

Abstract
The idea that some phenotypes bear a closer relationship to the biological processes that give rise to psychiatric illness than diagnostic categories has attracted considerable interest. Much effort has been devoted to finding such endophenotypes, partly because it is believed that the genetic basis of endophenotypes will be easier to analyse than that of psychiatric disease. This belief depends in part on the assumption that the effect sizes of genetic loci contributing to endophenotypes are larger than those contributing to disease susceptibility, hence increasing the chance that genetic linkage and association tests will detect them. We examine this assumption by applying meta-analytical techniques to genetic association studies of endophenotypes. We find that the genetic effect sizes of the loci examined to date are no larger than those reported for other phenotypes. A review of the genetic architecture of traits in model organisms also provides no support for the view that the effect sizes of loci contributing to phenotypes closer to the biological basis of disease is any larger than those contributing to disease itself. While endophenotype measures may afford greater reliability, it should not be assumed that they will also demonstrate simpler genetic architecture.
Introduction
This review examines what is known about the genetic architecture of endophenotypes and assesses the claim that endophenotypes are more suitable than psychiatric diagnostic categories for genetic dissection, by which we mean the application of genetic linkage and association strategies to identify genes associated with psychiatric disease and component mechanisms. We start with a brief introduction to endophenotypes and then proceed to examine examples where sufficient genetic data have accumulated for our purpose. We do not provide a comprehensive review of all endophenotypes employed in psychiatric genetics.
The concept of the endophenotype was introduced to psychiatry over 30 years ago by Gottesman & Shields (1973), but its popularity is more recent: there are eight PubMed entries before 2000 compared to 150 in the current century. Gottesman & Shields (1973) adapted the term from a 1966 paper that attributed the geographical distribution of grasshoppers to the insects’ ‘endophenotype’ (John & Lewis, 1966), a neologism alluding to a phenotype that was microscopic and internal, and therefore obscure to casual observation. Endophenotypes in psychiatry retain the notion of an internal process, but one that can be objectively measured, ideally in a robust and reliable fashion, a characteristic often lacking in the diseases with which they are associated.
Gottesman’s definition of an endophenotype is that it should be heritable, co-segregate with a psychiatric illness, yet be present even when the disease is not (i.e. state independent), and be found in non-affected family members at a higher rate than in the population (Gottesman & Gould, 2003). The criterion of state independence was modified to take into account the importance of epigenetic and developmental factors so that the endophenotype can be manifest only at a certain age and/or after a challenge (in the same way that a glucose challenge is used for a glucose tolerance test) (Hasler et al. 2006). Others have added criteria that require endophenotypes to be part of the causal process by which disease arises (Lavori et al. 2002), or at least be involved in a biologically plausible mechanism of pathogenesis (Tsuang et al. 1993; Castellanos & Tannock, 2002), or, following Almasy & Blangero (2001), require that an endophenotype ‘should be continuously quantifiable, should predict disorder probabilistically and should be closer to the site of primary causative agent (whether genetic or environmental) than to diagnostic categories’. It has also been suggested, that ‘priority should be given to endophenotypes that are based or anchored in neuroscience’ (Doyle et al. 2005).
Table 1 lists commonly investigated endophenotypes in psychiatry and reveals a number of features. First, a small number of endophenotypes have attracted disproportionate attention. Second, work on many endophenotypes often predates the development of the endophenotype concept and goes beyond its concern with genetics. Interest in endophenotypes reflects a longstanding interest in investigating the biological correlates of psychiatric disease. Third, endophenotypes can be categorized into six groups: anatomical, developmental, electrophysiological, metabolic, sensory or psychological/cognitive. Fourth, we identified papers claiming endophenotypes for just seven psychiatric disorders. Together, these observations indicate that there is plenty of scope to find new endophenotypes in psychiatry, both by analysis of other psychiatric disorders and by searching for new features that have the relevant characteristics.
Table 1.
Endophenotype measures in psychiatric disease
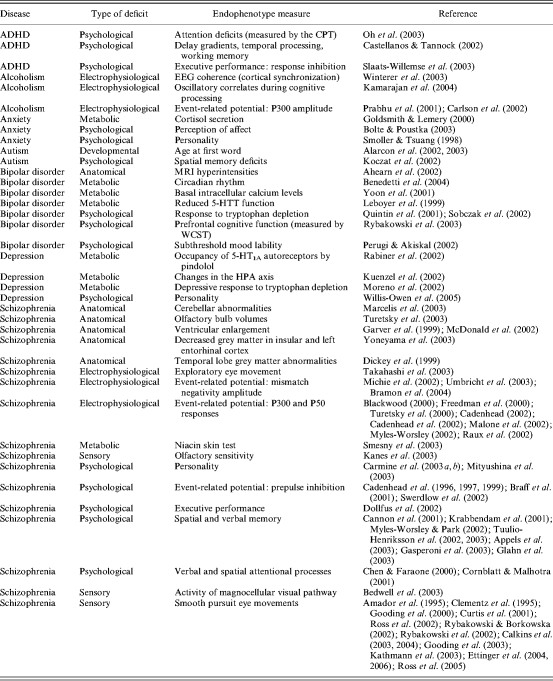
ADHD, Attention deficit hyperactivity disorder; CPT, Continuous Performance Test; WCST, Wisconsin Card Sorting Task.
One important reason for the popularity of endophenotypes is that they are believed to improve the chances of detecting at a molecular level the genetic variants that contribute to disease susceptibility (Freimer & Sabatti, 2003, 2004). We can summarize the argument as follows: endophenotypes provide a genetically tractable target, one that lies in the gap between gene and disease process, so that by dissecting their genetic basis we would understand something about the biology of a psychiatric disease. An example will clarify the idea.
Consider the difficulties of determining the molecular basis of schizophrenia. We have convincing evidence from twin, family and adoption studies that there is a substantial genetic contribution to the illness (Kendler & Diehl, 1993). Although diagnosis is not easy, the limitations of the diagnostic categories and the potential overlaps with other disorders are appreciated. Consequently we can categorize patients in ways that allow genetic analysis. Nevertheless, mapping genetic determinants by linkage or identifying genetic variants by association has proved extremely difficult, an observation that can be explained by assuming that schizophrenia has a complex genetic architecture, consisting of multiple genetic effects that individually alter susceptibility by a small degree and operate via interactions with each other and the environment (Riley & Kendler, 2006). The complex genetic architecture means that gene finding, using current methodologies, is extremely hard. Despite an enormous and rapidly growing molecular genetic literature, only a handful of candidate genes have been identified and there remains considerable controversy regarding the reliability of these findings (Riley & Kendler, 2006).
An attractive alternative is to work with endophenotypes because ‘endophenotypes represent more defined and quantifiable measures that are envisioned to involve fewer genes, fewer interacting levels and ultimately activation of a single set of neuronal circuits. The fewer the pathways that give rise to an endophenotype, the better the chances of efficiently discovering its genetic and neurobiological underpinnings’ (Gottesman & Gould, 2003). In short, the genetic basis of endophenotypes is assumed to be less complicated than that of their cognate psychiatric illness, their genetic determination is more direct, and, in consequence, they are potentially more tractable to genetic dissection than the disease states themselves. This presumption is made in most papers that seek to document endophenotypes, but has not so far been rigorously tested, due in large part to the paucity of relevant genetic studies. Our aim in this review is to find out whether the genetic basis of endophenotypes is indeed more tractable to genetic dissection than that of psychiatric disease. In order to do so, we need to establish criteria by which to assess whether a phenotype is likely to be genetically tractable.
Genetic effect sizes in psychiatric disorders
A critical measure of the genetic architecture of a phenotype is the effect size of a locus, by which we mean the extent to which a locus contributes to the variance of a phenotype or, as in the case here when disease outcome is involved, the extent to which it increases the risk of disease susceptibility. The power to detect an allele with a given relative risk depends on the size of the odds ratio, or percentage of phenotypic variance explained, and also on the allele frequency. For a locus with two alleles, power to detect is maximal when the allele frequency is 0·5 and the relative risk is large. This is the characteristic we expect to find at loci that contribute to disease susceptibility in a genetically tractable phenotype.
Can we establish the effect size that would make detection relatively straightforward? Instead of an absolute criterion for effect size, we can instead establish a relative one: we can compare the effect sizes of loci that contribute to psychiatric disease with those of loci contributing to variation in endophenotypes. Are the latter larger? Our problem is that there are very few robust estimates of the effect sizes for susceptibility loci in either psychiatric disease or endophenotypes. One approach to obtain good estimates is to combine studies using meta-analytical techniques.
Table 2 lists results from those meta-analyses of candidate genes for susceptibility to schizophrenia that found evidence of a positive genetic association. The summary presented suggests that genetic effects are small. The average odds ratio for the associations is 1·2, equivalent to ∼0·2% of the phenotypic variance, a figure consistent with that obtained from meta-analyses of other complex traits (Ioannidis et al. 2001).
Table 2.
Meta-analyses of case-control genetic association studies indicating possible association with schizophrenia
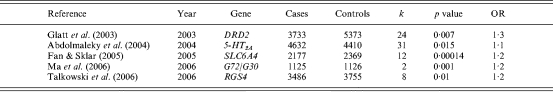
OR, Odds ratio.
This odds ratio is useful as it allows us also to estimate the size of studies needed to detect such effects and thereby assess the value of individual studies. Table 3 illustrates the total number of cases required (assuming that the total sample would in addition include an equal number of controls) in order to achieve 80% power to detect a range of effect sizes for a range of minor allele frequencies. As we can see, the majority of individual primary studies conducted are likely to be grossly under-powered to detect effect sizes of the magnitude indicated by the meta-analyses.
Table 3.
Power analysis
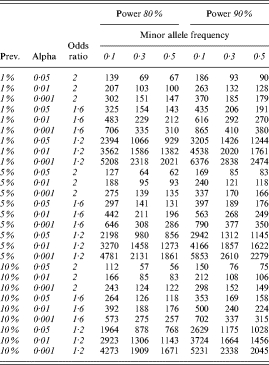
Prev., Prevalence.
For the discussion that follows next, it is important to consider the results of one negative meta-analysis. We recently conducted a meta-analysis of case-control genetic association studies of the COMT Val158/108 Met polymorphism in healthy control groups and clinically diagnosed schizophrenia patients (Munafo et al. 2005). The COMT gene has been regarded as a promising candidate gene for schizophrenia for some time, as it is the gene coding for the catechol O-methyltransferase enzyme, which inactivates catechols at post-synaptic sites in the human brain. COMT contains a functional polymorphism, a single nucleotide polymorphism at position 158/108 that results in a change from valine (Val) to methionine (Met) (Chen et al. 2004).
