

Abstract
Alkaptonuria (AKU) is a rare autosomal recessive metabolic disorder, characterized by accumulation of homogentisic acid, leading to darkened urine, pigmentation of connective tissue (ochronosis), joint and spine arthritis, and destruction of cardiac valves. AKU is due to mutations in the homogentisate dioxygenase gene, HGD, that converts homogentisic acid to maleylacetoacetic acid in the tyrosine catabolic pathway. Here we report a comprehensive mutation analysis of 93 patients enrolled in our study, as well as an extensive update of all previously published HGD mutations associated with AKU. Within our patient cohort, we identified 52 HGD variants, of which 22 were novel. This yields a total of 91 identified HGD variations associated with AKU to date, including 62 missense, 13 splice site, 10 frameshift, 5 nonsense and 1 no-stop mutation. Most HGD variants reside in exons 3, 6, 8 and 13. We assessed the potential effect of all missense variations on protein function, using 5 bioinformatic tools specifically designed for interpretation of missense variants (SIFT, POLYPHEN, PANTHER, PMUT and SNAP). We also analyzed the potential effect of splice site variants using two different tools (BDGP and NetGene2). This study provides valuable resources for molecular analysis of alkaptonuria and expands our knowledge of the molecular basis of this disease.
Keywords: Alkaptonuria, AKU, homogentisic acid, homogentisate dioxygenase, HGD, mutation analysis, missense variation, pathogenicity prediction
Introduction
Alkaptonuria (AKU; MIM# 203500) is a rare autosomal recessive disorder affecting 1 in 250,000 to 1 million people worldwide. AKU is caused by deficiency of homogentisic acid oxidase (HGD, EC 1.13.11.5), which leads to the accumulation of homogentisic acid (HGA), since it cannot be converted to maleylacetoacetic acid in the tyrosine catabolic pathway [Fernandez-Canon et al., 1996; Scriver et al., 2001; Phornphutkul et al., 2002]. The clinical findings of AKU result from the reaction of homogentisic acid and its homopolymeric oxidation products, i.e., benzoquinones, with connective tissue components. The benzoquinones form melanin-like polymers in a process that requires a copper-dependent enzyme, homogentisic acid polyphenol oxidase, that is present in mammalian skin and cartilage [Zannoni et al., 1969]. The oxidation of HGA cause darkening of the urine on standing or upon alkalinization (Fig. 1A) [Bodeker, 1859; Virchow, 1866; Scriver et al., 2001] and pigmentation (ochronosis) of cartilage and other connective tissues (Fig. 1B, 1C); patients later develop joint and spine arthritis in their thirties (Fig. 1D) and destruction of cardiac valves. AKU does not appear to reduce the lifespan of affected subjects [Srsen et al., 1985], but there is a high rate of disability, especially late in life [Perry et al., 2006]. Nitisinone, an inhibitor of the enzyme that forms HGA (i.e., para-hydroxyphenylpyruvic acid dioxygenase), lowers HGA production by 95% [Anikster et al., 1998; Phornphutkul et al., 2002; Suwannarat et al., 2005]; a clinical trial of nitisinone for AKU is underway with 40 patients, 20 receiving nitisinone and 20 control subjects (http://clinicaltrials.gov/; identifier: NCT00107783).
FIGURE 1.
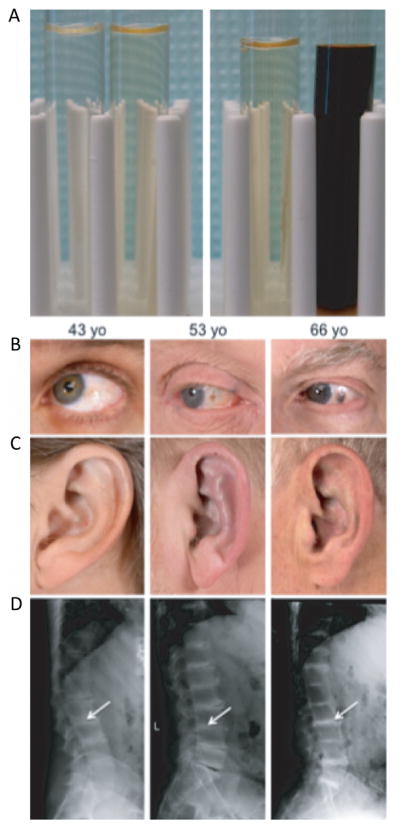
Clinical features of AKU. Detection of excess of homogentisic acid in urine on standing or artificially by adding a base. A: Left panel: Control urine. Left without base added, right with base added. Right panel: AKU urine. Left without base added, right with base added. Three AKU patients with increasing severity (left to right) of clinical symptoms. B: Ochronotic pigmentation of sclerae. C: Auricular cartilages with ochronosis deposition. D: Radiography of dorsolumbar spines: reduction and calcifications of the intervertebral discs (Arrows).
AKU patients generally present with either dark urine or early onset arthritis [Martin et al., 1952; Yules, 1954; Smith and Smith, 1955]. In our previous study [Phornphutkul et al., 2002], the diagnosis was based upon dark urine in 55% of 58 cases and upon chronic joint pain in 45%. AKU is definitively diagnosed by detecting gram quantities of homogentisic acid in the urine. This is easily achieved by gas chromatography-mass spectrometry (GC-MS) analysis as part of a urinary organic acid analysis. Other products of tyrosine metabolism are not generally elevated, and urinary amino acids are normal [Neuberger, 1947].
The gene responsible for AKU is HGD, coding for homogentisate dioxygenase (also called AKU or HGO, GenBank NM_000187). It is located on chromosome 3q21-q23. HGD contains 14 exons and covers 60 kb of genomic DNA [Fernandez-Canon et al., 1996]. It encodes a 49,973 dalton, 445 amino acid protein that forms a dimer of two trimers giving rise to a functional hexamer. The crystalline structure of the HGD protein has been resolved [Titus et al., 2000]. The 280 residue N-terminal domain has a central β-sandwich structure flanked by a β-sheet that interacts with the 140 residue C-terminal domain of neighboring subunits. The active site binds iron in a domain defined by His335, Glu341, and His371 side-chains [Titus et al., 2000].
An AKU mouse model (Hgdaku/aku) was identified from an ENU (ethylnitrosourea) mutagenesis screen [Montagutelli et al., 1994]. This mouse harbors a recessive splice site mutation, c.1006+2T>A, in its Hgd gene (GenBank NM_013547) located on mouse chromosome 16 (27.3cM). This mutation leads to skipping of one or two exons [Kress et al., 1999; Manning et al., 1999] and the generation of a premature stop codon [Kress et al., 1999]. Hgdaku/aku mice have high levels of HGA in the urine and plasma but have no signs of ochronosis [Kamoun et al., 1992]. A trial of nitisinone in this mouse model showed a significant reduction of HGA excretion in the urine [Suzuki et al., 1999].
AKU has also been described in a rabbit [Lewis, 1926] and in different primates, chimpanzee [Watkins et al., 1970] and orangutan [Keeling et al., 1973].
Interestingly, AKU was discovered in some Egyptian mummies. In 1961, Simon and Zorab [Simon and Zorab, 1961] and in 1962, Wells and Maxwell [Wells and Maxwell, 1962] showed, by radiography, that some mummies had suffered of ochronosis and that AKU might have occurred frequently in ancient Egypt. In 1967, Gray presumed that the dystrophic calcification of ochronosis seen on the mummies’ radiographies was an artifact of the embalming process and therefore that AKU was not frequent in ancient Egypt [Gray, 1967]. However, in 1977, Stenn showed that the ochronotic Egyptian mummy Harwa, dating from 1500 b.c., had probably suffered from AKU [Stenn et al., 1977] by demonstrating that the chemical characteristics of the black pigment found on the articular surface of the pelvic bone was identical to air-oxidized homogentisic acid.
