’Malley BW, Katzenellenbogen BS. Regulation of progesterone receptor messenger ribonucleic acid and protein levels in MCF-7 cells by estradiol: analysis of estrogen’s effect on progesterone receptor synthesis and degradation. Endocrinology. 1988;122(3):935–944. doi: 10.1210/endo-122-3-935. [DOI] [PubMed] [Google Scholar]
- 22.Read LD, Snider CE, Miller JS, Greene GL, Katzenellenbogen BS. Ligand-modulated regulation of progesterone receptor messenger ribonucleic acid and protein in human breast cancer cell lines. Mol Endocrinol. 1988;2(3):263–271. doi: 10.1210/mend-2-3-263. [DOI] [PubMed] [Google Scholar]
- 23.Schultz JR, Petz LN, Nardulli AM. Estrogen receptor alpha and Sp1 regulate progesterone receptor gene expression. Mol Cell Endocrinol. 2003;201(1–2):165–175. doi: 10.1016/s0303-7207(02)00415-x. [DOI] [PubMed] [Google Scholar]
- 24.Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002;143(12):4583–4591. doi: 10.1210/en.2002-220369. [DOI] [PubMed] [Google Scholar]
- 25.Petz LN, Nardulli AM. Sp1 binding sites and an estrogen response element half-site are involved in regulation of the human progesterone receptor A promoter. Mol Endocrinol. 2000;14(7):972–985. doi: 10.1210/mend.14.7.0493. [DOI] [PubMed] [Google Scholar]
- 26.Petz LN, Ziegler YS, Schultz JR, Kim H, Kemper JK, Nardulli AM. Differential regulation of the human progesterone receptor gene through an estrogen response element half site and Sp1 sites. J Steroid Biochem Mol Biol. 2004;88(2):113–122. doi: 10.1016/j.jsbmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Petz LN, Ziegler YS, Schultz JR, Nardulli AM. Fos and Jun inhibit estrogen-induced transcription of the human progesterone receptor gene through an activator protein-1 site. Mol Endocrinol. 2004;18(3):521–532. doi: 10.1210/me.2003-0105. [DOI] [PubMed] [Google Scholar]
- 28.Graham JD, Roman SD, McGowan E, Sutherland RL, Clarke CL. Preferential stimulation of human progesterone receptor B expression by estrogen in T-47D human breast cancer cells. J Biol Chem. 1995;270(51):30693–30700. doi: 10.1074/jbc.270.51.30693. [DOI] [PubMed] [Google Scholar]
- 29.Vienonen A, Syvala H, Miettinen S, Tuohimaa P, Ylikomi T. Expression of progesterone receptor isoforms A and B is differentially regulated by estrogen in different breast cancer cell lines. J Steroid Biochem Mol Biol. 2002;80(3):307–313. doi: 10.1016/s0960-0760(02)00027-4. [DOI] [PubMed] [Google Scholar]
- 30.Aupperlee M, Haslam S. Differential hormonal regulation and function of PR isoforms in normal adult mouse mammary gland. Endocrinology. 2007 doi: 10.1210/en.2006–1721. [DOI] [PubMed] [Google Scholar]
- 31.Aupperlee MD, Smith KT, Kariagina A, Haslam SZ. Progesterone receptor isoforms A and B: temporal and spatial differences in expression during murine mammary gland development. Endocrinology. 2005 Aug;146(8):3577–3588. doi: 10.1210/en.2005-0346. [DOI] [PubMed] [Google Scholar]
- 32.Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci U S A. 2003;100(17):9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mote PA, Johnston JF, Manninen T, Tuohimaa P, Clarke CL. Detection of progesterone receptor forms A and B by immunohistochemical analysis. J Clin Pathol. 2001;54(8):624–630. doi: 10.1136/jcp.54.8.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72(2):163–172. doi: 10.1023/a:1014820500738. [DOI] [PubMed] [Google Scholar]
- 35.Aupperlee MD, Smith KT, Kariagina A, Haslam SZ. Progesterone Receptor Isoforms A and B: Temporal and Spatial Differences in Expression During Murine Mammary Gland Development. Endocrinology. 2005;146(8):3577–3588. doi: 10.1210/en.2005-0346. [DOI] [PubMed] [Google Scholar]
- 36.Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res Treat. 1999;53(3):217–227. doi: 10.1023/a:1006186719322. [DOI] [PubMed] [Google Scholar]
- 37.Kariagina A, Aupperlee MD, Haslam SZ. Progesterone Receptor Isoforms and Proliferation in the Rat Mammary Gland During Development. Endocrinology. 2007 doi: 10.1210/en.2006-1493. [DOI] [PubMed] [Google Scholar]