The results of our meta-analysis, which included data from 18 published studies, did not support an association between the COMT Val allele and schizophrenia case status. We concluded that if a genuine association exists between the COMT gene and schizophrenia, the risk conferred is likely to be extremely small, with the largest (albeit non-significant) effect size observed in our meta-analysis being an odds ratio of 1·13, accounting for less than 0·2% of the phenotypic variance. Therefore, given a Val allele frequency of 50% and a disease prevalence of 1%, a sample consisting of in excess of 900 cases and an equal number of controls would be required to detect this effect with 80% power at a relatively non-conservative alpha of 0·05 (Table 3).
Can endophenotype measures afford larger effect sizes?
We need next to examine the genetic architecture of endophenotypes. We start by considering some of the most heavily investigated endophenotypes in schizophrenia, measures of neuropsychological dysfunction, and particularly those that reflect activity in the prefrontal and temporo-limbic systems (Cannon et al. 2000). Abnormalities of working memory, episodic learning, attention and reaction time aggregate in the families of schizophrenia patients and probably arise from a common genetic susceptibility (Cannon et al. 2000), as expected for an endophenotype of schizophrenia.
One reason for the interest in working memory is because it may reflect function in the meso-cortical dopamine (DA) pathway. Experimental manipulation of DA metabolism by pharmacological challenge in humans and animals supports the assumption that the DA system is involved in cognition: reduced DA transmission in the rat prefrontal cortex impairs cortical-dependent cognition (Jentsch et al. 1997), administration of a selective inhibitor of COMT improves cognitive function (including working memory) in patients with Parkinson’s disease (Gasparini et al. 1997), while injection of D1 antagonists into the dorsolateral prefrontal cortex impairs tasks that require working memory (Sawaguchi & Goldman-Rakic, 1991; Williams & Goldman-Rakic, 1995). As mentioned above, COMT plays an important role in DA metabolism in the frontal cortex and the Val/Met polymorphism is a critical factor in determining COMT efficiency: the Val isoform has higher activity which may lead to lower synaptic DA levels in the prefrontal cortex (Chen et al. 2004). Thus there is a strong prior hypothesis that implicates DA regulation in prefrontal cortical function, and particularly that variation in COMT genotype could be associated with an endophenotype of schizophrenia, even if it is not associated with schizophrenia case status (Munafo et al. 2005). Consequently a number of investigators have carried out genetic studies of COMT genotype and cognitive function. These studies allow us to compare the effect sizes observed in case-control studies of psychiatric disease with those observed in endophenotype studies.
COMT genotype and Wisconsin Card Sorting Task performance
We carried out a meta-analysis of published studies reporting data on the association between COMT genotype and Wisconsin Card Sorting Task (WCST) performance (in particular, perserverative errors), a recognized measure of precortical function (Goldberg & Weinberger, 1988). Recall that our recent meta-analysis of COMT genotype and schizophrenia case status (Munafo et al. 2005) described above did not offer any evidence in support of association between the gene and the disease, so a positive association with the endophenotype would be evidence that the endophenotype strategy is more powerful, regardless of the effect size. Analysis of the association between COMT genotype and WCST performance should provide a good test of the value of the endophenotype approach in genetics.
We searched for relevant publications in PubMed, PsycInfo and Medline, up to 31 May 2006, using the search terms ‘Wisconsin’, ‘card sorting’, ‘WCST’, ‘COMT’, ‘catechol O-methyl transferase’. Bibliographies of all articles obtained in this way were hand-searched for additional references. Data extraction and analysis procedures were similar to those employed in our previous meta-analyses (Munafo et al. 2005). Briefly, a fixed-effects framework, using inverse variance methods, was employed initially, and the assumption that the effect of allele frequency is constant across samples checked using a χ2 test of goodness of fit for homogeneity. When there was evidence of significant association in the presence of significant between-sample heterogeneity, the analysis was re-run using a random-effects framework, using Der Simonian and Laird methods.
A total of 16 studies (Egan et al. 2001; Bilder et al. 2002; Joober et al. 2002; Malhotra et al. 2002; Mattay et al. 2003; Tsai et al. 2003; Rosa et al. 2004; Taerk et al. 2004; Bruder et al. 2005; Galderisi et al. 2005; Ho et al. 2005; Lipsky et al. 2005; Minzenberg et al. 2006; Ohnishi et al. 2006; Rybakowski et al. 2006; Szoke et al. 2006) published between 2001 and 2006 and comprising 26 independent samples were identified by the search strategy and met the inclusion criteria (Table 4). Where data were not available in an appropriate format for inclusion, we attempted to contact authors directly. Data from two studies (Egan et al. 2001; Taerk et al. 2004) comprised normalized t scores (i.e. higher scores reflecting better performance) and were reversed for consistency with other studies. One study (Ohnishi et al. 2006), comprising two independent samples, had to be excluded because we could not obtain appropriate data.
Table 4.
Studies of COMT genotype and WCST perseverative errors
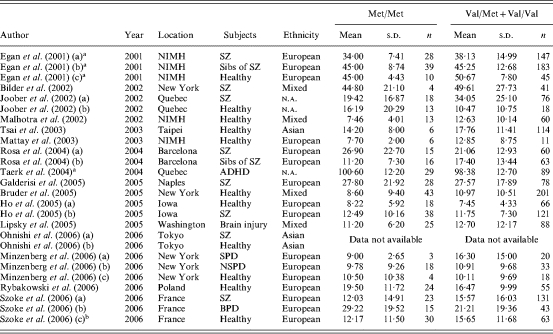
WCST, Wisconsin Card Sorting Task; NIMH, National Institute of Mental Health; SZ, schizophrenia; ADHD, attention deficit hyperactivity disorder; SPD, schizotypal personality disorder; NSPD, non-schizotypal personality disorder; BPD, bipolar disorder; n.a., not available.
Scoring of normalized t scores reversed to give consistent effect size estimate.
Includes healthy controls and unaffected first-degree relatives of SZ and BPD patients.
A fixed-effects model indicated evidence of association between COMT genotype and WCST performance (k=24, Z=1·97, p=0·048), with increased perseverative errors in the Val/Met+Val/Val group compared to the Met/Met group (d=0·10). There was no evidence of between-sample heterogeneity [χ2(23)=30·06, p=0·148]. These data are presented graphically in Fig. 1.
Fig. 1.

Meta-analysis of association of COMT genotype with WCST performance (perseverative errors). Meta-analysis indicates marginal evidence of association between COMT genotype and WCST performance, with a direction of effect consistent with increased perseverative errors in the Val/Met+Val/Val group compared to the Met/Met group. Bars represent individual study 95% confidence intervals, with a central block proportional to study size. The summary diamond bar represents the summary effect size estimate and 95% confidence interval.
Given evidence that the first published study of a genetic association frequently represents an over-estimation of the true effect size (Trikalinos et al. 2004), we re-ran our analysis excluding data from the three samples reported in the first published study (Egan et al. 2001). This resulted in non-significant evidence for association (p=0·166) and a reduction in the pooled effect-size estimate (d=0·08).
Our meta-analysis shows that if a genuine association exists between the COMT gene and WCST performance, the likely effect size is a standardized mean difference of 0·10, accounting for less than 0·5% of the phenotypic variance. Therefore, given a Val allele frequency of 50%, a sample consisting of in excess of 1700 subjects would be required to detect this effect with 80% power at a relatively non-conservative alpha of 0·05.
One potential criticism of the use of WCST performance as a schizophrenia endophenotype is that it reflects a complex behavioural phenotype which may only partially recruit prefrontal function. A more cognitively ‘pure’ task which more specifically recruits the prefrontal cortex may, therefore, represent a better assay for the purposes of genetic dissection. We consider next one such phenotype, the N-Back working memory task.
It is well-established that schizophrenia is associated with a deficit in working memory (Lee & Park, 2005), and is regarded as a cardinal cognitive symptom. The N-Back task requires participants to monitor a series of stimuli and to respond whenever a stimulus is presented that is the same as the one presented N trials previously, where N is a pre-specified integer, usually 1, 2, or 3. The task requires online monitoring, updating, and manipulation of remembered information and therefore places demands on key processes within working memory. Across studies, many different types of stimuli have been used via various input modalities (visuospatial, auditory, and olfactory) making demands on different processing systems.
A large number of studies have investigated working-memory performance in schizophrenia. Meta-analysis of these studies confirms that task performance is impaired in schizophrenia more than in controls (Lee & Park, 2005), and furthermore, working-memory tasks such as the N-Back have been demonstrated in fMRI investigations to heavily recruit the prefrontal cortex (Carpenter et al. 2000); working-memory performance is also highly heritable (Ando et al. 2001). N-Back task performance phenotypes satisfy the criteria for an endophenotype of schizophrenia.
COMT genotype and N-Back performance
We carried out a meta-analysis of published studies reporting data on the association between COMT genotype and N-Back performance. Recall that a potential criticism of our analysis of WCST data is that the latter represent a complex behavioural phenotype which only partially recruits prefrontal function. Analysis of the association between COMT genotype and N-Back performance allows us to investigate an endophenotype that is less subject to this criticism.
We searched for relevant publications in PubMed, PsycInfo and Medline, up to 31 May 2006, using the search terms ‘N-Back’, ‘working memory’, ‘COMT’, ‘catechol O-methyl transferase’. Data extraction and analysis procedures were identical to those described above.
A total of four studies (Goldberg et al. 2003; Stefanis et al. 2004; Bruder et al. 2005; Bertolino et al. 2006) published between 2003 and 2006 and comprising six independent samples were identified by the search strategy and met the inclusion criteria (Table 5). Where data were not available in an appropriate format for inclusion, we attempted to contact authors directly.
Table 5.
Studies of COMT genotype and N-Back 2 performance (% accuracy)
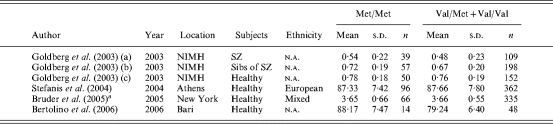
NIMH, National Institute of Mental Health; SZ, schizophrenia; n.a., not available.
Overall measure of sensitivity for detecting targets and avoiding non-targets (d′).
A fixed-effects model indicated marginal evidence of association between COMT genotype and N-Back performance (k=6, Z=1·96, p=0·050), with a direction of effect consistent with decreased accuracy in the Val/Met+Val/Val group compared to the Met/Met group (d=0·12). There was evidence of significant between-sample heterogeneity [χ2(4)=18·44, p<0·002] and when the analysis was re-run using a random-effects model the evidence for association was no longer significant (p=0·085). These data are presented graphically in Fig. 2.