Here we present a comprehensive mutation analysis of 93 AKU patients (79 probands and 14 affected relatives) enrolled in a natural history study of AKU at the NIH. In addition, we list all previously published HGD gene mutations. Of 91 human identified HGD potentially disease-causing variants, 22 were novel. Because the pathogenic consequences of variations in DNA sequence cannot always be determined unambiguously, we assessed the potential pathogenicity of all HGD missense and splice site variants using a variety of prediction programs. Hence, in this paper the term mutation will be used when the variant causes a nonsense, a frameshift or modification of the canonical sites for splicing. Potentially disease-causing variation will be used for any variation in the DNA sequence with unclear effect on the protein. However, the terms variant and variation will be used for any sequence modification.
Molecular analysis of the NIH patient cohort
Our patient population included 79 probands (158 independent alleles), their HGD variations and daily urinary excretions of HGA are listed in Table 1. Mutation analysis resulted in a total of 157 total sequence variations of 52 types including 6 insertions and/or deletions that resulted in a frame-shift (3 novel), 7 splice site modifications (3 novel), 2 nonsense mutations (1 novel), a no-stop mutation (novel), and 36 missense variations (14 novel) (Table 2). These variations were scattered over all 14 exons of HGD, with the largest number of variants in exons 3, 6, 8, and 13 (Fig. 2, Table 3).
Table 1.
HGD variants and HGA urinary excretion in all NIH probands and affected siblings (gray)
| #a | Variant 1b | Variant 2b | Variant 3b | HGA urine |
|
|---|---|---|---|---|---|
| g/24hc | Nd | ||||
| 1 | c.468+2T>C | c.1102A>G; p.M368V | 5.65 | 3.66 | |
| 2 | c.174delA; p.S59AfsX52 | c.481G>A; p.G161R | 4.69 | 2.82 | |
| 3 | c.174delA; p.S59AfsX52 | c.473C>T; p.P158L | 2.92 | 1.41 | |
| 5 | c.1076C>T; p.P359L | c.1102A>G; p.M368V | 2.07 | 2.17 | |
| 6 | c.342+1G>T | c.342+1G>T | 6.33 | 2.34 | |
| 7 | c.502G>A; p.E168K | c.502G>A; p.E168K | 8. 7 | 3.68 | |
| 8 | c.342+1G>T | c.342+1G>T | 3.99 | 1.82 | |
| 9 | c.125A>C; p.E42A | c.125A>C; p.E42A | 4.28 | 2.76 | |
| 71 | c.125A>C; p.E42A | c.125A>C; p.E42A | 7.41 | 4.81 | |
| 78 | c.125A>C; p.E42A | c.125A>C; p.E42A | 5.36 | 4.92 | |
| 10 | c.808G>A; p.G270R | c.808G>A; p.G270R | 7.3 | 3.01 | |
| 11 | c.174delA; p.S59AfsX52 | c.174delA; p.S59AfsX52 | 2.84 | 2.62 | |
| 12 | c.481G>A; p.G161R | c.1102A>G; p.M368V | 4.44 | 2.51 | |
| 72 | c.481G>A; p.G161R | c.1102A>G; p.M368V | 5.03 | 2.62 | |
| 13 | c.359G>T; p.C120F | c.1336T>C; p.X446RextX24 | 8.52 | 2.49 | |
| 14 | c.1007-2A>T | c.1201G>C; p.E401Q | 0.68 | 1.38 | |
| 15 | c.342+1G>A | c.1017_1019delGAGinsTA; p.M339IfsX30 | 2.7 | 2.23 | |
| 16 | c.182T>C; p.L61P | c.182T>C; p.L61P | 3.8 | 2.21 | |
| 73 | c.182T>C; p.L61P | c.182T>C; p.L61P | 6.23 | 1.61 | |
| 17 | c.174delA; p.S59AfsX52 | c.410T>C; p.L137P | 1.92 | 5.3 | |
| 18 | c.359G>T; p.C120F | c.1102A>G; p.M368V | 6.65 | 2.96 | |
| 19 | c.342+1G>T | c.1102A>G; p.M368V | 2.01 | 1.35 | |
| 20 | c.413_434+35del57 | c.1102A>G; p.M368V | 6.2 | 2.78 | |
| 21 | c.1078G>C; p.G360R | c.1102A>G; p.M368V | 3.38 | 2.95 | |
| 22 | c.125A>C; p.E42A | c.125A>C; p.E42A | 3 | 4.24 | |
| 23 | c.481G>A; p.G161R | c.559C>G; p.R187G | 2.59 | 1.21 | |
| 25 | c.174delA; p.S59AfsX52 | c.174delA; p.S59AfsX52 | 0.84 | 1.7 | |
| 26 | c.178T>G; p.W60G | c.1102A>G; p.M368V | 6.36 | 5.15 | |
| 76 | c.178T>G; p.W60G | c.1102A>G; p.M368V | 7.6 | 3.78 | |
| 75 | c.899T>G; p.V300G | c.1078G>C; p.G360R | 4.44 | 3.35 | |
| 27 | c.174delA; p.S59AfsX52 | c.649G>T; p.G217W | 7.58 | 3.62 | |
| 28 | c.359G>T; p.C120F | c.1102A>G; p.M368V | 3.71 | 2.98 | |
| 29 | c.11T>A; p.L4X | c.1102A>G; p.M368V | 4.38 | 2.46 | |
| 30 | c.11T>C; p.L4S | c.359G>T; p.C120F | 2.98 | 2.19 | |
| 56 | c.11T>C; p.L4S | c.359G>T; p.C120F | 4.85 | 2.84 | |
| 31 | c.899T>G; p.V300G | c.1078G>C; p.G360R | 7.37 | 4.01 | |
| 32 | c.11T>C; p.L4S | c.359G>T; p.C120F | 9.34 | 2.42 | |
| 33 | c.899T>G; p.V300G | c.1102A>G; p.M368V | 6.5 | 3.61 | |
| 34 | c.125A>C; p.E42A | c.125A>C; p.E42A | 5.34 | 2.79 | |
| 35 | c.549+1G>A | c.1102A>G; p.M368V | 1.64 | 1.64 | |
| 36 | c.447T>A; p.N149K | c.502G>T; p.E168X | c.1007-2A>T | 2.93 | 1.66 |
| 77 | c.217T>C; p.F73L | c.1078G>C; p.G360R | 5.13 | 2.36 | |
| 79 | c.481G>A; p.G161R | c.688C>T; p.P230S | 7.46 | 3.21 | |
| 38 | c.457dupG; p.D153GfsX26 | c.481G>A; p.G161R | 6.6 | 2.66 | |
