

Abstract
Fyn null mice display reduced adiposity associated with increased fatty acid oxidation, energy expenditure and activation of the AMP-dependent protein kinase (AMPK) in skeletal muscle and adipose tissue. The acute pharmacological inhibition of Fyn kinase activity with SU6656 in wild type mice reproduces these metabolic effects and induced a specific reduction in fat mass with no change in lean mass. LKB1, the main upstream AMPK kinase (AMPKK) in peripheral tissues was redistributed from the nucleus into the cytoplasm of cells treated with SU6656 and in cells expressing a kinase-deficient but not a constitutively kinase active Fyn mutant. Moreover, Fyn kinase directly phosphorylated LKB1 on tyrosine 261 and 365 residues and mutations of these sites resulted in LKB1 export into the cytoplasm and increased AMPK phosphorylation. These data demonstrate a novel crosstalk between Fyn tyrosine kinase and the AMPK energy-sensing pathway through Fyn-dependent regulation of the AMPK upstream activator LKB1.
Keywords: Fyn, AMP-dependent protein kinase, LKB1, energy expenditure, weight loss
Introduction
Fyn is one member of the large Src family of non-receptor tyrosine kinases that share conserved structural domains. The Src Homology 1 (SH1) domain contains the catalytic tyrosine kinase activity and the SH2 domain binds to tyrosine-phosphorylated substrates. In particular, Fyn SH2 domain binds the tyrosine 528 residue in the carboxyl terminal tail of the protein, stabilizing the structure into an inactive conformation, thereby inhibiting the tyrosine kinase SH1 domain (Sicheri and Kuriyan, 1997; Sicheri et al., 1997; Songyang et al., 1995). The dephosphorylation of this site is required to release the SH2 domain and to activate the tyrosine kinase activity of Fyn.
Several studies have implicated Src kinase family members in mediating a subset of insulin signaling events. For example, Fyn was reported to directly associates with insulin-stimulated tyrosine phosphorylated IRS and c-Cbl proteins (Myers et al., 1996; Ribon et al., 1998; Sun et al., 1996). In addition, Src family kinases have been found to activate the phosphatidylinositol (PI) 3-kinase signaling pathway, an established link to the stimulation of glucose transport in skeletal muscle and adipocytes (Choudhury et al., 2006). Upon post-translational modifications such as palmitoylation and/or N-myristoylation, the Fyn kinase dynamically and reversibly redistributes between the cell interior and the plasma membrane (Alland et al., 1994; Filipp et al., 2003; Shenoy-Scaria et al., 1994). Several studies have also implicated Fyn in the regulation of insulin signaling through lipid raft microdomains. For example, Fyn was reported to be the kinase responsible for 3T3L1 adipocyte insulin-stimulated caveolin tyrosine phosphorylation and to associate with lipid raft proteins flotilin and CD36 (Bull et al., 1994; Huang et al., 1991; Mastick and Saltiel, 1997). In this regard, CD36, also known as FAT (Fatty Acid Translocase), facilitates long-chain fatty acid uptake in skeletal muscle and adipose tissue and is linked to phenotypic features of the metabolic syndrome, including insulin resistance and dyslipidemia (Drover and Abumrad, 2005; Drover et al., 2005; Meex et al., 2005; Pravenec et al., 2003). Thus, the physical association of Fyn with CD36 further suggests a functional coupling between lipid raft organization and the regulation of fatty acid translocation and potentially fatty acid metabolism. More recently, we found that Fyn null mice display markedly improved insulin sensitivity, improved plasma and tissue triglyceride/non-esterified fatty acid levels coupled with higher rates of energy expenditure and fatty acid oxidation in the fasted state (Bastie et al., 2007). This was directly correlated with increased AMP-dependent protein kinase (AMPK) T172 α subunit phosphorylation, increased AMPK activity and inhibition of acetylCoA carboxylase (ACC) function.