- 38.Leonhardt SA, Boonyaratanakornkit V, Edwards DP. Progesterone receptor transcription and non-transcription signaling mechanisms. Steroids. 2003;68(10–13):761–770. doi: 10.1016/s0039-128x(03)00129-6. [DOI] [PubMed] [Google Scholar]
- 39.Roemer SC, Donham DC, Sherman L, Pon VH, Edwards DP, Churchill ME. Structure of the progesterone receptor-deoxyribonucleic acid complex: novel interactions required for binding to half-site response elements. Mol Endocrinol. 2006;20(12):3042–3052. doi: 10.1210/me.2005-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arnett-Mansfield RL, Graham JD, Hanson AR, Mote PA, Gompel A, Scurr LL, Gava N, de Fazio A, Clarke CL. Focal subnuclear distribution of progesterone receptor is ligand dependent and associated with transcriptional activity. Mol Endocrinol. 2007;21(1):14–29. doi: 10.1210/me.2006-0041. [DOI] [PubMed] [Google Scholar]
- 41.Yang Z, Wolf IM, Chen H, Periyasamy S, Chen Z, Yong W, Shi S, Zhao W, Xu J, Srivastava A, Sanchez ER, Shou W. FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor a isoform. Mol Endocrinol. 2006;20(11):2682–2694. doi: 10.1210/me.2006-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chadli A, Graham JD, Abel MG, Jackson TA, Gordon DF, Wood WM, Felts SJ, Horwitz KB, Toft D. GCUNC-45 is a novel regulator for the progesterone receptor/hsp90 chaperoning pathway. Mol Cell Biol. 2006;26(5):1722–1730. doi: 10.1128/MCB.26.5.1722-1730.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han SJ, DeMayo FJ, Xu J, Tsai SY, Tsai MJ, O’Malley BW. Steroid receptor coactivator (SRC)-1 and SRC-3 differentially modulate tissue-specific activation functions of the progesterone receptor. Mol Endocrinol. 2006;20(1):45–55. doi: 10.1210/me.2005-0310. [DOI] [PubMed] [Google Scholar]
- 44.Mukherjee A, Amato P, Allred DC, Fernandez-Valdivia R, Nguyen J, O’Malley BW, DeMayo FJ, Lydon JP. Steroid receptor coactivator 2 is essential for progesterone-dependent uterine function and mammary morphogenesis: insights from the mouse--implications for the human. J Steroid Biochem Mol Biol. 2006;102(1–5):22–31. doi: 10.1016/j.jsbmb.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 45.Jones MC, Fusi L, Higham JH, Abdel-Hafiz H, Horwitz KB, Lam EW, Brosens JJ. Regulation of the SUMO pathway sensitizes differentiating human endometrial stromal cells to progesterone. Proc Natl Acad Sci U S A. 2006;103(44):16272–16277. doi: 10.1073/pnas.0603002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Man JH, Li HY, Zhang PJ, Zhou T, He K, Pan X, Liang B, Li AL, Zhao J, Gong WL, Jin BF, Xia Q, Yu M, Shen BF, Zhang XM. PIAS3 induction of PRB sumoylation represses PRB transactivation by destabilizing its retention in the nucleus. Nucleic Acids Res. 2006;34(19):5552–5566. doi: 10.1093/nar/gkl691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore NL, Narayanan R, Weigel NL. Cyclin dependent kinase 2 and the regulation of human progesterone receptor activity. Steroids. 2007 doi: 10.1016/j.steroids.2006.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weigel NL, Moore NL. Cyclins, cyclin dependent kinases, and regulation of steroid receptor action. Mol Cell Endocrinol. 2007 doi: 10.1016/j.mce.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narayanan R, Edwards DP, Weigel NL. Human progesterone receptor displays cell cycle-dependent changes in transcriptional activity. Mol Cell Biol. 2005;25(8):2885–2898. doi: 10.1128/MCB.25.8.2885-2898.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lange CA. Making sense of cross-talk between steroid hormone receptors and intracellular signaling pathways: who will have the last word? Mol Endocrinol. 2004;18(2):269–278. doi: 10.1210/me.2003-0331. [DOI] [PubMed] [Google Scholar]
- 51.Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60(9):520–528. doi: 10.1016/j.biopha.2006.07.082. [DOI] [PubMed] [Google Scholar]