| 80 | c.457dupG; p.D153GfsX26 | c.481G>A; p.G161R | 5.05 | 2.26 | |
| 39 | c.1102A>G; p.M368V | c.1102A>G; p.M368V | 4.75 | 5.55 | |
| 40 | c.274C>A; p.P92T | NDe | 4.1 | 2.03 | |
| 41 | c.185A>G; p.Y62C | NDe | 3.44 | 3.05 | |
| 81 | c.1017_1019delGAGinsTA; p.M339IfsX30 | c.1017_1019delGAGinsTA; p.M339IfsX30 | 6.81 | 2.59 | |
| 82 | c.1017_1019delGAGinsTA; p.M339IfsX30 | c.1017_1019delGAGinsTA; p.M339IfsX30 | 3.31 | 1.58 | |
| 74 | c.1017_1019delGAGinsTA; p.M339IfsX30 | c.1017_1019delGAGinsTA; p.M339IfsX30 | 10.14 | 2.54 | |
| 43 | c.481G>A; p.G161R | c.481G>A; p.G161R | 1.43 | 0.85 | |
| 44 | c.773A>C; p.Q258P | c.1336T>C; p.X446RextX24 | 4.75 | 3.34 | |
| 45 | c.504G>C; p.E168D | c.504G>C; p.E168D | 2.32 | 1.77 | |
| 46 | c.365C>T; p.A122V | c.1078G>C; p.G360R | 4.46 | 2.52 | |
| 47 | c.365C>T; p.A122V | c.365C>T; p.A122V | 6.4 | 6.03 | |
| 48 | c.360T>G; p.C120W | c.360T>G; p.C120W | 3.45 | 3.4 | |
| 49 | c.289T>C; p.W97R | c.359G>T; p.C120F | 3.91 | 2.05 | |
| 50 | c.376_377delAA; p.K126VfsX2 | c.376_377delAA; p.K126VfsX2 | 2.99 | 1.8 | |
| 51 | c.289T>C; p.W97R | c.359G>T; p.C120F | 2.49 | 1.81 | |
| 83 | c.504G>C; p.E168D | c.504G>C; p.E168D | 4.31 | 5.01 | |
| 84 | c.457dupG; p.D153GfsX26 | c.457dupG; p.D153GfsX26 | 2.38 | 1.22 | |
| 52 | c.178T>G; pW60G | NDe | 2.69 | 3.61 | |
| 53 | c.481G>A; p.G161R | c.962G>A; p.R321P | 0.39 | 0.98 | |
| 85 | c.413_434+35del57 | c.1102A>G; p.M368V | 2.25 | 2.78 | |
| 54 | c.342+1G>T | c.1102A>G; p.M368V | 4.09 | 1.91 | |
| 55 | c.359G>T; p.C120F | c.1102A>G; p.M368V | 4.06 | 2.99 | |
| 57 | c.16-1G>A | c.481G>A; p.G161R | 6.26 | 3.25 | |
| 86 | c.16-1G>A | c.481G>A; p.G161R | 6.34 | 2.2 | |
| 37 | c.16-1G>A | c.481G>A; p.G161R | 2.52 | 2.11 | |
| 58 | c.1102A>G; p.M368V | c.1102A>G; p.M368V | 1.68 | 1.79 | |
| 59 | c.1017_1019delGAGinsTA; p.M339IfsX30 | c.1118C>T; p.P373L | 7.55 | 4.79 | |
| 60 | c.1102A>G; p.M368V | c.1111dupC; p.H371PfsX4 | 8.21 | 2.61 | |
| 61 | c.410T>C; p.L137P | c.410T>C; p.L137P | 3.4 | 3.07 | |
| 62 | c.674G>T; p.R225L | c.1111dupC; p.H371PfsX4 | 8.48 | 5.5 | |
| 87 | c.367G>A; p.G123R | c.367G>A; p.G123R | 6.37 | 4.6 | |
| 63 | c.688C>T; p.P230S | c.1102A>G; p.M368V | 4.84 | 3.61 | |
| 64 | c.549G>T; p.Q183R | c.1102A>G; p.M368V | 6.44 | 2.91 | |
| 65 | c.359G>T; p.C120F | c.1078G>C; p.G360R | 5.29 | 2.85 | |
| 88 | c.174delA; p.S59AfsX52 | c.1102A>G; p.M368V | 4.96 | 1.84 | |
| 89 | c.502G>A; p.E168K | c.502G>A; p.E168K | 9.79 | 4.19 | |
| 90 | c.502G>A; p.E168K | c.502G>A; p.E168K | 6.4 | 2.92 | |
| 66 | c.1085G>A; p.G362E | c.1102A>G; p.M368V | 4.87 | 2.23 | |
| 67 | c.899T>G; p.V300G | c.1102A>G; p.M368V | 3.36 | 3.59 | |
| 68 | c.359G>T; p.C120F | c.1078G>C; p.G360R | 3.36 | 5.11 | |
| 69 | c.1102A>G; p.M368V | NDe | 3.09 | 1.89 | |
| 91 | c.1102A>G; p.M368V | NDe | 2.68 | 1.9 | |
| 70 | c.174delA; p.S59AfsX52 | c.1006+2T>A | 4.34 | 3.15 | |
| 92 | c.174delA; p.S59AfsX52 | c.1006+2T>A | 3.67 | 2.51 | |
| 93 | c.8A>C; p.E3A | c.773A>C; p.Q258P | c.914C>T; p.S305F | 7.82 | 5.96 |
| 94 | c.16-1G>A | c.481G>A; p.G161R | c.1017_1019delGAGinsTA; p.M339IfsX30 | 4.64 | 5.54 |
| 95 | c.174delA; p.S59AfsX52 | c.1078G>C; p.G360R | 7.34 | 3.96 | |
| 96 | c.502G>A; p.E168K | c.806A>G; p.H269R | 9.09 | 3.03 | |
Patient numbers correspond to a master file of all NHGRI patients with AKU
DNA variant numbering system is based on cDNA (NM_000187.2), with +1 corresponding to the A of the ATG. The initiation codon is codon 1
Normal: <0.03g/24h
Urinary HGA normalized to urinary creatinine (μmoles HGA/μmoles creatinine), normal: <0.01
ND: Not Determined
Table 2.
Summary of identified HGD variants
| Missense | Splice site | Indel a | Other b | Total | |
|---|---|---|---|---|---|
| NIH Study (Novel) | 36 (14) | 7 (3) | 6 (3) | 3 (2) | 52 (22) |
| Other published | 26 | 6 | 4 | 3 | 39 |
| Total | 62 | 13 | 10 | 6 | 91 |
Indel: includes insertions, deletions and insertion-deletions
Other: Includes nonsense and no-stop mutations
FIGURE 2.

Distribution of <i>HGD</i> variants over the 14 exons of the homogentisic acid oxidase gene. The symbols above the gene represent the 52 variants found in this study, each symbol representing one of the 157 variants. Previously reported variants are filled in black or grey (if a different mutation occurred at the same position) and novel ones are filled in red. The positions of other previously reported variants (not found in the current study) are indicated by unfilled symbols under the gene.
Table 3.