AMPK is a heterotrimeric complex composed of one catalytic α plus two regulatory subunits β and γ. Each functional AMPK complex is composed of multiple isoforms with overlapping tissue distributions (Cheung et al., 2000; Daval et al., 2006). Skeletal muscle primarily expresses the α2 subunit as well as both β and all three γ isoforms whereas adipose tissue primarily expresses the α1 subunit with both β and γ1 and γ2 isoforms (Daval et al., 2006; Towler and Hardie, 2007). The regulation of AMPK activity depends on the type of subunits assembled and cellular energy status, being activated when the AMP/ATP ratio increases which occurs in states of cellular nutritional deficiency. Binding of AMP to the γ subunit results in a conformational change that may decrease AMPK as a substrate for the PP2C phosphatase (Steinberg, 2007). Alternatively, it was reported that AMP binding increases the ability of upstream kinases (AMPK kinases) to phosphorylate the activating threonine residue (T172) in the α subunit (Towler and Hardie, 2007). In neurons, the calcium-stimulated activation of AMPK is dependent upon the Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) family that phosphorylates the α subunit T172 residue (Hawley et al., 2005). Although CaMKKs have also been shown to activate AMPK in the skeletal muscle under mild tetanic contraction, CAMKK expression is very low in peripheral tissues and is primarily restricted to brain, testis, thymus and T-cells (Jensen et al., 2007)(Anderson et al., 1998). In contrast, LKB1 is expressed in insulin-responsive tissues and muscle specific LKB1 knockout mice are unable to activate AMPK (Alessi et al., 2006; Sakamoto et al., 2005).
LKB1 is a serine/threonine kinase originally identified as a tumor suppressor protein mutated in Peutz-Jeghers syndrome that controls diverse cellular processes, including cellular polarity, cancer and metabolism (Hemminki et al., 1997; Jenne et al., 1998). Regulation of LKB1 appears to be a complex process that involves phosphorylation on diverse residues (S31, S325, T366 and S431) and association into a ternary complex with MO25 and STRADα or STRADβ. Serine 431 in LKB1 is highly conserved in all organisms except Caenorhabditis elegans and is phosphorylated by p90 ribosomal S6 protein kinase (RSK) and protein kinase A (PKA). Although the phosphorylation of S431 was initially described as critical for LKB1 activity, more recent studies have suggested that it might not be necessary and other activation mechanisms might occur (Fogarty and Hardie, 2009). In particular, LKB1 subcellular localization is an important event regulating LKB1 activity as LKB1 functions as a tumor suppressor only when it localizes in the cytoplasm and appears to be inactive when restricted to the nucleus of cells (Alessi et al., 2006). Recent studies have demonstrated that MO25 stabilizes the interactions between LKB1 and STRADα, and that the ternary complex is cytoplasmically localized whereas the monomeric LKB1 protein and/or the dimeric LKB1/MO25 complex are primarily nuclear localized (Boudeau et al., 2003). In addition, LKB1 was reported to undergo sirtuin-mediated deacetylation with the acetylated form restricted to the nucleus and was redistributed to the cytoplasm following deacetylation (Lan et al., 2008).
We recently reported that conventional Fyn knockout mice displayed increased energy expenditure and fatty acid oxidation due to increased activation of adipose tissue and skeletal muscle AMPK activity, resulting in reduced adipose tissue mass and enhanced insulin sensitivity (Bastie et al., 2007). Importantly, the enhanced catabolism predominantly occurred in the fasted state while the Fyn null mice were able to convert to a normal anabolic state during the feeding cycle. These data suggested that pharmacological inhibition Fyn function could induce selective weight loss by decreasing fat mass and simultaneously increasing insulin sensitivity. In addition, this approach would identify the acute role of the Fyn tyrosine kinase activity in adult animals without regard to putative developmental/adaptive changes present in the conventional Fyn null mice. In this study, we examined the physiologic and molecular signaling events regulating energy metabolism using the selective Src family kinase inhibitor, SU6656 (Blake et al., 2000). We observed that acute treatment with SU6656 rapidly increases energy expenditure, fatty acid oxidation and AMPK α subunit T172 phosphorylation in wild type mice, mimicking the metabolic phenotype of the Fyn null mice. Mechanistically, AMPK activation resulted from a Fyn dependent tyrosine phosphorylation of LKB1 that regulates the subcellular localization of LKB1 and activation of AMPK independent of STRADα association. Together, these data provide a novel signaling pathway accounting for the control of energy expenditure by Fyn and support the therapeutic potential of acute Fyn inhibition in vivo.