Fig. 2.

Meta-analysis of association of COMT genotype with N-Back performance (2-Back). Meta-analysis indicates marginal evidence of association between COMT genotype and N-Back 2 performance, with a direction of effect consistent with decreased accuracy in the Val/Met+Val/Val group compared to the Met/Met group. Bars represent individual study 95% confidence intervals, with a central block proportional to study size. The summary diamond bar represents the summary effect size estimate and 95% confidence interval.
Assuming that a genuine association exists between the COMT gene and N-Back performance, the likely effect size indicated in our meta-analysis is a standardized mean difference of 0·12, accounting for less than 0·5% of the phenotypic variance. Therefore, given a Val allele frequency of 50%, a sample consisting of in excess of 1700 subjects would be required to detect this effect with 80% power at a relatively non-conservative alpha of 0·05.
One further criticism of the data which we present above is that the genetic architecture of a WCST and N-Back performance may be more complex than that of other endophenotypes. Perhaps other endophenotypes, for example those that involve physiological or anatomical measures, are more proximal to their genetic antecedents. We consider next one such phenotype, the P300 event-related potential.
There has been considerable interest in event-related potentials as a biological marker of schizophrenia from the early 1970s (Roth & Cannon, 1972) when reduced amplitude and delayed latency of the P300 waveform were first reported. An auditory, visual, or somatosensory random and salient stimulus is presented to subjects who must make a response, such as pressing a button. Around 300 ms after the stimulus, an event-related potential can be detected across the scalp. This P300 waveform recorded from the scalp has several components that reflect selective attention and working memory in response to an unexpected change in the environment.
A large number of studies have investigated the P300 in schizophrenia. Meta-analysis of these studies confirms that P300 amplitude reductions and latency delays occur in schizophrenia more than in controls (Bramon et al. 2005); furthermore P300 phenotypes are heritable (van Beijsterveldt & van Baal, 2002). P300 phenotypes satisfy the criteria for an endophenotype of schizophrenia.
COMT genotype and schizophrenia P300 amplitude and latency
We carried out a meta-analysis of published studies reporting data on the association between COMT genotype and P300 amplitude and latency. Recall that a potential criticism of our analysis of WCST and N-Back data is that they do not represent a truly biological endophenotype marker. Analysis of the association between COMT genotype and P300 amplitude and latency should provide a best-case scenario for the value of the endophenotype approach in genetics.
We searched for relevant publications in PubMed, PsycInfo and Medline, up to 31 May 2006, using the search terms ‘P300’, ‘ERP’, ‘event-related potential’, ‘COMT’, ‘catechol O-methyl transferase’. Data extraction and analysis procedures were identical to those described above.
A total of three studies (Gallinat et al. 2003; Tsai et al. 2003; Bramon et al. 2006) published between 2003 and 2006 and comprising six independent samples were identified by the search strategy and met the inclusion criteria (Table 6). Where data were not available in an appropriate format, we attempted to contact authors directly, which resulted in all available data being included in the meta-analysis.
Table 6.
Studies of COMT genotype and P300 amplitude and latency
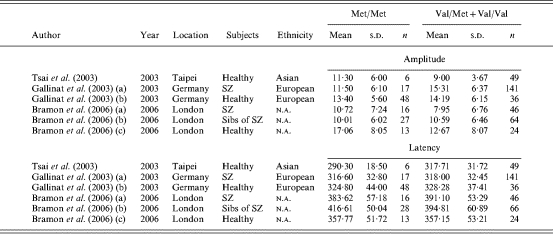
SZ, Schizophrenia; n.a., not available.
A fixed-effects model indicated no evidence of association between COMT genotype and P300 amplitude (k=6, Z=0·14, p=0·887) and latency (Z=0·14, p=0·892), although the direction of effect was consistent with increased amplitude and latency in the Val/Met+Val/Val group compared to the Met/Met group (d=0·02 and d=0·05 respectively). There was evidence of significant between-sample heterogeneity in the case of amplitude [χ2(5)=11·99, p=0·035] but not latency [χ2(5)=7·22, p=0·204]. These data are presented graphically in Fig. 3.
Fig. 3.

Meta-analysis of association of COMT genotype with P300 amplitude and latency. Meta-analysis indicates no evidence of association between COMT genotype and P300 amplitude (a) and latency (b), with a direction of effect consistent with increased amplitude and latency in the Val/Met+Val/Val group compared to the Met/Met group. Bars represent individual study 95% confidence intervals, with a central block proportional to study size. The summary diamond bar represents the summary effect size estimate and 95% confidence interval.
The likely effect size attributable to the COMT gene, if genuine, is therefore a standardized mean difference of 0·02, accounting for ∼0·01% of the phenotypic variance.
We can summarize the results of the meta-analyses by noting that the sample sizes required to detect the genetic loci contributing to endophenotypes are of equivalent size to those required to analyse the cognate psychiatric disease. In short, we find no evidence that the genetic architecture of endophenotypes is any simpler than that of psychiatric illness.
Conclusions
We have so far considered examples where we consider there to be sufficient genetic data available to arrive at an estimate of the effect size. Our conclusions, that the effect sizes found in genetic studies of endophenotypes are not considerably greater than those found in psychiatric diseases, may be biased because we have been limited to studying the COMT locus, or because so far no large effects have been found. The first issue is difficult to tackle. Significant associations reported for other genetic loci are based on either a single, or a very small number, of studies, so that we do not yet know how robust these findings are.
Two examples illustrate the point. The first concerns fMRI measures which are difficult to obtain in large numbers of people. Hariri and colleagues reported that a polymorphism in the serotonin transporter gene (5-HTT) was associated with the response of the amygdala to fearful stimuli (Hariri et al. 2002). In a comparison of two groups of 14 individuals, carriers of the s allele at the 5-HTT gene promoter were found to exhibit an increased amygdala fearful response compared with those homozygous for the l allele (Hariri et al. 2002): the means of the two groups were 0·28 (s.d.=0·22) and 0·03 (s.d.=0·19) with respect to % blood-oxygen-level-dependent (BOLD) signal change for fearful stimuli compared to neutral stimuli. In comparison with the data we have reviewed, this represents an enormous effect size (equivalent to ∼40% of phenotypic variance) that could be detected at a significance threshold of 0·05 with just 18 subjects. This finding could be due to chance statistical fluctuation. Indeed, a subsequent study of 92 individuals (including 19 from the first study) carried out by the same group again showed a significant effect, but with a reduction in the effect size. The reported mean values were this time 0·16 and 0·03 for the two groups (Hariri et al. 2005), equivalent to an effect size of just over 10% of the phenotypic variance. Additional studies are needed to confirm whether the effect is indeed this large.
The second example is the investigation into the genetic basis of an event-related potential, the P50. This is another endophenotype for schizophrenia, in which two stimuli are presented. At interstimulus intervals exceeding 8 s, two event-related potentials are detected. If the second stimulus occurs within 8 s of the first, normal subjects inhibit the response to the second stimulus. Schizophrenia subjects have a deficit in such inhibition, a finding supported by a recent meta-analysis (Bramon et al. 2005). The difference in inhibition is maximal when the second stimulus follows the first by 500 ms (Adler et al. 1982).
Interest in the molecular basis of the P50 phenotype was spurred by a 1997 report that a locus on chromosome 15 had been identified by linkage mapping (Freedman et al. 1997). The report was important because the locus contained a gene, the alpha 7-nicotinic acid receptor, which was later reported by the same group to be significantly associated with failure to inhibit the P50 auditory-evoked potential (Leonard et al. 2002). Furthermore, the group has presented evidence that functional variants in the promoter of the gene are present at significantly greater frequencies in schizophrenia subjects compared to controls (Leonard et al. 2002). A second group has independently reported that a 2 bp deletion in exon 6 of the gene and a polymorphism in the promoter are associated with the P50 phenotype (Raux et al. 2002; Houy et al. 2004) but not with schizophrenia. Clearly, given the findings for other complex traits, these results must be replicated in large samples if they are to be regarded as genuine.
The two examples encourage the view that some endophenotypes may be more genetically tractable than the ones we have discussed. Our concern is that as additional data accumulate these findings may turn out to be false positives, or at least the effect sizes will be much smaller than initially reported, as has often been found with genetic association studies (Trikalinos et al. 2004). Indeed, removing samples from the first published study in our analysis of WCST data rendered the association with COMT genotype non-significant and reduced the pooled effect size estimate by 20%.
The second issue is whether the current failure to find large genetic effects in endophenotypes can be read as a general indication of the complexity of genetic architecture for all phenotypes. Endophenotypes are assumed to have a relatively simple genetic architecture because there are relatively few pathways from gene to phenotype. The consequence is that sequence variants interact relatively directly with the phenotype so the correlation should be easier to detect. We have so far examined this assumption by investigating what is known about the genetic architecture of commonly investigated endophenotypes, and have shown that there is little evidence that it is considerably simpler than that of psychiatric disease. Perhaps we happen to have selected those phenotypes that have a complex genetic architecture. In the absence of detailed genetic analyses of multiple endophenotypes we cannot gainsay this point. However, we are able to approach this question from another point of view. We can ask what is known about the genetic architecture of phenotypes which have a much closer relationship to their genetic basis than endophenotypes for psychiatric disease.
Analyses of model organisms provide the relevant information. We will discuss two examples. The first is genetic analysis of phenotypes in the mouse, from which we have robust genome-wide association data for multiple phenotypes, behavioural and physiological, and associated estimates of locus effect sizes. These data allow us to compare the genetic architecture of behavioural phenotypes with those that would qualify as endophenotypes: for instance measures of electrophysiology, biochemistry, haematology and immunology. The drawback is that mouse models of psychiatric disease are imperfect, so that inferences drawn from the mouse data may be misleading. Nevertheless, we have no reason to expect the relationship between endophenotypes and behavioural phenotypes to be different in rodents and humans.
Reviews of the distribution of locus effect sizes show no difference between physiological and behavioural phenotypes (Flint et al. 2005). Moreover, in the most detailed analysis to date of the genetic architecture of complex traits in the mouse, among phenotypes there was no significant difference in the number or effect size of loci detected (Valdar et al. 2006). Intriguingly, regardless of the phenotype, the genetic effects that were detected explained about the same proportion of the additive variance, suggesting considerable similarity in the genetic architecture of many phenotypes (Valdar et al. 2006). Fig. 4 shows the effect sizes of 843 quantitative trait loci (QTL). The phenotypes include measures of anxiety and learning and memory, as well as haematology, immunology, biochemistry, physiology and anatomy. A full description is given in Valdar et al. (2006) and on a website (http://gscan.well.ox.ac.uk). Fig. 4 shows that preponderance of small effects. For each of the 100 phenotypes analysed, many loci contribute a small proportion to the variance. Large effect QTL are rare: only ten account for greater than 5% of phenotypic variance, and the mean is 2·2%.