Repartition of location and occurrence of the HGD variants
| VARIANTa,b | Number of times detectedc (#) | Frequency (%) | #/% per exon | |||
|---|---|---|---|---|---|---|
| # | Nucleotide modification | Protein effect | ||||
| Ex1 | 1 | c.8A>C | p.E3A | 1 | 0.64 | 4/2.5 |
| 2 | c.11T>C | p.L4S | 2 | 1.27 | ||
| 3 | c.11T>A | p.L4X | 1 | 0.64 | ||
| Ex2 | 4 | c.16-1G>A | Exon skipping | 2 | 1.27 | 2/1.3 |
| 5 | c.32_33delGGinsATT | p.G11DfsX2 | ND | 0 | ||
| 6 | c.74T>C | p.L25P | ND | 0 | ||
| Ex3 | 7 | c.125A>C | p.E42A | 6 | 3.82 | 17/10.8 |
| 8 | c.140C>T | pS47L | ND | 0 | ||
| 9 | c.157C>T | p.R53W | ND | 0 | ||
| 10 | c.171G>C | p.K57N | ND | 0 | ||
| 11 | c.174delA | p.S59AfsX52 | 11 | 7.01 | ||
| 12 | c.?d | p.S59X | ND | 0 | ||
| Ex4 | 13 | c.177-2G>Ae | Exon skipping | ND | 0 | 7/4.5 |
| 14 | c.178T>G | p.W60G | 2 | 1.27 | ||
| 15 | c.182T>C | p.L61P | 2 | 1.27 | ||
| 16 | c.185A>G | p.Y62C | 1 | 0.64 | ||
| 17 | c.217T>C | p.F73L | 1 | 0.64 | ||
| 18 | c.274C>A | p.P92T | 1 | 0.64 | ||
| Ex5 | 19 | c.289T>C | p.W97R | 2 | 1.27 | 9/5.7 |
| 20 | c.289T>G | p.W97G | ND | 0 | ||
| 21 | c.342+1G>T | Exon skipping | 6 | 3.82 | ||
| 22 | c.342+1G>A | Exon skipping | 1 | 0.64 | ||
| Ex6 | 23 | c.357dupG | p.C120VfsX9 | ND | 0 | 24/15.3 |
| 24 | c.359G>T | p.C120F | 10 | 6.37 | ||
| 25 | c.360T>G | p.C120W | 2 | 1.27 | ||
| 26 | c.365C>T | p.A122V | 3 | 1.91 | ||
| 27 | c.365C>A | p.A122D | ND | 0 | ||
| 28 | c.367G>A | p.G123R | 2 | 1.27 | ||
| 29 | c.376_377delAA | p.K126VfsX2 | 2 | 1.27 | ||
| 30 | c.407T>A | p.F136Y | ND | 0 | ||
| 31 | c.410T>C | p.L137P | 3 | 1.91 | ||
| 32 | c.429G>?d | p.E143D | ND | 0 | ||
| 33 | c.413_434+35del57 | Exon skipping | 2 | 1.27 | ||
| 34 | c.433A>T | p.R145X | ND | 0 | ||
| Ex7 | 35 | c.447T>A | p.N149K | 1 | 0.64 | 5/3.2 |
| 36 | c.457dupG | p.D153GfsX26 | 3 | 1.91 | ||
| 37 | c.458A>G | p.D153G | ND | 0 | ||
| 38 | c.468+2T>C | Exon skipping | 1 | 0.64 | ||
| 39 | c.468+5G>A | Exon skipping | ND | 0 | ||
| Ex8 | 40 | c.473C>T | p.P158L | 1 | 0.64 | 23/14.7 |
| 41 | c.473C>G | p.P158R | ND | 0 | ||
| 42 | c.477G>?d | p.Q159H | ND | 0 | ||
| 43 | c.481G>A | p.G161R | 10 | 6.37 | ||
| 44 | c.502G>A | p.E168K | 5 | 3.18 | ||
| 45 | c.502G>T | p.E168X | 1 | 0.64 | ||
| 46 | c.504G>C | p.E168D | 4 | 2.55 | ||
| 47 | c.513G>?d | p.K171N | ND | 0 | ||
| 48 | c.534G>?d | p.E178D | ND | 0 | ||
| 49 | c.541G>T | p.V181F | ND | 0 | ||
| 50 | c.549G>T | p.Q183R | 1 | 0.64 | ||
| 51 | c.549+1G>A | Exon skipping | 1 | 0.64 | ||
| Ex9 | 52 | c.550-2A>C | Exon skipping | ND | 0 | 2/1.3 |
| 53 | c.559C>G | p.R187G | 1 | 0.64 | ||
| 54 | c.566G>T | p.S189I | ND | 0 | ||
| 55 | c.588delC | p.R197GfsX32 | ND | 0 | ||
| 56 | c.593G>A | p.G198D | ND | 0 | ||
| 57 | c.647T>C | p.I216T | ND | 0 | ||
| 58 | c.649G>T | p.G217W | 1 | 0.64 | ||
| Ex10 | 59 | c.650-56G>A | Exon skipping | ND | 0 | 5/3.2 |
| 60 | c.650-17G>A | Exon skipping | ND | 0 | ||
| 61 | c.674G>T | p.R225L | 1 | 0.64 | ||
| 62 | c.674G>A | p.R225H | ND | 0 | ||
| 63 | c.680T>C | p.F227S | ND | 0 | ||
| 64 | c.688C>T | p.P230S | 2 | 1.27 | ||
| 65 | c.688C>A | p.P230T | ND | 0 | ||
| 66 | c.743A>G | p.K248R | ND | 0 | ||
| 67 | c.773A>C | p.Q258P | 2 | 1.27 | ||
| Ex11 | 68 | c.806A>G | p.H269R | 1 | 0.64 | 3/1.9 |
| 69 | c.808G>A | p.G270R | 2 | 1.27 | ||
| 70 | c.873C>A | p.D291E | ND | 0 | ||
| 71 | c.875A>G | p.H292R | ND | 0 | ||
| Ex12 | 72 | c.899T>G | p.V300G | 4 | 2.55 | 7/4.5 |
| 73 | c.914C>T | p.S305F | 1 | 0.64 | ||
| 74 | c.961C>T | p.R321X | ND | 0 | ||
| 75 | c.962G>A | p.R321P | 1 | 0.64 | ||
| 76 | c.964T>C | p.W322R | ND | 0 | ||
| 77 | c.990G>T | p.R330S | ND | 0 | ||
| 78 | c.1006+2T>A | Exon skipping | 1 | 0.64 | ||
| Ex13 | 79 | c.1007-2A>T | Exon skipping | 2 | 1.27 | 46/29.3 |
| 80 | c.1016delT | p.M339RfsX30 | ND | 0 | ||
| 81 | c.1017_1019delGAGinsTA | p.M339IfsX30 | 5 | 3.18 | ||
| 82 | c.1076C>T | p.P359L | 1 | 0.64 | ||
| 83 | c.1078G>C | p.G360R | 8 | 5.1 | ||
| 84 | c.1085G>A | p.G362E | 1 | 0.64 | ||
| 85 | c.1102A>G | p.M368V | 26 | 16.56 | ||
| 86 | c.1111dupC | p.H371PfsX4 | 2 | 1.27 | ||
| 87 | c.1112A>G | p.H371R | ND | 0 | ||
| 88 | c.1118C>T | p.P373L | 1 | 0.64 | ||
| 89 | c.1188+1G>T | Exon skipping | ND | 0 | ||
| Ex14 | 90 | c.1201G>C | p.E401Q | 1 | 0.64 | 3/1.9 |
| 91 | c.1336T>C | p.X446RextX24 | 2 | 1.27 | ||
| Total | 157 | 100 | 157/100 | |||
DNA variant numbering system is based on cDNA (NM_000187.2), with +1 corresponding to the A of the ATG. The initiation codon is codon 1
Novel variant
ND: Not detected in our cohort
Exact nucleotide change was not indicated and not reconstructable
Published nucleotide change is inconsistent with gene sequence
Note that patients 45, 46, 47 and 83 have Indian ancestry and are the only patients in whom variants 26 (c.365C>T; p.A122V) and 46 (c.504G>C; p.E168D) occurred.
In 72 probands, two candidate disease-causing HGD variations were identified (Table 1). In the 10 cases in which DNA from affected siblings was available (10 sibships, 13 affected siblings), the same variants were found (grey shaded in Table 1). In 4 patients, only one candidate disease-causing variation was detected (4 of 158 alleles, 2.5%, which is significantly lower than other studies (Beltran-Valero de Bernabe et al. 1998 7%, Phornphutkul et al. 2002, 9%); the same variant was found in the case with an affected sibling. In three patients, three candidate disease-causing HGD variations were present (Table 1). No apparent correlation existed between a patient’s genotype and the level of excreted homogentisic acid. Moreover, excretion of homogentisic acid in urine varied within sibships (Table 1).