Results
Fyn activity inhibition increases energy expenditure and lipid utilization
Loss of Fyn kinase protein in the conventional Fyn knockout mice leads to increased lipid utilization and energy expenditure, particularly in the fasting/resting state (Bastie et al., 2007). To examine the effects of acute Fyn inhibition, we investigated the in vivo effects of the pharmacological selective Src family kinase inhibitor SU6656. Wild type mice were placed into metabolic chambers and allowed to acclimatize for 48 hours. Intraperitoneal injection of SU6656 or vehicle was performed at the beginning of the light cycle (fasting/resting state) and the respiratory quotient (RQ), oxygen consumption (VO2), energy expenditure (EE) and physical activity were monitored for the following 12 hours. Both animal groups displayed identical carbohydrate utilization during the dark cycle preceding the injection as shown by similarly high respiratory quotients (Fig. 1A). As expected, the RQ gradually decreased in the vehicle-treated mice during the light cycle as the mice normally switched to lipid utilization. However, the decrease of RQ was more pronounced in SU6656-treated mice, and was particularly more effective during the first three hours following the injection (Fig. 1A). Oxygen consumption and energy expenditure were similar in both groups before the injection and were decreased during the light period as the animals were resting with reduced basal metabolism. However, both oxygen consumption and energy expenditure remained 14% more elevated in the SU6656-injected group (Fig. 1B and C) without any alteration in physical activity in either the dark or light cycle (Fig. 1D).
Figure 1. Acute pharmacological inhibition of Fyn increases energy expenditure.
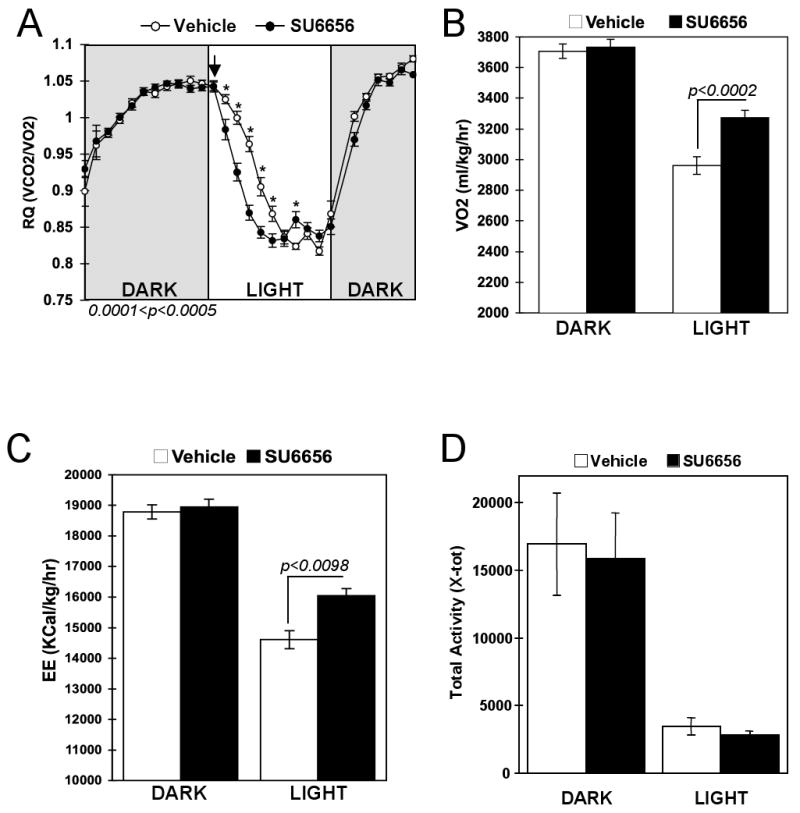
Three month old C57/B6 males received an injection of vehicle or SU6656 (4mg/kg) at 0700h and were placed into metabolic chambers without access to food. Respiratory quotient (RQ) and Oxygen consumption (VO2) were recorded during the dark period preceding the injection and during the light period following the injection. A) Respiratory quotient (RQ) from the average of 4 mice injected with vehicle (open circles) and 4 mice injected with SU6656 (dark circles). B) VO2 recorded before (dark period) and after the injection (light period). C) Energy expenditure (EE) was calculated using the equation of Weir: EE (Kcal/kg/hr)= (3.815 × VO2) + (1.232 × VO2). D) Physical activity was recorded during the dark (before injection) and light (after injection) periods. 0.001< p < 0.01.