Fig. 4.

Distribution of effect sizes of 843 mouse quantitative trait loci (QTL).
The mouse data indicate that we would not have obtained a simpler genetic architecture by working with any of the physiological, immunological, biochemical or haematological phenotypes in place of the behavioural phenotypes. We would still face the currently demanding challenge of having to identify the molecular basis of many small genetic effects.
The second example addresses the question of the relationship between genetic variants and phenotypes at an extremely immediate level, namely the correlation between DNA sequence variants and variation in the relative abundance of mRNA. Variation in transcript abundance is heritable and it is reasonable to expect that the variation in expression of some genes may correlate with psychiatric disease. Thus a gene expression profile could act as an endophenotype, although currently we do not know which genes show expression patterns correlated with psychiatric disease. Compared to any of the endophenotypes so far analysed (Table 1), variation in the amount of transcript is more proximal to DNA sequence variation and, if the assumptions about the nature of an endophenotype are correct, the genetic architecture of transcript variation should be relatively simple.
Analyses of gene expression variation in yeast, rodents and humans concur in finding that the genetic architecture of gene expression is polygenic and that the genetic effects are relatively small (Morley et al. 2004; Brem & Kruglyak, 2005; Chesler et al. 2005; Hubner et al. 2005). In yeast, where we have the most reliable estimates, relatively few transcripts have large effects: the median effect size was equivalent to 27% of variation in transcript level, only 16% of loci accounted for more than 60% of variation, and a quarter explained less than 10% (Brem & Kruglyak, 2005). Although estimates of effect size are not as robust in rodents, the available data indicate that the effect sizes are comparable to those found in yeast (Morley et al. 2004; Chesler et al. 2005; Hubner et al. 2005).
Although the effect sizes of loci contributing to variation in mRNA transcript abundance are larger than the effects found in complex traits (which explain less than 1% of the phenotypic variance) effect sizes are relative to the population in which they are measured. A reasonable comparison for the rodent mRNA data is with the effect sizes of QTL found in crosses between inbred strains of mice. Remarkably, the median effect size of QTL is 12% (Flint et al. 2005), just under half that of the expression phenotypes. Therefore, even when we consider a phenotype that is directly linked to the genetic constitution of the organism, genetic architecture is not radically different from complex phenotypes.
We have reviewed what is known about the genetic basis of endophenotypes and shown that their genetic architecture may be as complex as that of psychiatric disease. This does not mean there is no advantage to the use of endophenotypes for genetic studies. We have pointed out that the robust, quantitative measures that are typical of many endophenotypes means that they may be suitable for collecting the large samples needed for genetic analysis of complex traits, and may afford more statistically reliable data. We suggest that, along with the frequency and penetrance of a disease-causing allele given in Table 3, the ease and reliability of phenotyping should be factored into power calculations.
There are important limitations to our analysis. First, we have little reliable data about the genetic basis of complex traits in general and psychiatric endophenotypes in particular. Assumptions about the genetic architecture of complex traits depend so far largely on negative findings: our inability to detect robust linkage and association signals is due to lack of power and we have not sufficiently appreciated the genetic complexity. It is possible that, with the completion of the first whole genome association studies when estimates of effect size across the genome are available, a different picture will emerge. A second important limitation is that our review has concentrated on the effect of COMT on endophenotypes. Unfortunately there are no other examples where sufficient data have accumulated to be included in meta-analyses. Again, the availability of additional data might alter our results.
However, we think that our conclusions are unlikely to change much: first, studies in genetically more tractable organisms, such as yeast, flies and rodents, confirm the finding of genetic complexity for all phenotypes. The results are not here based on negative results: we have definite evidence of complexity. Second, as we have shown in the example of the genetic analysis of transcriptional abundance, there is no indication that alternative phenotypes will be any easier to deal with. Thus, while endophenotypes may be useful for many reasons, such as providing trait markers of susceptibility to psychiatric illness, for providing biological markers of disease and models for investigating disease process, we do not think they are likely to be any easier to dissect at a genetic level than the disorders to which they are related.
Acknowledgements
Jonathan Flint is supported by the Wellcome Trust. The authors are grateful to Taane Clark for assistance with the power calculations, and to Alessandro Bertolino, Elvira Bramon, Gerard Bruder, Jürgen Gallinat, Terry Goldberg, Beng Ho, Ridha Joober, Venkata Mattay, Andreas Meyer-Lindenberg, Michael Minzenberg, Araceli Rosa, Nicholas Stefanis and Daniel Weinberger, who kindly agreed to release their data to us in a format which enabled inclusion in the meta-analyses we report.
Declaration of Interest
None.
References
- Abdolmaleky H. M., Faraone S. V., Glatt S. J., Tsuang M. T.. Meta-analysis of association between the T102C polymorphism of the 5HT2a receptor gene and schizophrenia. Schizophrenia Research. 2004;67:53–62. doi: 10.1016/s0920-9964(03)00183-x. [DOI] [PubMed] [Google Scholar]
- Adler L. E., Pachtman E., Franks R. D., Pecevich M., Waldo M. C., Freedman R.. Neurophysiological evidence for a defect in neuronal mechanisms involved in sensory gating in schizophrenia. Biological Psychiatry. 1982;17:639–654. [PubMed] [Google Scholar]
- Ahearn E. P., Speer M. C., Chen Y. T., Steffens D. C., Cassidy F., Van Meter S., Provenzale J. M., Weisler R. H., Krishnan K. R.. Investigation of Notch3 as a candidate gene for bipolar disorder using brain hyperintensities as an endophenotype. American Journal of Medical Genetics. 2002;114:652–658. doi: 10.1002/ajmg.10512. [DOI] [PubMed] [Google Scholar]
- Alarcon M., Cantor R. M., Geschwind D. H.. Genetic etiology of autism endophenotypes. American Journal of Physical Anthropology. 2003;S36 , 57. [Google Scholar]
- Alarcon M., Cantor R. M., Liu J., Gilliam T. C., Geschwind D. H.. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. American Journal of Human Genetics. 2002;70:60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy L., Blangero J.. Endophenotypes as quantitative risk factors for psychiatric disease: rationale and study design. American Journal of Medical Genetics. 2001;105:42–44. [PubMed] [Google Scholar]
- Amador X. F., Malaspina D., Sackeim H. A., Coleman E. A., Kaufmann C. A., Hasan A., Gorman J. M.. Visual fixation and smooth pursuit eye movement abnormalities in patients with schizophrenia and their relatives. Journal of Neuropsychiatry and Clinical Neuroscience. 1995;7:197–206. doi: 10.1176/jnp.7.2.197. [DOI] [PubMed] [Google Scholar]
- Ando J., Ono Y., Wright M. J.. Genetic structure of spatial and verbal working memory. Behavior Genetics. 2001;31:615–624. doi: 10.1023/a:1013353613591. [DOI] [PubMed] [Google Scholar]
- Appels M. C., Sitskoorn M. M., Westers P., Lems E., Kahn R. S.. Cognitive dysfunctions in parents of schizophrenic patients parallel the deficits found in patients. Schizophrenia Research. 2003;63:285–293. doi: 10.1016/s0920-9964(02)00342-0. [DOI] [PubMed] [Google Scholar]
- Bedwell J. S., Brown J. M., Miller L. S.. The magnocellular visual system and schizophrenia: what can the color red tell us. Schizophrenia Research. 2003;63:273–284. doi: 10.1016/s0920-9964(02)00356-0. [DOI] [PubMed] [Google Scholar]
- Benedetti F., Bernasconi A., Lorenzi C., Pontiggia A., Serretti A., Colombo C., Smeraldi E.. A single nucleotide polymorphism in glycogen synthase kinase 3-beta promoter gene influences onset of illness in patients affected by bipolar disorder. Neuroscience Letters. 2004;355:37–40. doi: 10.1016/j.neulet.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Bertolino A., Blasi G., Latorre V., Rubino V., Rampino A., Sinibaldi L., Caforio G., Petruzzella V., Pizzuti A., Scarabino T., Nardini M., Weinberger D. R., Dallapiccola B.. Additive effects of genetic variation in dopamine regulating genes on working memory cortical activity in human brain. Journal of Neuroscience. 2006;26:3918–3922. doi: 10.1523/JNEUROSCI.4975-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder R. M., Volavka J., Czobor P., Malhotra A. K., Kennedy J. L., Ni X., Goldman R. S., Hoptman M. J., Sheitman B., Lindenmayer J. P., Citrome L., McEvoy J. P., Kunz M., Chakos M., Cooper T. B., Lieberman J. A.. Neurocognitive correlates of the COMT Val(158)Met polymorphism in chronic schizophrenia. Biological Psychiatry. 2002;52:701–707. doi: 10.1016/s0006-3223(02)01416-6. [DOI] [PubMed] [Google Scholar]
- Blackwood D.. P300, a state and a trait marker in schizophrenia. Lancet. 2000;355:771–772. doi: 10.1016/S0140-6736(99)00261-5. [DOI] [PubMed] [Google Scholar]