Review of previously reported HGD variants
After the first report that HGD mutations cause alkaptonuria [Fernandez-Canon et al., 1996], other molecular studies described a variety of HGD mutations and polymorphisms, including haplotype and mutation analysis of the HGD gene in 29 previously unstudied AKU patients, identifying 12 novel potentially disease-causing variations, 5 polymorphic sites, and mutation hotspots [Beltran-Valero de Bernabe et al., 1998; Beltran-Valero de Bernabe et al., 1999a]. In 1999, the molecular defects of 30 AKU patients from central Europe were described, including 5 novel HGD potentially disease-causing variations [Muller et al., 1999]. In Slovakia, the country with the highest frequency of AKU, 2 recurrent variations, c.16-1G>A (INV1-1G>A) and p.G161R, were identified in more than 50% of the patients’ chromosomes, indicating that two independent founders contributed to the region’s high prevalence of AKU [Zatkova et al., 2000a; Zatkova et al., 2000b; Zatkova et al., 2003]. In addition, case reports of sequence modifications associated with AKU were described in patients of Japanese [Higashino et al., 1998], Finnish [Beltran-Valero de Bernabe et al., 1999b], Spanish [Rodriguez et al., 2000], Italian [Porfirio et al., 2000; Mannoni et al., 2004], Dominican [Goicoechea De Jorge et al., 2002], Algerian [Ladjouze-Rezig et al., 2006] and other descents [Beltran-Valero de Bernabe et al., 1998; Beltran-Valero de Bernabe et al., 1999a; Felbor et al., 1999; Phornphutkul et al., 2002; Srsen et al., 2002; Grasko et al., 2009] (Table 3).
We combined HGD variants reported in the Human Genome Mutation Database (HGMD) [Stenson et al., 2003] with an extensive literature search for HGD variations. This yielded 69 HGD variants (65 found in HGMD) described as AKU-associated mutations from individuals of diverse ethnicities (Spanish, Slovakian, Japanese, Finnish, French, Italian, German, Dominican, Algerian, Indian, Turkish and other patients from central Europe). Of these variations, forty-eight (69.5%) were missense, 10 (14.5%) splice site, 4 (5.5%) nonsense, 3 (4.5%) insertion, 3 (4.5%) deletion and 1 (1.5%) insertion/deletion.
Variants with unclear pathogenicity
Here we describe a total of 91 HGD variants (62 missense, 13 splice site, 10 frameshift, 5 nonsense and a no-stop), 52 from our study (22 novels) and 39 previously reported. The frameshift variants, the no-stop variant, and the variants modifying the canonical sites for splicing have a high likelihood to be pathogenic and result in a deleterious HGD protein. However, the pathogenicity of the missense mutations is less clear. To investigate the effect of the 62 HGD missense variants, we evaluated their possible pathogenicity using different bioinformatic prediction tools: BLOSUM [Henikoff and Henikoff, 1992], SIFT [Ng and Henikoff, 2003; Xi et al., 2004], POLYPHEN [Ramensky et al., 2002], PANTHER [Thomas et al., 2003; Thomas and Kejariwal, 2004; Thomas et al., 2006], PMUT [Ferrer-Costa et al., 2005] and SNAP [Bromberg and Rost, 2007; Bromberg et al., 2008] (Supp. Methods). All the results of these different tools are indexed the Supp. Table S1. Based upon the substitution matrix BLOSUM 62, 31 of 62 missense variations (50%) had a value <-2, classifying them as deleterious. Of the remaining 31, 25 (40%) were ambiguous (score between −1 and +1) and 6 (10%) were classified as benign (score > +1) (Supp. Table S1).
Among the 62 candidate missense variations submitted to the SIFT program, 55 (88.5%) were identified to be deleterious with a tolerance index score <0.05, one (1.5%) was ambiguous, and 6 (10%) were benign (Supp. Table S1). SNAP predicted 55 (90%) of these variants to have a non-neutral effect on the protein and 6 (10%) to have a neutral effect. When the same variants were submitted to POLYPHEN, 50 (80.5%) were called probably damaging, 46 based on the alignment and 4 based on their effect on the 3D structure of the protein. Eight (13%) were considered possibly damaging and only four (6.5%) were called benign (Supp. Table S1).
The results obtained using PANTHER and PMUT were less significant. PANTHER predicted 39 variants (63%) to be mutations (subPSEC <-3) and 23 (37%) to be nsSNP. PMUT called only 35 variants (56.5%) pathological, with a neuronal score over 0.5; only 14 (22.5%) had a con dence index over 5. The other 27 (43.5%) were considered neutral (neuronal score under 0.5) and only 10 (16%) had a confidence index over 5 (Supp. Table S1).
We examined the possible pathogenicity of all the splice site variations using different analysis programs, including the splice site prediction tool from the Berkeley Drosophila Genome Project (BDGP) web site (http://www.fruitfly.org/seq_tools/splice.html) [Reese et al., 1997] and NetGene2, available from the Center of Biological Sequence analysis (CBS), http://www.cbs.dtu.dk/services/NetGene2/ [Hebsgaard et al., 1996]. The results of these two programs are indexed the Supp. Table S2. Both programs showed a drastic decrease of the score for 11 (84.5%) of the 13 variants. These variants all affected the canonical site for splicing. Interestingly, for 4 variants, BDGP predicted that the alteration may create a new intronic splice site further downstream in the intron (Supp. Table S2). The two other variants, c.650-56G>A and c.650-17G>A, for BDGP do not modify the score (0.84) and for NetGene2 the NN score does not vary (0.662) or varies only slightly (0.635). Therefore, this seems to predict these two previously reported variants not to affect the splicing site.
We identified three probands (Patients 36, 93 and 94), each with three HGD candidate disease-causing mutations. Patient 36 carried a nonsense mutation p.E168X and a splice site modification, c.1007-2A>T, strongly predicted to affect the splicing by BDGP and NetGene2, since it modifies the AG of the acceptor site. Patient 36 also carried a missense variant p.N149K which was predicted to be deleterious by POLYPHEN, SIFT and SNAP, ambiguous by PMUT and benign by PANTHER. None of these three variants was previously reported.
Patient 93 had three missense variations. One, p.S305F, is universally predicted as deleterious by all prediction tools and has been previously described [Phornphutkul et al., 2002]. POLYPHEN, PMUT, PANTHER and SNAP predicted the second variant in Patient 93, p.Q258P, to be deleterious; SIFT considered it benign. This variant was also previously reported [Phornphutkul et al., 2002]. POLYPHEN, PMUT and PANTHER predicted the third variant in Patient 93, p.E3A, to be benign and SIFT predicted it to be deleterious.
The third patient with three candidate disease-causing mutations, Patient 94, carried a previously reported missense variant p.G161R [Gehrig et al., 1997]. Functional analysis of this variant showed a loss of 99% of HGD enzymatic activity [Rodriguez et al., 2000]. The second variant, which was previously reported [Beltran-Valero de Bernabe et al., 1999a], in Patient 94, c.16-1G>A, represents a strongly predicted splice site modification, since it affects the GT of the donor site. The third variant of Patient 94 was an indel, c.1387_1389delGAGinsTA, which creates a frameshift and is expected to produce a truncated protein, p.M339IfsX368. It is a novel mutation, and was detected in three other unrelated probands in this study (Patients 15, 59, 81 and siblings).