- 52.Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci U S A. 2000;97(3):1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pierson-Mullany LK, Lange CA. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol. 2004;24(24):10542–10557. doi: 10.1128/MCB.24.24.10542-10557.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vicent GP, Ballare C, Zaurin R, Saragueta P, Beato M. Chromatin remodeling and control of cell proliferation by progestins via cross talk of progesterone receptor with the estrogen receptors and kinase signaling pathways. Ann N Y Acad Sci. 2006;1089:59–72. doi: 10.1196/annals.1386.025. [DOI] [PubMed] [Google Scholar]
- 55.Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27(2):466–480. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lange CA, Richer JK, Horwitz KB. Hypothesis: Progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol. 1999;13(6):829–836. doi: 10.1210/mend.13.6.0290. [DOI] [PubMed] [Google Scholar]
- 57.Boonyaratanakornkit V, Edwards DP. Receptor mechanisms of rapid extranuclear signalling initiated by steroid hormones. Essays Biochem. 2004;40:105–120. doi: 10.1042/bse0400105. [DOI] [PubMed] [Google Scholar]
- 58.Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997;11(2):179–186. doi: 10.1101/gad.11.2.179. [DOI] [PubMed] [Google Scholar]
- 59.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277(7):5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- 60.Jacobsen BM, Richer JK, Sartorius CA, Horwitz KB. Expression profiling of human breast cancers and gene regulation by progesterone receptors. J Mammary Gland Biol Neoplasia. 2003;8(3):257–268. doi: 10.1023/b:jomg.0000010028.48159.84. [DOI] [PubMed] [Google Scholar]
- 61.Jacobsen BM, Schittone SA, Richer JK, Horwitz KB. Progesterone-Independent Effects of Human Progesterone Receptors (PRs) in Estrogen Receptor-Positive Breast Cancer: PR Isoform-Specific Gene Regulation and Tumor Biology. Mol Endocrinol. 2005;19(3):574–587. doi: 10.1210/me.2004-0287. [DOI] [PubMed] [Google Scholar]
- 62.Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE. Estrogen-Related Receptor {alpha}1 Transcriptional Activities Are Regulated in Part via the ErbB2/HER2 Signaling Pathway. Mol Cancer Res. 2007;5(1):71–85. doi: 10.1158/1541-7786.MCR-06-0227. [DOI] [PubMed] [Google Scholar]
- 63.Graham JD, Yager ML, Hill HD, Byth K, O’Neill GM, Clarke CL. Altered progesterone receptor isoform expression remodels progestin responsiveness of breast cancer cells. Mol Endocrinol. 2005;19(11):2713–2735. doi: 10.1210/me.2005-0126. [DOI] [PubMed] [Google Scholar]
- 64.Graham JD, Yeates C, Balleine RL, Harvey SS, Milliken JS, Bilous AM, Clarke CL. Progesterone receptor A and B protein expression in human breast cancer. J Steroid Biochem Mol Biol. 1996;56(1–6):93–98. doi: 10.1016/0960-0760(95)00226-x. [DOI] [PubMed] [Google Scholar]
- 65.Bray JD, Jelinsky S, Ghatge R, Bray JA, Tunkey C, Saraf K, Jacobsen BM, Richer JK, Brown EL, Winneker RC, Horwitz KB, Lyttle CR. Quantitative analysis of gene regulation by seven clinically relevant progestins suggests a highly similar mechanism of action through progesterone receptors in T47D breast cancer cells. J Steroid Biochem Mol Biol. 2005;97(4):328–341. doi: 10.1016/j.jsbmb.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 66.Groshong SD, Owen GI, Grimison B, Schauer IE, Todd MC, Langan TA, Sclafani RA, Lange CA, Horwitz KB. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1) Mol Endocrinol. 1997;11(11):1593–1607. doi: 10.1210/mend.11.11.0006. [DOI] [PubMed] [Google Scholar]
- 67.Han EK, Ng SC, Arber N, Begemann M, Weinstein IB. Roles of cyclin D1 and related genes in growth inhibition, senescence and apoptosis. Apoptosis. 1999;4(3):213–219. doi: 10.1023/a:1009618824145. [DOI] [PubMed] [Google Scholar]
- 68.Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol. 2007;21(2):359–375. doi: 10.1210/me.2006-0337. [DOI] [PubMed] [Google Scholar]