- Bolte S., Poustka F.. The recognition of facial affect in autistic and schizophrenic subjects and their first-degree relatives. Psycholigcal Medicine. 2003;33:907–915. doi: 10.1017/s0033291703007438. [DOI] [PubMed] [Google Scholar]
- Braff D. L., Geyer M. A., Light G. A., Sprock J., Perry W., Cadenhead K. S., Swerdlow N. R.. Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophrenia Research. 2001;49:171–178. doi: 10.1016/s0920-9964(00)00139-0. [DOI] [PubMed] [Google Scholar]
- Bramon E., Croft R. J., McDonald C., Virdi G. K., Gruzelier J. G., Baldeweg T., Sham P. C., Frangou S., Murray R. M.. Mismatch negativity in schizophrenia: a family study. Schizophrenia Research. 2004;67:1–10. doi: 10.1016/s0920-9964(03)00132-4. [DOI] [PubMed] [Google Scholar]
- Bramon E., Dempster E., Frangou S., McDonald C., Schoenberg P., MacCabe J. H., Walshe M., Sham P., Collier D., Murray R. M.. Is there an association between the COMT gene and P300 endophenotypes. European Psychiatry. 2006;21:70–73. doi: 10.1016/j.eurpsy.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Bramon E., McDonald C., Croft R. J., Landau S., Filbey F., Gruzelier J. H., Sham P. C., Frangou S., Murray R. M.. Is the P300 wave an endophenotype for schizophrenia? A meta-analysis and a family study. Neuroimage. 2005;27:960–968. doi: 10.1016/j.neuroimage.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Brem R. B., Kruglyak L.. The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proceedings of the National Academy of Sciences USA. 2005;102:1572–1577. doi: 10.1073/pnas.0408709102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruder G. E., Keilp J. G., Xu H., Shikhman M., Schori E., Gorman J. M., Gilliam T. C.. Catechol-O-methyltransferase (COMT) genotypes and working memory: associations with differing cognitive operations. Biological Psychiatry. 2005;58:901–907. doi: 10.1016/j.biopsych.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Cadenhead K. S.. Vulnerability markers in the schizophrenia spectrum: implications for phenomenology, genetics, and the identification of the schizophrenia prodrome. Psychiatric Clinics of North America. 2002;25:837–853. doi: 10.1016/s0193-953x(02)00021-7. [DOI] [PubMed] [Google Scholar]
- Cadenhead K. S., Carasso B. S., Swerdlow N. R., Geyer M. A., Braff D. L.. Prepulse inhibition and habituation of the startle response are stable neurobiological measures in a normal male population. Biological Psychiatry. 1999;45:360–364. doi: 10.1016/s0006-3223(98)00294-7. [DOI] [PubMed] [Google Scholar]
- Cadenhead K. S., Geyer M. A., Butler R. W., Perry W., Sprock J., Braff D. L.. Information processing deficits of schizophrenia patients: relationship to clinical ratings, gender and medication status. Schizophrenia Research. 1997;28:51–62. doi: 10.1016/s0920-9964(97)00085-6. [DOI] [PubMed] [Google Scholar]
- Cadenhead K. S., Light G. A., Geyer M. A., McDowell J. E., Braff D. L.. Neurobiological measures of schizotypal personality disorder: defining an inhibitory endophenotype. American Journal of Psychiatry. 2002;159:869–871. doi: 10.1176/appi.ajp.159.5.869. [DOI] [PubMed] [Google Scholar]
- Cadenhead K. S., Perry W., Braff D. L.. The relationship of information-processing deficits and clinical symptoms in schizotypal personality disorder. Biological Psychiatry. 1996;40:853–858. doi: 10.1016/0006-3223(95)00547-1. [DOI] [PubMed] [Google Scholar]
- Calkins M. E., Curtis C. E., Iacono W. G., Grove W. M.. Antisaccade performance is impaired in medically and psychiatrically healthy biological relatives of schizophrenia patients. Schizophrenia Research. 2004;71:167–178. doi: 10.1016/j.schres.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Calkins M. E., Iacono W. G., Curtis C. E.. Smooth pursuit and antisaccade performance evidence trait stability in schizophrenia patients and their relatives. International Journal of Psychophysiology. 2003;49:139–146. doi: 10.1016/s0167-8760(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Cannon T. D., Gasperoni T. L., van Erp T. G., Rosso I. M.. Quantitative neural indicators of liability to schizophrenia: implications for molecular genetic studies. American Journal of Medical Genetics. 2001;105:16–19. [PubMed] [Google Scholar]
- Cannon T. D., Huttunen M. O., Lonnqvist J., Tuulio-Henriksson A., Pirkola T., Glahn D., Finkelstein J., Hietanen M., Kaprio J., Koskenvuo M.. The inheritance of neuropsychological dysfunction in twins discordant for schizophrenia. American Journal of Human Genetics. 2000;67:369–382. doi: 10.1086/303006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson S. R., Iacono W. G., McGue M.. P300 amplitude in adolescent twins discordant and concordant for alcohol use disorders. Biological Psychology. 2002;61:203–227. doi: 10.1016/s0301-0511(02)00059-5. [DOI] [PubMed] [Google Scholar]
- Carmine A. Buervenich S. Galter D. Jonsson E. G. Sedvall G. C. Farde L. Gustavsson J. P. Bergman H. Chowdari K. V. Nimgaonkar V. L. Anvret M. Sydow O. Olson L. 2003aNURR1 promoter polymorphisms: Parkinson’s disease, schizophrenia, and personality traits American Journal of Medical Genetics. B: Neuropsychiatric Genetics 12051–57. [DOI] [PubMed] [Google Scholar]
- Carmine A. Chheda M. G. Jonsson E. G. Sedvall G. C. Farde L. Gustavsson J. P. Bergman H. Anvret M. Buervenich S. Olson L. 2003bTwo NOTCH4 polymorphisms and their relation to schizophrenia susceptibility and different personality traits Psychiatric Genetics 1323–28. [DOI] [PubMed] [Google Scholar]
- Carpenter P. A., Just M. A., Reichle E. D.. Working memory and executive function: evidence from neuroimaging. Current Opinion in Neurobiology. 2000;10:195–199. doi: 10.1016/s0959-4388(00)00074-x. [DOI] [PubMed] [Google Scholar]
- Castellanos F. X., Tannock R.. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nature Reviews Neuroscience. 2002;3:617–628. doi: 10.1038/nrn896. [DOI] [PubMed] [Google Scholar]
- Chen J., Lipska B. K., Halim N., Ma Q. D., Matsumoto M., Melhem S., Kolachana B. S., Hyde T. M., Herman M. M., Apud J., Egan M. F., Kleinman J. E., Weinberger D. R.. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. American Journal of Human Genetics. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. J., Faraone S. V.. Sustained attention deficits as markers of genetic susceptibility to schizophrenia. American Journal of Medical Genetics. 2000;97:52–57. doi: 10.1002/(sici)1096-8628(200021)97:1<52::aid-ajmg7>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Chesler E. J., Lu L., Shou S., Qu Y., Gu J., Wang J., Hsu H. C., Mountz J. D., Baldwin N. E., Langston M. A., Threadgill D. W., Manly K. F., Williams R. W.. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nature Genetics. 2005;37:233–242. doi: 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- Clementz B. A., Reid S. A., McDowell J. E., Cadenhead K. S.. Abnormality of smooth pursuit eye movement initiation: specificity to the schizophrenia spectrum. Psychophysiology. 1995;32:130–134. doi: 10.1111/j.1469-8986.1995.tb03304.x. [DOI] [PubMed] [Google Scholar]
- Cornblatt B. A., Malhotra A. K.. Impaired attention as an endophenotype for molecular genetic studies of schizophrenia. American Journal of Medical Genetics. 2001;105:11–15. [PubMed] [Google Scholar]
- Curtis C. E., Calkins M. E., Grove W. M., Feil K. J., Iacono W. G.. Saccadic disinhibition in patients with acute and remitted schizophrenia and their first-degree biological relatives. American Journal of Psychiatry. 2001;158:100–106. doi: 10.1176/appi.ajp.158.1.100. [DOI] [PubMed] [Google Scholar]
- Dickey C. C., McCarley R. W., Voglmaier M. M., Niznikiewicz M. A., Seidman L. J., Hirayasu Y., Fischer I., Teh E. K., Van Rhoads R., Jakab M., Kikinis R., Jolesz F. A., Shenton M. E.. Schizotypal personality disorder and MRI abnormalities of temporal lobe gray matter. Biological Psychiatry. 1999;45:1393–1402. doi: 10.1016/s0006-3223(99)00030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollfus S., Lombardo C., Benali K., Halbecq I., Abadie P., Marie R. M., Brazo P.. Executive/attentional cognitive functions in schizophrenic patients and their parents: a preliminary study. Schizophrenia Research. 2002;53:93–99. doi: 10.1016/s0920-9964(01)00156-6. [DOI] [PubMed] [Google Scholar]
- Doyle A. E., Faraone S. V., Seidman L. J., Willcutt E. G., Nigg J. T., Waldman I. D., Pennington B. F., Peart J., Biederman J.. Are endophenotypes based on measures of executive functions useful for molecular genetic studies of ADHD. Journal of Child Psychology and Psychiatry. 2005;46:774–803. doi: 10.1111/j.1469-7610.2005.01476.x. [DOI] [PubMed] [Google Scholar]
- Egan M. F., Goldberg T. E., Kolachana B. S., Callicott J. H., Mazzanti C. M., Straub R. E., Goldman D., Weinberger D. R.. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proceedings of the National Academy of Sciences USA. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger U., Kumari V., Crawford T. J., Corr P. J., Das M., Zachariah E., Hughes C., Sumich A. L., Rabe-Hesketh S., Sharma T.. Smooth pursuit and antisaccade eye movements in siblings discordant for schizophrenia. Journal of Psychiatric Research. 2004;38:177–184. doi: 10.1016/s0022-3956(03)00105-5. [DOI] [PubMed] [Google Scholar]
- Ettinger U., Picchioni M., Hall M. H., Schulze K., Toulopoulou T., Landau S., Crawford T. J., Murray R. M.. Antisaccade performance in monozygotic twins discordant for schizophrenia: the Maudsley twin study. American Journal of Psychiatry. 2006;163:543–545. doi: 10.1176/appi.ajp.163.3.543. [DOI] [PubMed] [Google Scholar]