We conclude that for these 3 patients, without further functional studies, it is impossible to indicate the disease-causing variants because all three variants have a potentially deleterious effect on protein function.
Polymorphisms
Few polymorphisms were described in HGD and were identified in non-AKU population. The database dbSNP reports 7 variants (Table 4) but these include only one synonymous SNP, c.477G>T. The other variants reported are 4 missense variants and 2 frameshifts. Out of the 6 variants that modify the protein sequence, only rs2255543:A>T was confirmed to be a polymorphism. Two of the 4 missense variants, rs28941783:G>A and rs28942100:C>T, were previously associated with AKU [Fernandez-Canon et al., 1996; Gehrig et al., 1997]. It is not excluded that the three other variants are disease-causing. Indeed, the two frameshift variants, rs34214309 and rs35952153, are both truncating the protein (p.V324GfsX3 and p.N337EfsX5) and therefore are likely pathogenic.
Table 4.
Summary of identified HGD variants
| Name | Position a | Genotype | AA Changeb | MAFc |
|---|---|---|---|---|
| rs17140398 | c.16−175 | C/G | None | 0.234 |
| rs2733825 | c.87+35 | A/T | None | 0.12 |
| rs17140396 | c.87+118 | A/G | None | 0.076 |
| Unknown | c.176+114 | T/A | None | 0.013 |
| rs2255543 | c.240 | A/T | Q>H | 0.297 |
| rs35702995 | c.260 | A/C | E>A | NDf |
| rs3732367 | c.283−145 | C/T | None | 0.032 |
| rs2551607 | c.343+25 | T/C | None | 0.127 |
| rs3817627 | c.434+46 | A/C | None | 0.62 |
| Unknown | c.468+24 | C/A | None | 0.013 |
| rs2293734 | c.474 | G/T | None | NDf |
| rs28941783d | c.481 | G/A | G>R | 0.064 |
| Unknown | c.549+31 | G/A | None | 0.006 |
| rs1862942 | c.649+104 | C/T | None | 0.095 |
| rs2075504 | c.650−86 | A/G | None | 0.057 |
| Unknown | c.650−38 | InsA | None | 0.013 |
| rs28942100d | c.688 | C/T | P>S | 0.013 |
| rs1800700 | c.879+18 | A/G | None | 0.013 |
| rs34214309 | c.970 | DupG | fse | NDf |
| Unknown | c.1007−170 | A/G | None | 0.006 |
| rs35952153 | c.1008 | DupG | fse | NDf |
| Unknown | c.1188+51 | A/G | None | 0.006 |
| Unknown | c.1188+89 | C/T | None | 0.025 |
| rs61799339 | c.1188+142 | G/T | None | 0.063 |
| rs804974 | c.1188+203 | G/A | None | 0.203 |
DNA variant numbering system is based on cDNA (NM_000187.2), with +1 corresponding to the A of the ATG.
Amino Acide (AA) modification of the variant
MAF: Minor Allele Frequency in our cohort
Variant found in dbSNP and also previously reported as a mutation
fs: Frameshift effect of the variant
ND: Not Detected in our cohort
During the sequencing of our cohort of AKU patient, we identified 19 polymorphisms (Table 4), only one was in the coding region, rs2255543:A>T. Seven of them have never been reported (Table 4). We looked for some known Copy Number Variations (CNV) on the Database of the Genomic Variants (http://projects.tcag.ca/variation/) [Iafrate et al., 2004] and no CNV has been identified in the coding region nor in the introns.
Clinical relevance and genotype/phenotype correlation
In 1584, Scribonius first described a child with black urine. In 1859, Bodeker [Bödeker, 1859] and Virchow, in 1866 [Virchow, 1866], reported several cases and introduced the terms alkapton and ochronosis. AKU was one of the four diseases, “inborn errors of metabolism”, Sir Archibald Edward Garrod used to describe “Chemical Individuality” and to explain the idea of inheritance [Garrod, 1902]. Over the past century, the clinical and laboratory features of AKU have been well established, making diagnosis straightforward. However, in sporadic cases a patient can be misdiagnosed with AKU. For example, the use of minocycline, a tetracycline used for treatment of dermatologic or rheumatologic disorders, can induce hyperpigmentation of the auricle and bluish discoloration of the sclera that can be confounded with ochronosis and AKU. Therefore, a definite diagnosis of AKU should be confirmed by quantitative measurement of HGA in the urine [Suwannarat et al., 2004]. In our patients, the mean urinary HGA excreted was 4.8± 2.2 g/day (2.9± 1.2 μmole of HGA/μmole of creatinine) which is 100 time normal excretion [Introne et al., 2007] (Available at http://www.genetests.org).
If two deleterious HGD variants are found in an AKU patient, they are likely disease-causing. However, the term deleterious should be used with recognition of the fact that a certain minimum reduction in enzyme activity is required before clinical disease occurs; a mildly deleterious allele may not be consistent with AKU. It is known that the human liver produces enough homogentisic acid oxidase to convert over 1.5 kg of homogentisic acid per day [Scriver et al., 2001]. Therefore, for a patient to display AKU symptoms, a loss of more than 99% of the enzyme activity is required. Variability in residual HGD enzymatic activities may explain the absence of a clear correlation between genotype and phenotype. This may be explained by the influence of the amount of protein into the diet, but it has not been demonstrated that the diet has a straight effect on the HGA excretion in urine [Phornphutkul et al., 2002; Introne et al., 2007]. Note that the variance of the excretion of the HGA is smaller within affected siblings (common genotype), in average 0.5μmoles HGA/μmoles creatinine, compared to the variance in the unrelated patients (different genotypes), 1.6μmoles HGA/μmoles creatinine.
In the AKU patients with only one identified HGD variant in the coding sequence or exon boundaries, the other disease-causing variant may occur in unsequenced regulatory regions or introns. Mosaicism could also explain the absence of mutation; sequencing DNA from other tissues may detect it. Therefore, if only one HGD variant is found studying the RNA or the protein may help finding mutation. Indeed if mis-splicing occurs it can be explain with a mutation in the intron. If the expression level is different than normal it can be explain by a mutation in regulatory regions. Haplotype analysis can also be performed and comparison of patients with lacking mutation. If two variants with unclear pathogenic significance are identified, the best way to determine the real pathogenicity of these variants resides in a functional assay.
Conclusion and future prospects
A total of 91 HGD variants (52 from our study, 39 previously reported) in patients with AKU are described in this report, including 62 missense, 13 splice site, 10 frameshift, 5 nonsense and a no-stop (Supp. Tables S1, S2 and S3). Of these, 22 were previously unreported. In our NIH AKU patient cohort, a total of 52 different potentially disease-causing variants were identified. Among these variations, 4 were highly recurrent; p.G161R was detected 10 times, c.174delA 11 times, p.C120F 10 times, and p.M368V 26 times. Some other variations were also detected more than once (2 to 8 times) in our cohort (Table 3). This may be due to the existence of founder effects. It was hypothesized that mutations in HGD are concentrated near or in CCC motifs (or GGG) [Beltran-Valero de Bernabe et al., 1999a]. Indeed, they found 10 of the 29 (34.5%) nucleotide changes in or near a CCC motif. However, our current study shows that only 18 of the 91 (20%) HGD variations are in or near CCC motifs, decreasing the importance of these motifs in HGD mutability.