To examine whether the increased energy expenditure resulted in weight loss in the SU6656-injected mice, total body mass was evaluated before (T= 0 h) and after (T= 12 h) vehicle and SU6656 injections (Fig. 2A, respectively left and right panels). As mice generally consume 80% of their calories during the dark cycle and have a limited food intake during the light cycle, they undergo a diurnal pattern of weight gain and loss as shown by the decreased body mass at T= 12 h in the vehicle-treated mice. The SU6656-treated animals also displayed decreased body weight 12 h after the injection (Fig. 2A, right panel) and the total weight loss was 40% greater than that of the vehicle-treated group (Fig. 2B). Lean mass was slightly reduced during the light cycle but there was no significant difference between vehicle and SU6656-injected mice (Fig. 2C). In contrast, fat mass was significantly reduced in the SU6656-treated mice compared to vehicle-injected control animals (Fig. 2D). To determine whether the lowered RQ in the mice treated with SU6656 was directly due to increased skeletal muscle fatty acid oxidation, red soleus and white gastrocnemius muscles were isolated 3 hours following the vehicle or SU6656 injection and incubated with 14C-palmitate. The oxidation of 14C-palmitate to 14CO2 was increased 25% and 33%, respectively in the soleus and the gastrocnemius of the SU6656-treated mice relative to vehicle treated controls (Fig. 3A and B). The SU6656-stimulated increase in fatty acid oxidation occurred concomitantly, with an approximate 3-fold increased AMPK α subunit T172 phosphorylation and increased acetyl-CoA carboxylase (ACC) S79 (ACC1) and S221 (ACC2) phosphorylation levels (Fig. 3C and D).
Figure 2. SU6656-induced Fyn inhibition promotes fat mass loss.
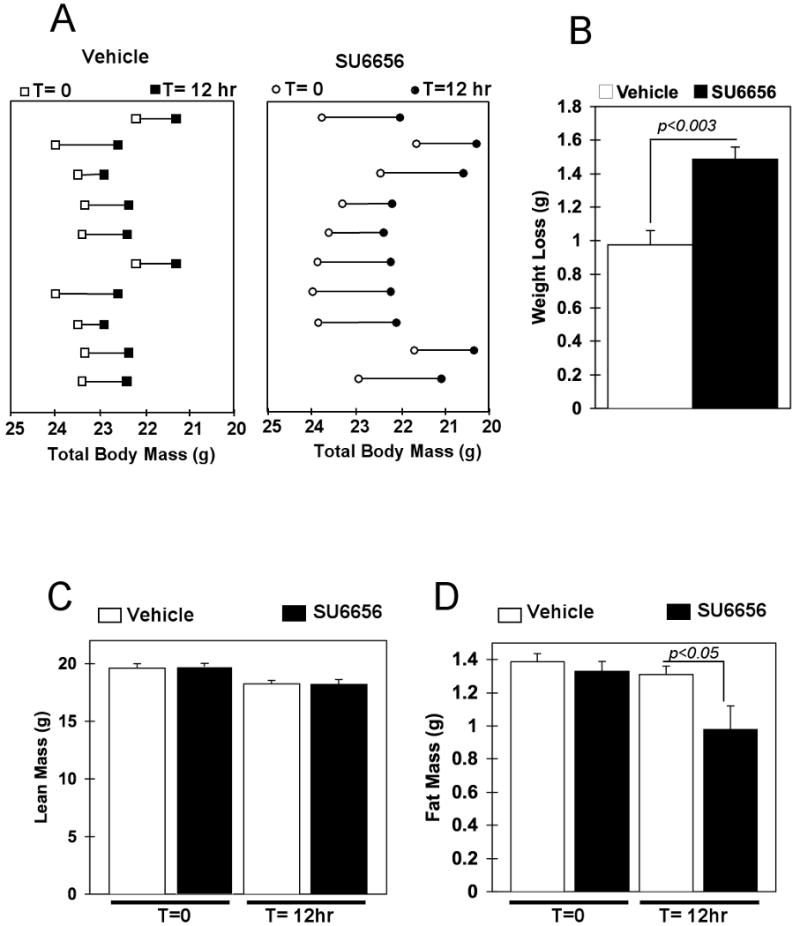
A) Body weight distribution of vehicle and SU6656-injected mice before (T=0) and after (T=12h) the injection. B) Total weight loss twelve hours after the injection of vehicle (open bar) and SU6656 (dark bar) treated animals. C) Fat mass before (T=0) and after (T= 12h) vehicle or SU6656 injection. D) Lean mass before (T=0) and after (T= 12h) vehicle or SU6656 injection. (see also Figure S1).