- Fan J. B., Sklar P.. Meta-analysis reveals association between serotonin transporter gene STin2 VNTR polymorphism and schizophrenia. Molecular Psychiatry. 2005;10:928–938. doi: 10.1038/sj.mp.4001690. , 891. [DOI] [PubMed] [Google Scholar]
- Flint J., Valdar W., Shifman S., Mott R.. Strategies for mapping and cloning quantitative trait genes in rodents. Nature Reviews Genetics. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- Freedman R., Adams C. E., Adler L. E., Bickford P. C., Gault J., Harris J. G., Nagamoto H. T., Olincy A., Ross R. G., Stevens K. E., Waldo M., Leonard S.. Inhibitory neurophysiological deficit as a phenotype for genetic investigation of schizophrenia. American Journal of Medical Genetics. 2000;97:58–64. doi: 10.1002/(sici)1096-8628(200021)97:1<58::aid-ajmg8>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Freedman R., Coon H., Myles-Worsley M., Orr-Urtreger A., Olincy A., Davis A., Polymeropoulos M., Holik J., Hopkins J., Hoff M., Rosenthal J., Waldo M. C., Reimherr F., Wender P., Yaw J., Young D. A., Breese C. R., Adams C., Patterson D., Adler L. E., Kruglyak L., Leonard S., Byerley W.. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proceedings of the National Academy of Sciences USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimer N., Sabatti C.. The human phenome project. Nature Genetics. 2003;34:15–21. doi: 10.1038/ng0503-15. [DOI] [PubMed] [Google Scholar]
- Freimer N., Sabatti C.. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nature Genetics. 2004;36:1045–1051. doi: 10.1038/ng1433. [DOI] [PubMed] [Google Scholar]
- Galderisi S., Maj M., Kirkpatrick B., Piccardi P., Mucci A., Invernizzi G., Rossi A., Pini S., Vita A., Cassano P., Stratta P., Severino G., Del Zompo M.. Catechol-O-methyltransferase Val158Met polymorphism in schizophrenia: associations with cognitive and motor impairment. Neuropsychobiology. 2005;52:83–89. doi: 10.1159/000087096. [DOI] [PubMed] [Google Scholar]
- Gallinat J., Bajbouj M., Sander T., Schlattmann P., Xu K., Ferro E. F., Goldman D., Winterer G.. Association of the G1947A COMT (Val(108/158)Met) gene polymorphism with prefrontal P300 during information processing. Biological Psychiatry. 2003;54:40–48. doi: 10.1016/s0006-3223(02)01973-x. [DOI] [PubMed] [Google Scholar]
- Garver D. L., Nair T. R., Christensen J. D., Holcomb J., Ramberg J., Kingsbury S.. Atrophic and static (neurodevelopmental) schizophrenic psychoses: premorbid functioning, symptoms and neuroleptic response. Neuropsychopharmacology. 1999;21:82–92. doi: 10.1016/S0893-133X(98)00138-9. [DOI] [PubMed] [Google Scholar]
- Gasparini M., Fabrizio E., Bonifati V., Meco G.. Cognitive improvement during Tolcapone treatment in Parkinson’s disease. Journal of Neural Transmission. 1997;104:887–894. doi: 10.1007/BF01285556. [DOI] [PubMed] [Google Scholar]
- Gasperoni T. L., Ekelund J., Huttunen M., Palmer C. G., Tuulio-Henriksson A., Lonnqvist J., Kaprio J., Peltonen L., Cannon T. D.. Genetic linkage and association between chromosome 1q and working memory function in schizophrenia. American Journal of Medical Genetics. B: Neuropsychiatric Genetics. 2003;116:8–16. doi: 10.1002/ajmg.b.10757. [DOI] [PubMed] [Google Scholar]
- Glahn D. C., Therman S., Manninen M., Huttunen M., Kaprio J., Lonnqvist J., Cannon T. D.. Spatial working memory as an endophenotype for schizophrenia. Biological Psychiatry. 2003;53:624–626. doi: 10.1016/s0006-3223(02)01641-4. [DOI] [PubMed] [Google Scholar]
- Glatt S. J., Faraone S. V., Tsuang M. T.. Meta-analysis identifies an association between the dopamine D2 receptor gene and schizophrenia. Molecular Psychiatry. 2003;8:911–915. doi: 10.1038/sj.mp.4001321. [DOI] [PubMed] [Google Scholar]
- Goldberg T. E., Egan M. F., Gscheidle T., Coppola R., Weickert T., Kolachana B. S., Goldman D., Weinberger D. R.. Executive subprocesses in working memory: relationship to catechol-O-methyltransferase Val158Met genotype and schizophrenia. Archives of General Psychiatry. 2003;60:889–896. doi: 10.1001/archpsyc.60.9.889. [DOI] [PubMed] [Google Scholar]
- Goldberg T. E., Weinberger D. R.. Probing prefrontal function in schizophrenia with neuropsychological paradigms. Schizophrenia Bulletin. 1988;14:179–183. doi: 10.1093/schbul/14.2.179. [DOI] [PubMed] [Google Scholar]
- Goldsmith H. H., Lemery K. S.. Linking temperamental fearfulness and anxiety symptoms: a behavior-genetic perspective. Biological Psychiatry. 2000;48:1199–1209. doi: 10.1016/s0006-3223(00)01003-9. [DOI] [PubMed] [Google Scholar]
- Gooding D. C., Grabowski J. A., Hendershot C. S.. Fixation stability in schizophrenia, bipolar, and control subjects. Psychiatry Research. 2000;97:119–128. doi: 10.1016/s0165-1781(00)00226-2. [DOI] [PubMed] [Google Scholar]
- Gooding D. C., Mohapatra L., Shea H. B.. Temporal stability of saccadic task performance in psychotic patients. Psychophysiology. 2003;40:S43. doi: 10.1017/s003329170300165x. [DOI] [PubMed] [Google Scholar]
- Gottesman I. I., Gould T. D.. The endophenotype concept in psychiatry: etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gottesman I. I., Shields J.. Genetic theorizing and schizophrenia. British Journal of Psychiatry. 1973;122:15–30. doi: 10.1192/bjp.122.1.15. [DOI] [PubMed] [Google Scholar]
- Hariri A. R., Drabant E. M., Munoz K. E., Kolachana B. S., Mattay V. S., Egan M. F., Weinberger D. R.. A susceptibility gene for affective disorders and the response of the human amygdala. Archives of General Psychiatry. 2005;62:146–152. doi: 10.1001/archpsyc.62.2.146. [DOI] [PubMed] [Google Scholar]
- Hariri A. R., Mattay V. S., Tessitore A., Kolachana B., Fera F., Goldman D., Egan M. F., Weinberger D. R.. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297:400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- Hasler G., Drevets W. C., Gould T. D., Gottesman I. I., Manji H. K.. Toward constructing an endophenotype strategy for biopolar disorders. Biological Psychiatry. 2006;60:93–105. doi: 10.1016/j.biopsych.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Ho B. C., Wassink T. H., O’Leary D. S., Sheffield V. C., Andreasen N. C.. Catechol-O-methyl transferase Val158Met gene polymorphism in schizophrenia: working memory, frontal lobe MRI morphology and frontal cerebral blood flow. Molecular Psychiatry. 2005;10:287–298. doi: 10.1038/sj.mp.4001616. , 229, [DOI] [PubMed] [Google Scholar]
- Houy E., Raux G., Thibaut F., Belmont A., Demily C., Allio G., Haouzir S., Fouldrin G., Petit M., Frebourg T., Campion D.. The promoter-194 C polymorphism of the nicotinic alpha 7 receptor gene has a protective effect against the P50 sensory gating deficit. Molecular Psychiatry. 2004;9:320–322. doi: 10.1038/sj.mp.4001443. [DOI] [PubMed] [Google Scholar]
- Hubner N., Wallace C. A., Zimdahl H., Petretto E., Schulz H., Maciver F., Mueller M., Hummel O., Monti J., Zidek V., Musilova A., Kren V., Causton H., Game L., Born G., Schmidt S., Muller A., Cook S. A., Kurtz T. W., Whittaker J., Pravenec M., Aitman T. J.. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nature Genetics. 2005;37:243–253. doi: 10.1038/ng1522. [DOI] [PubMed] [Google Scholar]
- Ioannidis J. P., Ntzani E. E., Trikalinos T. A., Contopoulos-Ioannidis D. G.. Replication validity of genetic association studies. Nature Genetics. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- Jentsch J. D., Tran A., Le D., Youngren K. D., Roth R. H.. Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology. 1997;17:92–99. doi: 10.1016/S0893-133X(97)00034-1. [DOI] [PubMed] [Google Scholar]
- John B., Lewis K. R.. Chromosome variability and geographical distribution in insects: chromosome rather than gene variation provide the key to differences among populations. Science. 1966;152:711–721. doi: 10.1126/science.152.3723.711. [DOI] [PubMed] [Google Scholar]
- Joober R., Gauthier J., Lal S., Bloom D., Lalonde P., Rouleau G., Benkelfat C., Labelle A.. Catechol-O-methyltransferase Val-108/158-Met gene variants associated with performance on the Wisconsin Card Sorting Test. Archives of General Psychiatry. 2002;59:662–663. doi: 10.1001/archpsyc.59.7.662. [DOI] [PubMed] [Google Scholar]
- Kamarajan C., Porjesz B., Jones K. A., Choi K., Chorlian D. B., Padmanabhapillai A., Rangaswamy M., Stimus A. T., Begleiter H.. The role of brain oscillations as functional correlates of cognitive systems: a study of frontal inhibitory control in alcoholism. International Journal of Psychophysiology. 2004;51:155–180. doi: 10.1016/j.ijpsycho.2003.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanes S. J., Tokarczyk J., Chinitz J., Turetsky B., Moberg P., Bucan M., Gur R.. Olfaction: towards a novel mouse endophenotypic model of schizophrenia. Schizophrenia Research. 2003;60:S109. [Google Scholar]
- Kathmann N., Hochrein A., Uwer R., Bondy B.. Deficits in gain of smooth pursuit eye movements in schizophrenia and affective disorder patients and their unaffected relatives. American Journal of Psychiatry. 2003;160:696–702. doi: 10.1176/appi.ajp.160.4.696. [DOI] [PubMed] [Google Scholar]
- Kendler K. S., Diehl S. R.. The genetics of schizophrenia: a current, genetic-epidemiologic perspective. Schizophrenia Bulletin. 1993;19:261–285. doi: 10.1093/schbul/19.2.261. [DOI] [PubMed] [Google Scholar]
- Koczat D. L., Rogers S. J., Pennington B. F., Ross R. G.. Eye movement abnormality suggestive of a spatial working memory deficit is present in parents of autistic probands. Journal of Autism and Developmental Disorders. 2002;32:513–518. doi: 10.1023/a:1021246712459. [DOI] [PubMed] [Google Scholar]