We tried to predict the potential effects on HGD protein function and/or enzyme activity for all the identified (previously reported and novel) missense variants. For this, five current prediction tools (Supp. Table S1) were employed to estimate their effect. In general, the performances of the prediction tools are estimated between 50% and 80% accurate [Ng and Henikoff, 2006; Bromberg and Rost, 2007]. In the case of AKU, as the crystal structure is known [Rodriguez et al., 2000; Titus et al., 2000] tools that use 3D structure of the protein, such as POLYPHEN and SNAP, may be more reliable. In addition, most missense substitution prediction tools today offer con dence scores or an estimate of the degree to which a substitution is deleterious. For only half of the identified missense variants, all five prediction-tools gave the same pathogenicity prediction (Table 5). Therefore, the creation of a functional assay for all the missense variants is required. In 2000, Rodriguez et al. characterized the effects of different HGD mutations on HGD enzyme activities. But, as mentioned before, a moderate decrease in HGD enzyme activity does not always explain the disease [Scriver et al., 2001]. Thus, correlation between a potential deleterious effect on the protein and the loss of enzyme activity needed to develop AKU is an important issue. Site-directed mutagenesis in mouse models or in human cell lines may be effective techniques to develop such a functional assay.
Table 5.
Predictions of the effect of the 62 identified missense variations on HGD protein function a
| Study | SIFT | SNAP | POLYPHEN | PANTHER | PMUT | Common b | |
| Deleterious | 33 | 33 | 32 | 24 | 21 | 17 | |
| Ambiguous | 1 | 0 | 3 | 0 | 0 | 0 | |
| Benign | 2 | 3 | 1 | 12 | 15 | 0 | |
| Published | SIFT | SNAP | POLYPHEN | PANTHER | PMUT | Common b | |
| Deleterious | 22 | 22 | 18 | 15 | 14 | 12 | |
| Ambiguous | 0 | 0 | 5 | 0 | 0 | 0 | |
| Benign | 4 | 4 | 3 | 11 | 12 | 2 |
The predictions were made using prediction software (SIFT, POLYPHEN, PANTHER, PMUT and SNAP), see Supporting Information for more details
Number of variants that have the same prediction in all the prediction tools
In sum, the current study provides a valuable resource for the molecular analysis of alkaptonuria. It is often difficult to predict the pathogenicity of each mutation, especially of missense variants. We demonstrate the use of pathogenicity prediction tools, and conclude that for an accurate prediction of the pathogenicity of some HGD variants a functional (enzyme) assay is required.
Supplementary Material
Acknowledgments
We thank Chanika Phornphutkul and David Adams for expert advice on this study and for assistance with mutation prediction tools and Maya Tuchman and Carla Ciccone for skillful laboratory assistance.
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, USA.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Anikster Y, Nyhan WL, Gahl WA. NTBC and alkaptonuria. Am J Hum Genet. 1998;63:920–921. doi: 10.1086/302027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Granadino B, Chiarelli I, Porfirio B, Mayatepek E, Aquaron R, Moore MM, Festen JJ, Sanmarti R, Penalva MA, de Cordoba SR. Mutation and polymorphism analysis of the human homogentisate 1, 2-dioxygenase gene in alkaptonuria patients. Am J Hum Genet. 1998;62:776–784. doi: 10.1086/301805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Jimenez FJ, Aquaron R, Rodriguez de Cordoba S. Analysis of alkaptonuria (AKU) mutations and polymorphisms reveals that the CCC sequence motif is a mutational hot spot in the homogentisate 1,2 dioxygenase gene (HGO) Am J Hum Genet. 1999a;64:1316–1322. doi: 10.1086/302376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Peterson P, Luopajarvi K, Matintalo P, Alho A, Konttinen Y, Krohn K, Rodriguez de Cordoba S, Ranki A. Mutational analysis of the HGO gene in Finnish alkaptonuria patients. J Med Genet. 1999b;36:922–923. [PMC free article] [PubMed] [Google Scholar]
- Bödeker C. Ueber das Alcapton. Ztschr f rat Med. 1859:130. [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics. 2008;24:2397–2398. doi: 10.1093/bioinformatics/btn435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felbor U, Mutsch Y, Grehn F, Muller CR, Kress W. Ocular ochronosis in alkaptonuria patients carrying mutations in the homogentisate 1,2-dioxygenase gene. Br J Ophthalmol. 1999;83:680–683. doi: 10.1136/bjo.83.6.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, Renedo M, Fernandez-Ruiz E, Penalva MA, Rodriguez de Cordoba S. The molecular basis of alkaptonuria. Nat Genet. 1996;14:19–24. doi: 10.1038/ng0996-19. [DOI] [PubMed] [Google Scholar]
- Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- Garrod AE. The incidence of alkaptonuria: a study in chemical individuality. The Lancet. 1902:1616–1620. [Google Scholar]
- Gehrig A, Schmidt SR, Muller CR, Srsen S, Srsnova K, Kress W. Molecular defects in alkaptonuria. Cytogenet Cell Genet. 1997;76:14–16. doi: 10.1159/000134501. [DOI] [PubMed] [Google Scholar]
- Goicoechea De Jorge E, Lorda I, Gallardo ME, Perez B, Perez De Ferran C, Mendoza H, Rodriguez De Cordoba S. Alkaptonuria in the Dominican Republic: identification of the founder AKU mutation and further evidence of mutation hot spots in the HGO gene. J Med Genet. 2002;39:E40. doi: 10.1136/jmg.39.7.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasko JM, Hooper AJ, Brown JW, McKnight CJ, Burnett JR. A novel missense HGD gene mutation, K57N, in a patient with alkaptonuria. Clin Chim Acta. 2009 doi: 10.1016/j.cca.2009.03.032. [DOI] [PubMed] [Google Scholar]
- Gray PH. Radiography of ancient Egyptian mummies. Med Radiogr Photogr. 1967;43:34–44. [PubMed] [Google Scholar]
- Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashino K, Liu W, Ohkawa T, Yamamoto T, Fukui K, Ohno M, Imanishi H, Iwasaki A, Amuro Y, Hada T. A novel point mutation associated with alkaptonuria. Clin Genet. 1998;53:228–229. doi: 10.1111/j.1399-0004.1998.tb02684.x. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Introne WI, Kayser MA, Gahl WA. GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Copyright, University of Washington; Seattle: 2007. Alkaptonuria; pp. 1997–2009. [Google Scholar]
- Kamoun P, Coude M, Forest M, Montagutelli X, Guenet JL. Ascorbic acid and alkaptonuria. Eur J Pediatr. 1992;151:149. doi: 10.1007/BF01958966. [DOI] [PubMed] [Google Scholar]
- Keeling ME, McClure HM, Kibler RF. Alkaptonuria in an orangutan (Pongo pygmaeus) Am J Phys Anthropol. 1973;38:435–438. doi: 10.1002/ajpa.1330380245. [DOI] [PubMed] [Google Scholar]
- Kress W, Schmidt SR, Halliger-Keller B, Montagutelli X, Muller CR. The genetic defect of the alkaptonuric mouse (aku) Mamm Genome. 1999;10:68–70. doi: 10.1007/s003359900945. [DOI] [PubMed] [Google Scholar]