Figure 3. Fyn specific inhibition increased skeletal muscle Fatty Acid Oxidation and T172 AMPK phosphorylation.
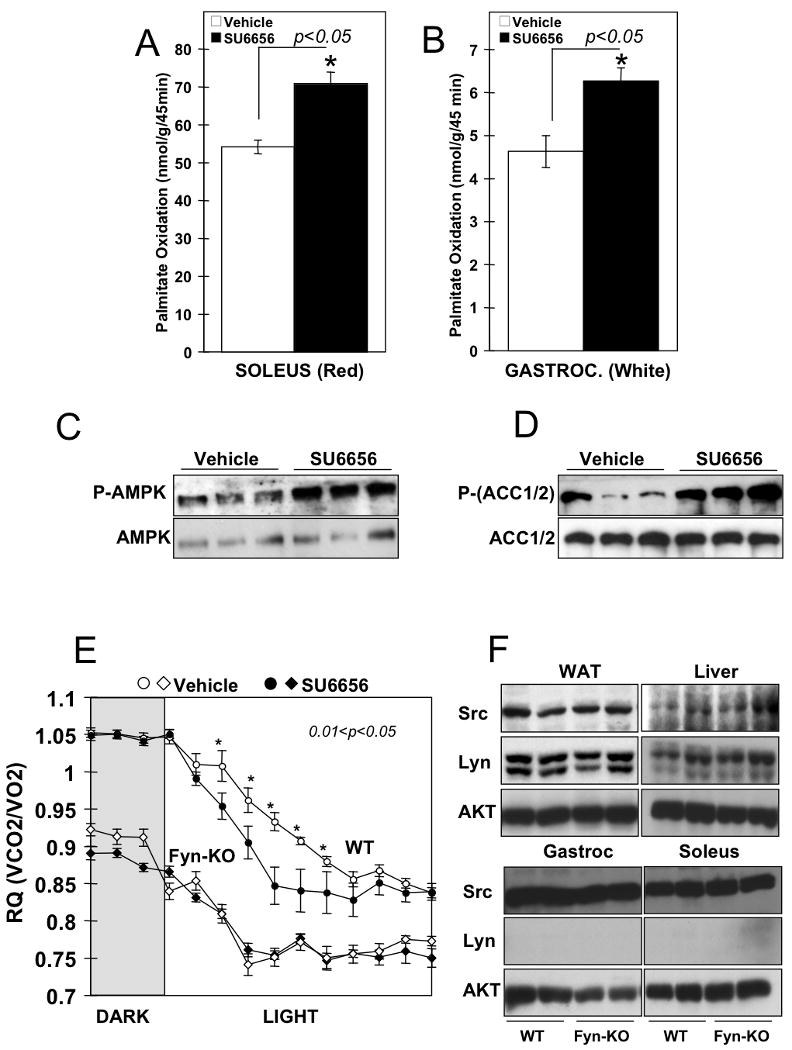
Palmitate oxidation was determined in (A) red muscle (soleus) and (B) white gastrocnemius muscle (White Gastroc.) of mice injected with vehicle or SU6656 (4mg/kg). Data are the mean ± SE of 5 independent experiments. C) Phospho-T(172)-AMPK and total AMPK protein expression levels in white gastrocnemius of vehicle or SU6656-treated mice. D) Phospho-ACC and total ACC protein expression levels in white gastrocnemius of vehicle or SU6656-treated mice. E) Three month old Fyn knockout (diamonds) mice and their controls (circles) were treated with vehicle (open symbols) or SU6656 (filled symbols) at the beginning of the light cycle. Respiratory quotient (RQ) was recorded during the preceding dark period and during the 12 h following the injection. F) Expression levels of Src and Lyn kinase in white adipose tissue (WAT), liver, gastrocnemius (Gastroc) and soleus muscle of Fyn null mice (Fyn-KO) and their controls (WT).