- Krabbendam L., Marcelis M., Delespaul P., Jolles J., van Os J.. Single or multiple familial cognitive risk factors in schizophrenia. American Journal of Medical Genetics. 2001;105:183–188. doi: 10.1002/ajmg.1197. [DOI] [PubMed] [Google Scholar]
- Kuenzel H. E., Binder E. B., Nickel T., Ising M., Fuchs B., Modell S., Holsboer F.. The combined dexamethasone-suppression/corticotropin-releasing- hormone(CRH) stimulation test, a possible endophenotype for major depression. American Journal of Medical Genetics. 2002;114B:P36. [Google Scholar]
- Lavori P. W., Krause-Steinrauf H., Brophy M., Buxbaum J., Cockroft J., Cox D. R., Fiore L., Greely H. T., Greenberg H., Holmes E. W., Nelson L. M., Sugarman J.. Principles, organization, and operation of a DNA bank for clinical trials: a Department of Veterans Affairs cooperative study. Controlled Clinical Trials. 2002;23:222–239. doi: 10.1016/s0197-2456(02)00193-9. [DOI] [PubMed] [Google Scholar]
- Leboyer M., Quintin P., Manivet P., Varoquaux O., Allilaire J. F., Launay J. M.. Decreased serotonin transporter binding in unaffected relatives of manic depressive patients. Biological Psychiatry. 1999;46:1703–1706. doi: 10.1016/s0006-3223(99)00178-x. [DOI] [PubMed] [Google Scholar]
- Lee J., Park S.. Working memory impairments in schizophrenia: a meta-analysis. Journal of Abnormal Psychology. 2005;114:599–611. doi: 10.1037/0021-843X.114.4.599. [DOI] [PubMed] [Google Scholar]
- Leonard S., Gault J., Hopkins J., Logel J., Vianzon R., Short M., Drebing C., Berger R., Venn D., Sirota P., Zerbe G., Olincy A., Ross R. G., Adler L. E., Freedman R.. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Archives of General Psychiatry. 2002;59:1085–1096. doi: 10.1001/archpsyc.59.12.1085. [DOI] [PubMed] [Google Scholar]
- Lipsky R. H., Sparling M. B., Ryan L. M., Xu K., Salazar A. M., Goldman D., Warden D. L.. Association of COMT Val158Met genotype with executive functioning following traumatic brain injury. Journal of Neuropsychiatry and Clinical Neuroscience. 2005;17:465–471. doi: 10.1176/jnp.17.4.465. [DOI] [PubMed] [Google Scholar]
- Ma J., Qin W., Wang X. Y., Guo T. W., Bian L., Duan S. W., Li X. W., Zou F. G., Fang Y. R., Fang J. X., Feng G. Y., Gu N. F., St Clair D., He L.. Further evidence for the association between G72/G30 genes and schizophrenia in two ethnically distinct populations. Molecular Psychiatry. 2006;11:479–487. doi: 10.1038/sj.mp.4001788. [DOI] [PubMed] [Google Scholar]
- Malhotra A. K., Kestler L. J., Mazzanti C., Bates J. A., Goldberg T., Goldman D.. A functional polymorphism in the COMT gene and performance on a test of prefrontal cognition. American Journal of Psychiatry. 2002;159:652–654. doi: 10.1176/appi.ajp.159.4.652. [DOI] [PubMed] [Google Scholar]
- Malone S. M., Iacono W. G., Mcgue M.. A behavior-genetic perspective on psychopathology: P300 amplitude as an endophenotype for substance abuse and related disorders in male and female adolescent twins. Psychophysiology. 2002;39:S3. [Google Scholar]
- Marcelis M., Suckling J., Woodruff P., Hofman P., Bullmore E., van Os J.. Searching for a structural endophenotype in psychosis using computational morphometry. Psychiatry Research. 2003;122:153–167. doi: 10.1016/s0925-4927(02)00125-7. [DOI] [PubMed] [Google Scholar]
- Mattay V. S., Goldberg T. E., Fera F., Hariri A. R., Tessitore A., Egan M. F., Kolachana B., Callicott J. H., Weinberger D. R.. Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proceedings of the National Academy of Sciences USA. 2003;100:6186–6191. doi: 10.1073/pnas.0931309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C., Grech A., Touopoulou T., Schulze K., Chapple B., Sham P., Walshe M., Sharma T., Sigmundsson T., Chitnis X., Murray R. M.. Brain volumes in familial and non-familial schizophrenic probands and their unaffected relatives. American Journal of Medical Genetics. 2002;114:616–625. doi: 10.1002/ajmg.10604. [DOI] [PubMed] [Google Scholar]
- Michie P. T., Innes Brown H., Todd J., Jablensky A. V.. Duration mismatch negativity in biological relatives of patients with schizophrenia spectrum disorders. Bioligcal Psychiatry. 2002;52:749–758. doi: 10.1016/s0006-3223(02)01379-3. [DOI] [PubMed] [Google Scholar]
- Minzenberg M. J., Xu K., Mitropoulou V., Harvey P. D., Finch T., Flory J. D., New A. S., Goldman D., Siever L. J.. Catechol-O-methyltransferase Val158Met genotype variation is associated with prefrontal-dependent task performance in schizotypal personality disorder patients and comparison groups. Psychiatric Genetics. 2006;16:117–124. doi: 10.1097/01.ypg.0000199448.00163.e6. [DOI] [PubMed] [Google Scholar]
- Mityushina N. G., Alfimova M. V., Lyashenko G. L., Asanov A. Y., Golimbet V. E.. Serotonin receptor gene 5-HTR2A polymorphism and schizotypic traits in mentally healthy subjects. Zhurnal Nevropatologii I Psikhiatrii Imeni SS Korsakova. 2003;103:53–57. [PubMed] [Google Scholar]
- Moreno F. A., Rowe D. C., Kaiser B., Chase D., Michaels T., Gelernter J., Delgado P. L.. Association between a serotonin transporter promoter region polymorphism and mood response during tryptophan depletion. Molecular Psychiatry. 2002;7:213–216. doi: 10.1038/sj.mp.4000962. [DOI] [PubMed] [Google Scholar]
- Morley M., Molony C. M., Weber T. M., Devlin J. L., Ewens K. G., Spielman R. S., Cheung V. G.. Genetic analysis of genome-wide variation in human gene expression. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo M. R., Bowes L., Clark T. G., Flint J.. Lack of association of the COMT (Val158/108 Met) gene and schizophrenia: a meta-analysis of case-control studies. Molecular Psychiatry. 2005;10:765–770. doi: 10.1038/sj.mp.4001664. [DOI] [PubMed] [Google Scholar]
- Myles-Worsley M.. P50 sensory gating in multiplex schizophrenia families from a Pacific Island isolate. American Journal of Psychiatry. 2002;159:2007–2012. doi: 10.1176/appi.ajp.159.12.2007. [DOI] [PubMed] [Google Scholar]
- Myles-Worsley M., Park S.. Spatial working memory deficits in schizophrenia patients and their first degree relatives from Palau, Micronesia. American Journal of Medical Genetics. 2002;114:609–615. doi: 10.1002/ajmg.10644. [DOI] [PubMed] [Google Scholar]
- Oh K. S., Shin D. W., Oh G. T., Noh K. S.. Dopamine transporter genotype influences the attention deficit in Korean boys with ADHD. Yonsei Medical Journal. 2003;44:787–792. doi: 10.3349/ymj.2003.44.5.787. [DOI] [PubMed] [Google Scholar]
- Ohnishi T., Hashimoto R., Mori T., Nemoto K., Moriguchi Y., Iida H., Noguchi H., Nakabayashi T., Hori H., Ohmori M., Tsukue R., Anami K., Hirabayashi N., Harada S., Arima K., Saitoh O., Kunugi H.. The association between the Val158Met polymorphism of the catechol-O-methyl transferase gene and morphological abnormalities of the brain in chronic schizophrenia. Brain. 2006;129:399–410. doi: 10.1093/brain/awh702. [DOI] [PubMed] [Google Scholar]
- Perugi G., Akiskal H. S.. The soft bipolar spectrum redefined: focus on the cyclothymic, anxious-sensitive, impulse-dyscontrol, and binge-eating connection in bipolar II and related conditions. Psychiatric Clinics of North America. 2002;25:713–737. doi: 10.1016/s0193-953x(02)00023-0. [DOI] [PubMed] [Google Scholar]
- Prabhu V. R., Porjesz B., Chorlian D. B., Wang K., Stimus A., Begleiter H.. Visual p3 in female alcoholics. Alcohol Clinical and Experimental Research. 2001;25:531–539. [PubMed] [Google Scholar]
- Quintin P., Benkelfat C., Launay J. M., Arnulf I., Pointereau-Bellenger A., Barbault S., Alvarez J. C., Varoquaux O., Perez-Diaz F., Jouvent R., Leboyer M.. Clinical and neurochemical effect of acute tryptophan depletion in unaffected relatives of patients with bipolar affective disorder. Biological Psychiatry. 2001;50:184–190. doi: 10.1016/s0006-3223(01)01140-4. [DOI] [PubMed] [Google Scholar]
- Rabiner E., Bhagwagar Z., Gunn R., Bench C., Cowen P., Grasby P.. Attenuation of preferential occupancy of 5-HT1A autoreceptors by pindolol in depressed patients: effect of SSRIs or an endophenotype of the depressed state. Neuroimage. 2002;16:S67. [Google Scholar]
- Raux G., Bonnet-Brilhault F., Louchart S., Houy E., Gantier R., Levillain D., Allio G., Haouzir S., Petit M., Martinez M., Frebourg T., Thibaut F., Campion D.. The -2 bp deletion in exon 6 of the ‘alpha 7-like’ nicotinic receptor subunit gene is a risk factor for the P50 sensory gating deficit. Molecular Psychiatry. 2002;7:1006–1011. doi: 10.1038/sj.mp.4001140. [DOI] [PubMed] [Google Scholar]
- Riley B., Kendler K. S.. Molecular genetic studies of schizophrenia. European Journal of Human Genetics. 2006;14:669–680. doi: 10.1038/sj.ejhg.5201571. [DOI] [PubMed] [Google Scholar]
- Rosa A., Peralta V., Cuesta M. J., Zarzuela A., Serrano F., Martinez-Larrea A., Fananas L.. New evidence of association between COMT gene and prefrontal neurocognitive function in healthy individuals from sibling pairs discordant for psychosis. American Journal of Psychiatry. 2004;161:1110–1112. doi: 10.1176/appi.ajp.161.6.1110. [DOI] [PubMed] [Google Scholar]
- Ross R. G., Heinlein S., Zerbe G. O., Radant A.. Saccadic eye movement task identifies cognitive deficits in children with schizophrenia, but not in unaffected child relatives. Journal of Child Psychology and Psychiatry. 2005;46:1354–1362. doi: 10.1111/j.1469-7610.2005.01437.x. [DOI] [PubMed] [Google Scholar]
- Ross R. G., Olincy A., Mikulich S. K., Radant A. D., Harris J. G., Waldo M., Compagnon N., Heinlein S., Leonard S., Zerbe G. O., Adler L., Freedman R.. Admixture analysis of smooth pursuit eye movements in probands with schizophrenia and their relatives suggests gain and leading saccades are potential endophenotypes. Psychophysiology. 2002;39:809–819. doi: 10.1111/1469-8986.3960809. [DOI] [PubMed] [Google Scholar]