- 69.Gizard F, Robillard R, Gervois P, Faucompre A, Revillion F, Peyrat JP, Hum WD, Staels B. Progesterone inhibits human breast cancer cell growth through transcriptional upregulation of the cyclin-dependent kinase inhibitor p27Kip1 gene. FEBS Lett. 2005;579(25):5535–5541. doi: 10.1016/j.febslet.2005.08.084. [DOI] [PubMed] [Google Scholar]
- 70.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273(17):10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 71.Gizard F, Robillard R, Gross B, Barbier O, Revillion F, Peyrat JP, Torpier G, Hum DW, Staels B. TReP-132 Is a Novel Progesterone Receptor Coactivator Required for the Inhibition of Breast Cancer Cell Growth and Enhancement of Differentiation by Progesterone. Mol Cell Biol. 2006;26(20):7632–7644. doi: 10.1128/MCB.00326-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clarke RB, Howell A, Potten CS, Anderson E. Dissociation between steroid receptor expression and cell proliferation in the human breast. Cancer Res. 1997;57(22):4987–4991. [PubMed] [Google Scholar]
- 73.Kim NS, Kim HJ, Koo BK, Kwon MC, Kim YW, Cho Y, Yokota Y, Penninger JM, Kong YY. Receptor activator of NF-kappaB ligand regulates the proliferation of mammary epithelial cells via Id2. Mol Cell Biol. 2006;26(3):1002–1013. doi: 10.1128/MCB.26.3.1002-1013.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gonzalez-Suarez E, Branstetter D, Armstrong A, Dinh H, Blumberg H, Dougall WC. RANK Overexpression in Transgenic Mice with Mouse Mammary Tumor Virus Promoter-Controlled RANK Increases Proliferation and Impairs Alveolar Differentiation in the Mammary Epithelia and Disrupts Lumen Formation in Cultured Epithelial Acini. Mol Cell Biol. 2007;27(4):1442–1454. doi: 10.1128/MCB.01298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ayyanan A, Civenni G, Ciarloni L, Morel C, Mueller N, Lefort K, Mandinova A, Raffoul W, Fiche M, Dotto GP, Brisken C. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc Natl Acad Sci. 2006;103(10):3799–3804. doi: 10.1073/pnas.0600065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cross SS, Harrison RF, Balasubramanian SP, Lippitt JM, Evans CA, Reed MW, Holen I. Expression of receptor activator of nuclear factor kappabeta ligand (RANKL) and tumour necrosis factor related, apoptosis inducing ligand (TRAIL) in breast cancer, and their relations with osteoprotegerin, oestrogen receptor, and clinicopathological variables. Journal of Clinical Pathology. 2006;59(7):716–720. doi: 10.1136/jcp.2005.030031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA, Jr, Shyamala G, Conneely OM, O’Malley BW. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9(18):2266–2278. doi: 10.1101/gad.9.18.2266. [DOI] [PubMed] [Google Scholar]
- 78.Masters JR, Drife JO, Scarisbrick JJ. Cyclic Variation of DNA synthesis in human breast epithelium. J Natl Cancer Inst. 1977;58(5):1263–1265. doi: 10.1093/jnci/58.5.1263. [DOI] [PubMed] [Google Scholar]
- 79.Meyer JS. Cell proliferation in normal human breast ducts, fibroadenomas, and other ductal hyperplasias measured by nuclear labeling with tritiated thymidine. Effects of menstrual phase, age, and oral contraceptive hormones. Hum Pathol. 1977;8(1):67–81. doi: 10.1016/s0046-8177(77)80066-x. [DOI] [PubMed] [Google Scholar]
- 80.Going JJ, Anderson TJ, Battersby S, MacIntyre CC. Proliferative and secretory activity in human breast during natural and artificial menstrual cycles. Am J Pathol. 1988;130(1):193–204. [PMC free article] [PubMed] [Google Scholar]
- 81.Russo J, Russo IH. Development of Human Mammary Gland. In: Daniel CM, editor. The Mammary Gland Development, Regulation and Function. New York: Plenum Pub. Corp; 1987. [Google Scholar]
- 82.Williams G, Anderson E, Howell A, Watson R, Coyne J, Roberts SA, Potten CS. Oral contraceptive (OCP) use increases proliferation and decreases oestrogen receptor content of epithelial cells in the normal human breast. Int J Cancer. 1991;48(2):206–210. doi: 10.1002/ijc.2910480209. [DOI] [PubMed] [Google Scholar]
- 83.Fendrick JL, Raafat AM, Haslam SZ. Mammary gland growth and development from the postnatal period to postmenopause: ovarian steroid receptor ontogeny and regulation in the mouse. J Mammary Gland Biol Neoplasia. 1998;3(1):7–22. doi: 10.1023/a:1018766000275. [DOI] [PubMed] [Google Scholar]