- Ladjouze-Rezig A, Rodriguez de Cordoba S, Aquaron R. Ochronotic rheumatism in Algeria: clinical, radiological, biological and molecular studies--a case study of 14 patients in 11 families. Joint Bone Spine. 2006;73:284–292. doi: 10.1016/j.jbspin.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Lewis JH. Alcaptonuria in a rabbit. J Biol Chem. 1926;70:659–661. [Google Scholar]
- Manning K, Fernandez-Canon JM, Montagutelli X, Grompe M. Identification of the mutation in the alkaptonuria mouse model. Mutations in brief no. 216. Online. Hum Mutat. 1999;13:171. doi: 10.1002/(SICI)1098-1004(1999)13:2<171::AID-HUMU15>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Mannoni A, Selvi E, Lorenzini S, Giorgi M, Airo P, Cammelli D, Andreotti L, Marcolongo R, Porfirio B. Alkaptonuria, ochronosis, and ochronotic arthropathy. Semin Arthritis Rheum. 2004;33:239–248. doi: 10.1053/s0049-0172(03)00080-5. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Underdahl LO, Mathieson DR. Alkaptonuria: report of 3 cases. Mayo Clin Proc. 1952;27:193–200. [PubMed] [Google Scholar]
- Montagutelli X, Lalouette A, Coude M, Kamoun P, Forest M, Guenet JL. aku, a mutation of the mouse homologous to human alkaptonuria, maps to chromosome 16. Genomics. 1994;19:9–11. doi: 10.1006/geno.1994.1004. [DOI] [PubMed] [Google Scholar]
- Muller CR, Fregin A, Srsen S, Srsnova K, Halliger-Keller B, Felbor U, Seemanova E, Kress W. Allelic heterogeneity of alkaptonuria in Central Europe. Eur J Hum Genet. 1999;7:645–651. doi: 10.1038/sj.ejhg.5200343. [DOI] [PubMed] [Google Scholar]
- Neuberger A. Studies on alcaptonuria: 1. The estimation of homogentisic acid. Biochem J. 1947;41:431–438. doi: 10.1042/bj0410431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Perry MB, Suwannarat P, Furst GP, Gahl WA, Gerber LH. Musculoskeletal findings and disability in alkaptonuria. J Rheumatol. 2006;33:2280–2285. [PubMed] [Google Scholar]
- Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y, Gerber LH, Gahl WA. Natural history of alkaptonuria. N Engl J Med. 2002;347:2111–2121. doi: 10.1056/NEJMoa021736. [DOI] [PubMed] [Google Scholar]
- Porfirio B, Chiarelli I, Graziano C, Mannoni A, Morrone A, Zammarchi E, De Bernabe DB, De Cordoba SR. Alkaptonuria in Italy: polymorphic haplotype background, mutational profile, and description of four novel mutations in the homogentisate 1,2-dioxygenase gene. J Med Genet. 2000;37:309–312. doi: 10.1136/jmg.37.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Rodriguez JM, Timm DE, Titus GP, Beltran-Valero De Bernabe D, Criado O, Mueller HA, Rodriguez De Cordoba S, Penalva MA. Structural and functional analysis of mutations in alkaptonuria. Hum Mol Genet. 2000;9:2341–2350. doi: 10.1093/oxfordjournals.hmg.a018927. [DOI] [PubMed] [Google Scholar]
- Scriver CR, Beaudet AL, Valle D, Sly WS. Alkaptonuria. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill, Inc; 2001. pp. 2109–2123. [Google Scholar]
- Simon G, Zorab PA. The radiographic changes in alkaptonuric arthritis: a report on three cases (one an Egyptian mummy) Brit J Radiol. 1961;34:384. [Google Scholar]
- Smith HP, Smith HP., Jr Ochronosis: report of two cases. Ann Intern Med. 1955;42:171–178. doi: 10.7326/0003-4819-42-1-171. [DOI] [PubMed] [Google Scholar]
- Srsen S, Muller CR, Fregin A, Srsnova K. Alkaptonuria in Slovakia: thirty-two years of research on phenotype and genotype. Mol Genet Metab. 2002;75:353–359. doi: 10.1016/S1096-7192(02)00002-1. [DOI] [PubMed] [Google Scholar]
- Srsen S, Vondracek J, Srsnova K, Svac J. Analysis of the life span of alkaptonuric patients. Cas Lek Cesk. 1985;124:1288–1291. [PubMed] [Google Scholar]
- Stenn FF, Milgram JW, Lee SL, Weigand RJ, Veis A. Biochemical identification of homogentisic acid pigment in an ochronotic egyptian mummy. Science. 1977;197:566–568. doi: 10.1126/science.327549. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- Suwannarat P, O’Brien K, Perry MB, Sebring N, Bernardini I, Kaiser-Kupfer MI, Rubin BI, Tsilou E, Gerber LH, Gahl WA. Use of nitisinone in patients with alkaptonuria. Metabolism. 2005;54:719–728. doi: 10.1016/j.metabol.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Suwannarat P, Phornphutkul C, Bernardini I, Turner M, Gahl WA. Minocycline-induced hyperpigmentation masquerading as alkaptonuria in individuals with joint pain. Arthritis Rheum. 2004;50:3698–3701. doi: 10.1002/art.20606. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Oda K, Yoshikawa Y, Maeda Y, Suzuki T. A novel therapeutic trial of homogentisic aciduria in a murine model of alkaptonuria. J Hum Genet. 1999;44:79–84. doi: 10.1007/s100380050114. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A, Guo N, Mi H, Campbell MJ, Muruganujan A, Lazareva-Ulitsky B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34:W645–650. doi: 10.1093/nar/gkl229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus GP, Mueller HA, Burgner J, Rodriguez De Cordoba S, Penalva MA, Timm DE. Crystal structure of human homogentisate dioxygenase. Nat Struct Biol. 2000;7:542–546. doi: 10.1038/76756. [DOI] [PubMed] [Google Scholar]
- Virchow R. Ein fall von allgemeiner ochronose der knorpel und knorpelähnlichen theile. Arch Pathol Anat. 1866:212. [Google Scholar]
- Watkins SP, Benley H, Shulman NR. Alkaptonuria in a chimpanzee. In: Goldsmith EI, Moor-Jankowski J, editors. Medical Primatology 1970. 1970. pp. 297–298. [Google Scholar]
- Wells C, Maxwell BM. Alkaptonuria in an Egyptian mummy. Brit J Radiol. 1962;35:679–682. doi: 10.1259/0007-1285-35-418-679. [DOI] [PubMed] [Google Scholar]
- Xi T, Jones IM, Mohrenweiser HW. Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics. 2004;83:970–979. doi: 10.1016/j.ygeno.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Yules JH. Ochronotic arthritis; report of a case. Bull New Engl Med Cent. 1954;16:168–173. [PubMed] [Google Scholar]
- Zannoni VG, Lomtevas N, Goldfinger S. Oxidation of homogentisic acid to ochronotic pigment in connective tissue. Biochim Biophys Acta. 1969;177:94–105. doi: 10.1016/0304-4165(69)90068-3. [DOI] [PubMed] [Google Scholar]
- Zatkova A, Chmelikova A, Polakova H, Ferakova E, Kadasi L. Rapid detection methods for five HGO gene mutations causing alkaptonuria. Clin Genet. 2003;63:145–149. doi: 10.1034/j.1399-0004.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- Zatkova A, de Bernabe DB, Polakova H, Zvarik M, Ferakova E, Bosak V, Ferak V, Kadasi L, de Cordoba SR. High frequency of alkaptonuria in Slovakia: evidence for the appearance of multiple mutations in HGO involving different mutational hot spots. Am J Hum Genet. 2000a;67:1333–1339. doi: 10.1016/s0002-9297(07)62964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatkova A, Polakova H, Micutkova L, Zvarik M, Bosak V, Ferakova E, Matusek J, Ferak V, Kadasi L. Novel mutations in the homogentisate-1,2-dioxygenase gene identified in Slovak patients with alkaptonuria. J Med Genet. 2000b;37:539–542. doi: 10.1136/jmg.37.7.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.