- Roth W. T., Cannon E. H.. Some features of the auditory evoked response in schizophrenics. Archives of General Psychiatry. 1972;27:466–471. doi: 10.1001/archpsyc.1972.01750280034007. [DOI] [PubMed] [Google Scholar]
- Rybakowski J. K., Borkowska A.. Eye movement and neuropsychological studies in first-degree relatives of schizophrenic patients. Schizophrenia Research. 2002;54:105–110. doi: 10.1016/s0920-9964(01)00357-7. [DOI] [PubMed] [Google Scholar]
- Rybakowski J. K., Borkowska A., Czerski P. M., Dmitrzak-Weglarz M., Skibinska M., Kapelski P., Hauser J.. Performance on the Wisconsin Card Sorting Test in schizophrenia and genes of dopaminergic inactivation (COMT, DAT, NET) Psychiatry Research. 2006;143:13–19. doi: 10.1016/j.psychres.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Rybakowski J. K., Borkowska A., Czerski P. M., Hauser J.. Eye movement disturbances in schizophrenia and a polymorphism of catechol-O-methyltransferase gene. Psychiatry Research. 2002;113:49–57. doi: 10.1016/s0165-1781(02)00245-7. [DOI] [PubMed] [Google Scholar]
- Rybakowski J. K., Borkowska A., Czerski P. M., Skibinska M., Hauser J.. Polymorphism of the brain-derived neurotrophic factor gene and performance on a cognitive prefrontal test in bipolar patients. Bipolar Disorder. 2003;5:468–472. doi: 10.1046/j.1399-5618.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T., Goldman-Rakic P. S.. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;251:947–950. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- Slaats-Willemse D., Swaab-Barneveld H., de Sonneville L., van der Meulen E., Buitelaar J.. Deficient response inhibition as a cognitive endophenotype of ADHD. Journal of the American Academy of Child and Adolescent Psychiatry. 2003;42:1242–1248. doi: 10.1097/00004583-200310000-00016. [DOI] [PubMed] [Google Scholar]
- Smesny S., Berger G., Rosburg T., Riemann S., Riehemann S., McGorry P., Sauer H.. Potential use of the topical niacin skin test in early psychosis – a combined approach using optical reflection spectroscopy and a descriptive rating scale. Journal of Psychiatric Research. 2003;37:237–247. doi: 10.1016/s0022-3956(03)00006-2. [DOI] [PubMed] [Google Scholar]
- Smoller J. W., Tsuang M. T.. Panic and phobic anxiety: defining phenotypes for genetic studies. American Journal of Psychiatry. 1998;155:1152–1162. doi: 10.1176/ajp.155.9.1152. [DOI] [PubMed] [Google Scholar]
- Sobczak S., Riedel W. J., Booij I., Aan Het Rot M., Deutz N. E., Honig A.. Cognition following acute tryptophan depletion: difference between first-degree relatives of bipolar disorder patients and matched healthy control volunteers. Psychological Medicine. 2002;32:503–515. doi: 10.1017/s0033291702005342. [DOI] [PubMed] [Google Scholar]
- Stefanis N. C., Van Os J., Avramopoulos D., Smyrnis N., Evdokimidis I., Hantoumi I., Stefanis C. N.. Variation in catechol-o-methyltransferase val158 met genotype associated with schizotypy but not cognition: a population study in 543 young men. Biological Psychiatry. 2004;56:510–515. doi: 10.1016/j.biopsych.2004.06.038. [DOI] [PubMed] [Google Scholar]
- Swerdlow N. R., Eastvold A., Karban B., Ploum Y., Stephany N., Geyer M. A., Cadenhead K., Auerbach P. P.. Dopamine agonist effects on startle and sensorimotor gating in normal male subjects: time course studies. Psychopharmacology (Berlin) 2002;161:189–201. doi: 10.1007/s00213-002-1040-3. [DOI] [PubMed] [Google Scholar]
- Szoke A., Schurhoff F., Meary A., Mathieu F., Chevalier F., Trandafir A., Alter C., Roy I., Bellivier F., Leboyer M.. Lack of influence of COMT and NET genes variants on executive functions in schizophrenic and bipolar patients, their first-degree relatives and controls. American Journal of Medical Genetics. B: Neuropsychiatric Genetics. 2006;141:504–512. doi: 10.1002/ajmg.b.30352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taerk E., Grizenko N., Ben Amor L., Lageix P., Mbekou V., Deguzman R., Torkaman-Zehi A., Ter Stepanian M., Baron C., Joober R.. Catechol-O-methyltransferase (COMT) Val108/158 Met polymorphism does not modulate executive function in children with ADHD. BMC Medical Genetics. 2004;5:30. doi: 10.1186/1471-2350-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S., Ohtsuki T., Yu S. Y., Tanabe E., Yara K., Kamioka M., Matsushima E., Matsuura M., Ishikawa K., Minowa Y., Noguchi E., Nakayama J., Yamakawa-Kobayashi K., Arinami T., Kojima T.. Significant linkage to chromosome 22q for exploratory eye movement dysfunction in schizophrenia. American Journal of Medical Genetics. B: Neuropsychiatric Genetics. 2003;123:27–32. doi: 10.1002/ajmg.b.10046. [DOI] [PubMed] [Google Scholar]
- Talkowski M. E., Seltman H., Bassett A. S., Brzustowicz L. M., Chen X., Chowdari K. V., Collier D. A., Cordeiro Q., Corvin A. P., Deshpande S. N., Egan M. F., Gill M., Kendler K. S., Kirov G., Heston L. L., Levitt P., Lewis D. A., Li T., Mirnics K., Morris D. W., Norton N., O’Donovan M. C., Owen M. J., Richard C., Semwal P., Sobell J. L., St Clair D., Straub R. E., Thelma B. K., Vallada H., Weinberger D. R., Williams N. M., Wood J., Zhang F., Devlin B., Nimgaonkar V. L.. Evaluation of a susceptibility gene for schizophrenia: genotype based meta-analysis of RGS4 polymorphisms from thirteen independent samples. Biological Psychiatry. 2006;60:152–162. doi: 10.1016/j.biopsych.2006.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikalinos T. A., Ntzani E. E., Contopoulos-Ioannidis D. G., Ioannidis J. P.. Establishment of genetic associations for complex diseases is independent of early study findings. European Journal of Human Genetics. 2004;12:762–769. doi: 10.1038/sj.ejhg.5201227. [DOI] [PubMed] [Google Scholar]
- Tsai S. J., Yu Y. W., Chen T. J., Chen J. Y., Liou Y. J., Chen M. C., Hong C. J.. Association study of a functional catechol-O-methyltransferase-gene polymorphism and cognitive function in healthy females. Neuroscience Letters. 2003;338:123–126. doi: 10.1016/s0304-3940(02)01396-4. [DOI] [PubMed] [Google Scholar]
- Tsuang M. T., Faraone S. V., Lyons M. J.. Identification of the phenotype in psychiatric genetics. European Archives of Psychiatry and Clinical Neuroscience. 1993;243:131–142. doi: 10.1007/BF02190719. [DOI] [PubMed] [Google Scholar]
- Turetsky B. I., Cannon T. D., Gur R. E.. P300 subcomponent abnormalities in schizophrenia: III. Deficits in unaffected siblings of schizophrenic probands. Biological Psychiatry. 2000;47:380–390. doi: 10.1016/s0006-3223(99)00290-5. [DOI] [PubMed] [Google Scholar]
- Turetsky B. I., Moberg P. J., Arnold S. E., Doty R. L., Gur R. E.. Low olfactory bulb volume in first-degree relatives of patients with schizophrenia. American Journal of Psychiatry. 2003;160:703–708. doi: 10.1176/appi.ajp.160.4.703. [DOI] [PubMed] [Google Scholar]
- Tuulio-Henriksson A., Arajarvi R., Partonen T., Haukka J., Varilo T., Schreck M., Cannon T., Lonnqvist J.. Familial loading associates with impairment in visual span among healthy siblings of schizophrenia patients. Biological Psychiatry. 2003;54:623–628. doi: 10.1016/s0006-3223(03)00232-4. [DOI] [PubMed] [Google Scholar]
- Tuulio-Henriksson A., Haukka J., Partonen T., Varilo T., Paunio T., Ekelund J., Cannon T. D., Meyer J. M., Lonnqvist J.. Heritability and number of quantitative trait loci of neurocognitive functions in families with schizophrenia. American Journal of Medical Genetics. 2002;114:483–490. doi: 10.1002/ajmg.10480. [DOI] [PubMed] [Google Scholar]
- Umbricht D., Koller R., Schmid L., Skrabo A., Grubel C., Huber T., Stassen H.. How specific are deficits in mismatch negativity generation to schizophrenia. Biological Psychiatry. 2003;53:1120–1131. doi: 10.1016/s0006-3223(02)01642-6. [DOI] [PubMed] [Google Scholar]
- Valdar W., Solberg L. C., Gauguier D., Burnett S., Klenerman P., Cookson W. O., Taylor M. S., Rawlins J. N. P., Mott R., Flint J.. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nature Genetics. 2006;38:879–887. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- van Beijsterveldt C. E., van Baal G. C.. Twin and family studies of the human electroencephalogram: a review and a meta-analysis. Biological Psychology. 2002;61:111–138. doi: 10.1016/s0301-0511(02)00055-8. [DOI] [PubMed] [Google Scholar]
- Williams G. V., Goldman-Rakic P. S.. Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature. 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- Willis-Owen S. A., Turri M. G., Munafo M. R., Surtees P. G., Wainwright N. W., Brixey R. D., Flint J.. The serotonin transporter length polymorphism, neuroticism, and depression: a comprehensive assessment of association. Biological Psychiatry. 2005;58:451–456. doi: 10.1016/j.biopsych.2005.04.050. [DOI] [PubMed] [Google Scholar]
- Winterer G., Enoch M. A., White K. V., Saylan M., Coppola R., Goldman D.. EEG phenotype in alcoholism: increased coherence in the depressive subtype. Acta Psychiatrica Scandinavica. 2003;108:51–60. doi: 10.1034/j.1600-0447.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- Yoneyama E., Matsui M., Kawasaki Y., Nohara S., Takahashi T., Hagino H., Suzuki M., Seto H., Kurachi M.. Gray matter features of schizotypal disorder patients exhibiting the schizophrenia-related code types of the Minnesota Multiphasic Personality Inventory. Acta Psychiatrica Scandinavica. 2003;108:333–340. doi: 10.1034/j.1600-0447.2003.00202.x. [DOI] [PubMed] [Google Scholar]
- Yoon I. S., Li P. P., Siu K. P., Kennedy J. L., Cooke R. G., Parikh S. V., Warsh J. J.. Altered IMPA2 gene expression and calcium homeostasis in bipolar disorder. Molecular Psychiatry. 2001;6:678–683. doi: 10.1038/sj.mp.4000901. [DOI] [PubMed] [Google Scholar]