Although SU6656 is a highly selective Src family kinase inhibitor it is not specific for Fyn. Thus, these data do not exclude a role for other Src family kinases in mediating these metabolic effects. To address this issue, we examined the metabolic effect of SU6656 on wild type versus Fyn knockout mice. As previously reported (Bastie et al., 2007), Fyn null mice display a marked reduction in RQ compared to wild type mice (Fig. 3E). Similarly to Figure 1, SU6656 treatment of wild type mice resulted in an enhanced conversion to fatty acid utilization characterized by a lower RQ (Fig. 3E, filled versus open circles). In contrast, SU6656 treatment had no significant effect on the rate of change or the RQ level in the Fyn knockout mice (Fig. 3E, filled versus open diamonds). In addition, we did not observe any alterations of body mass or physical activity after SU6656 treatment in the Fyn null mice (Fig. S1). As the two most closely related kinases to Fyn are Src and Lyn we examined the protein expression levels of Src and Lyn proteins in the Fyn null mice. As shown in Figure 3F, the levels of Src were unchanged in adipose tissue, liver or skeletal muscle of the Fyn null mice. Although Lyn is not expressed in skeletal muscle, there was no change in expression in either adipose tissue or liver. Thus, the lack of effect of SU6656 was not due to altered expression of Src or Lyn expression in the Fyn null mice. Taken together, these results demonstrate that an acute treatment with SU6656 increases whole body energy expenditure, skeletal muscle fatty acid oxidation, decreases adiposity and promotes weight loss most likely through a Fyn kinase dependent mechanism.
Fyn regulates LKB1 subcellular localization
Previous studies have demonstrated that LKB1 is predominantly nuclear localized in cultured cells whereas its substrate target, AMPK, is primarily localized to the cytoplasm (Alessi et al., 2006). To determine if Fyn regulates LKB1 localization, we transfected the C2C12 muscle cell line with GFP-LKB1 and examined the effect of acute SU6656 treatment on subcellular LKB1 localization (Fig. 4). As previously reported (Nezu et al., 1999; Tiainen et al., 2002), in vehicle-treated cells the majority of the LKB1 was nuclear localized (Fig. 4A and B). However, treatment with SU6656 resulted in the redistribution of LKB1 into the cytoplasm in approximately 65% of the cells. Figure S2 shows representative images of multiple C2C12 cells treated with Vehicle (panels A) or SU6656 (panels B). Since the concentration of SU6656 (10 μM) is sufficient to inactivate Fyn kinase activity (Blake et al., 2000), this suggests that LKB1 subcellular localization was controlled by the catalytic activity of Fyn. We therefore transfected C2C12 and 3T3L1 adipocytes with a constitutively active Fyn mutant (Fyn-CA) in which the negative regulatory tyrosine (Y528) site was mutated to phenylalanine and a kinase-defective mutant (Fyn-KD) in which the catalytic lysine (K299) residue was mutated to methionine. Expression of Fyn-CA in C2C12 appeared to alter the morphology to a more rounded phenotype but there was no significant effect on the nuclear localization of LKB1 whereas expression of Fyn-KD had no morphology effect but resulted in a redistribution of more than 50% of the LKB1 out of the nucleus and into the cytoplasm (Fig. 4C and D). Figure S3 shows representative images of multiple C2C12 cells transfected with constitutively active Fyn (Fyn-CA, panels A) or kinase-defective Fyn (Fyn-KD, panels B).