- 84.Haslam SZ. The ontogeny of mouse mammary gland responsiveness to ovarian steroid hormones. Endocrinology. 1989;125(5):2766–2772. doi: 10.1210/endo-125-5-2766. [DOI] [PubMed] [Google Scholar]
- 85.Russo IH, Russo J. Role of hormones in mammary cancer initiation and progression. J Mammary Gland Biol Neoplasia. 1998;3(1):49–61. doi: 10.1023/a:1018770218022. [DOI] [PubMed] [Google Scholar]
- 86.Russo J, Russo IH. Molecular Basis of Breast Cancer: Prevention and Treatment. Berlin, Heidelberg: Springer-Verlag; 2004. [Google Scholar]
- 87.Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, Fuqua SA. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res. 2004;10(8):2751–2760. doi: 10.1158/1078-0432.ccr-03-0141. [DOI] [PubMed] [Google Scholar]
- 88.King TA, Gemignani ML, Li W, Giri DD, Panageas KS, Bogomolniy F, Arroyo C, Olvera N, Robson ME, Offit K, Borgen PI, Boyd J. Increased progesterone receptor expression in benign epithelium of BRCA1-related breast cancers. Cancer Res. 2004;64(15):5051–5053. doi: 10.1158/0008-5472.CAN-04-1283. [DOI] [PubMed] [Google Scholar]
- 89.Mote PA, Leary JA, Avery KA, Sandelin K, Chenevix-Trench G, Kirk JA, Clarke CL. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer. 2004;39(3):236–248. doi: 10.1002/gcc.10321. [DOI] [PubMed] [Google Scholar]
- 90.Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA, Rosen EM. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006;20(1):14–34. doi: 10.1210/me.2004-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sartorius CA, Shen T, Horwitz KB. Progesterone receptors A and B differentially affect the growth of estrogen-dependent human breast tumor xenografts. Breast Cancer Res Treat. 2003;79(3):287–299. doi: 10.1023/a:1024031731269. [DOI] [PubMed] [Google Scholar]
- 92.Sartorius CA, Harvell DM, Shen T, Horwitz KB. Progestins initiate a luminal to myoepithelial switch in estrogen-dependent human breast tumors without altering growth. Cancer Res. 2005;65(21):9779–9788. doi: 10.1158/0008-5472.CAN-05-0505. [DOI] [PubMed] [Google Scholar]
- 93.Pechoux C, Gudjonsson T, Ronnov-Jessen L, Bissell MJ, Petersen OW. Human mammary luminal epithelial cells contain progenitors to myoepithelial cells. Dev Biol. 1999;206(1):88–99. doi: 10.1006/dbio.1998.9133. [DOI] [PubMed] [Google Scholar]
- 94.Van de Rijn M, Perou CM, Tibshirani R, Haas P, Kallioniemi O, Kononen J, Torhorst J, Sauter G, Zuber M, Kochli OR, Mross F, Dieterich H, Seitz R, Ross D, Botstein D, Brown P. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am J Pathol. 2002;161(6):1991–1996. doi: 10.1016/S0002-9440(10)64476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lamb CA, Helguero LA, Giulianelli S, Soldati R, Vanzulli SI, Molinolo A, Lanari C. Antisense oligonucleotides targeting the progesterone receptor inhibit hormone-independent breast cancer growth in mice. Breast Cancer Res. 2005;7(6):R1111–1121. doi: 10.1186/bcr1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lydon JP, Ge G, Kittrell FS, Medina D, O’Malley BW. Murine mammary gland carcinogenesis is critically dependent on progesterone receptor function. Cancer Res. 1999;59(17):4276–4284. [PubMed] [Google Scholar]
- 97.Chou YC, Uehara N, Lowry JR, Shyamala G. Mammary epithelial cells of PR-A transgenic mice exhibit distinct alterations in gene expression and growth potential associated with transformation. Carcinogenesis. 2003;24(3):403–409. doi: 10.1093/carcin/24.3.403. [DOI] [PubMed] [Google Scholar]
- 98.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314(5804):1467–1470. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 99.Jung YW, Hong EJ, Choi KC, Jeung EB. Novel progestogenic activity of environmental endocrine disruptors in the upregulation of calbindin-D9k in an immature mouse model. Toxicol Sci. 2005;83(1):78–88. doi: 10.1093/toxsci/kfi015. [DOI] [PubMed] [Google Scholar]