Figure 4. Fyn kinase activity regulates LKB1 subcellular distribution.
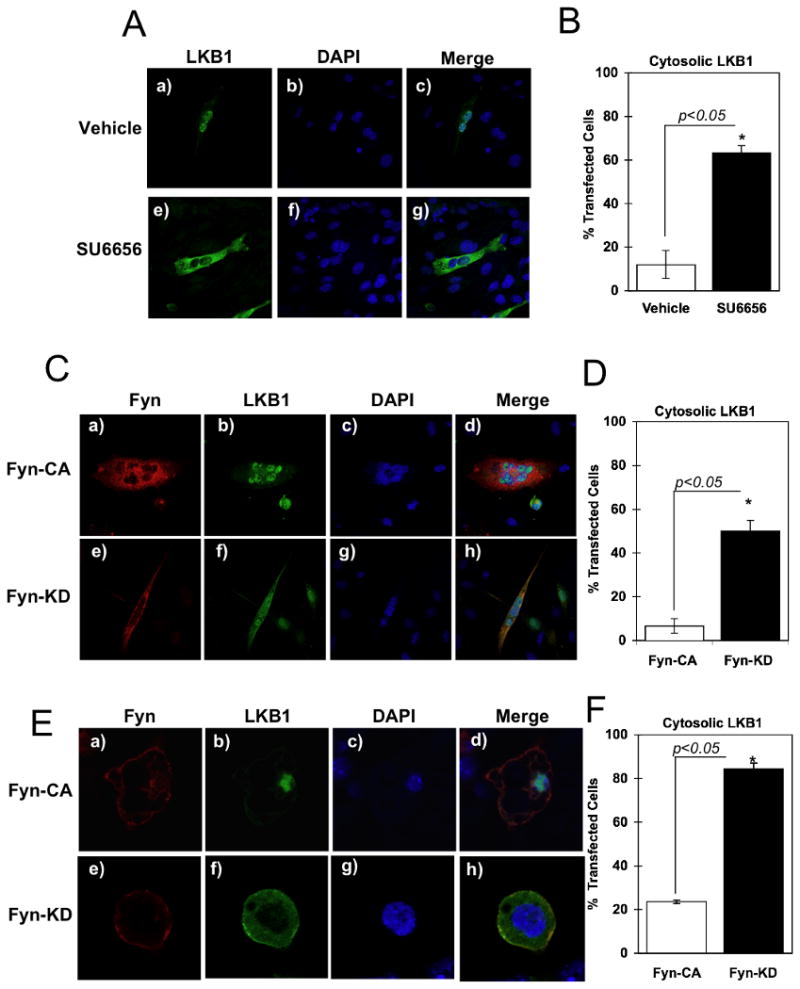
A) C2C12 myotubes were transfected with pEGFP-LKB1 and incubated with vehicle or SU6656 (10μM) for 2 h. Cells were fixed and mounted with proQ diamond with DAPI solution. B) Percentage of cells with pEGFP-LKB1 signal detected in the cytoplasm. C) C2C12 myotubes were co-transfected with pEGFP-LKB1 and pcDNA3-Fyn-KD or pcDNA3-Fyn-CA. Cells were fixed and incubated with the Mouse Fyn monoclonal antibody. Immunofluorescence was performed using the Alexa Fluor 594 Anti-mouse IgG. D) Percentage of C2C12 cells with pEGFP-LKB1 signal detected in the cytoplasm. Data are representative of n = 5 experiments. E) Fully differentiated 3T3L1 adipocytes were co-transfected with pcDNA3-LKB1 and pcDNA3-Fyn-KD or pcDNA3-Fyn-CA. Immunofluorescence was performed using the rabbit LKB1 polyclonal antibody and mouse Fyn monoclonal antibody followed by Alexa Fluor 488 Anti-Rabbit IgG and Alexa Fluor 564 Anti-Mouse IgG. F) Percentage of 3T3L1 cells with LKB1 signal detected in the cytoplasm. Data are representative of n = 5 experiments. (see also Figure S2, S3, S4 and S5).
In contrast to C2C12 cells, subcellular localization is more readily visualized in adipocytes due to the presence of large lipid droplets. Expression of Fyn-CA or Fyn-KD has no significant effect on the morphology of the adipocytes (Fig. 4E). LKB1 remained nuclear localized in the presence of Fyn-CA whereas expression of Fyn-KD resulted in decreased nuclear LKB1 with an increased cytosolic localization (Fig. 4E and F). Figure S4 shows representative images of multiple 3T3L1 adipocytes transfected with constitutive active Fyn (Fyn-CA, panels A) or kinase-defective Fyn (Fyn-KD, panels B).
Since STRAD〈 has been reported to regulate intracellular LKB1 distribution (Boudeau et al., 2003; Boudeau et al., 2004), we co-transfected 3T3L1 adipocytes with STRAD〈 and either Fyn-CA or Fyn-KD and examined LKB1 subcellular localization. As previously observed, LKB1 alone was nuclear localized in adipocytes (Fig. S5A, panels a-c) but following co-expression with STRAD〈 there was an efficient redistribution of LKB1 into the cytoplasm (panels d-g). However, LKB1 remained nuclear localized when co-expressed with the constitutively active form of Fyn kinase even in presence of STRAD〈 (Fig. S5B, panels a-e). As a control, the expression of the kinase-deficient Fyn (Fyn-KD) with STRADα had no additional effect on the cytoplasmic localization of LKB1 (Fig. S2B, panels f-j). The quantification of these data is presented in supplementary Fig. 5C. Due to the high expression levels of endogenous MO25 in adipocytes, identical results were also obtained when STRADα and MO25 were co-expressed with LKB1 (data not shown).
Figure 5. Fyn phosphorylates LKB1 on tyrosine residues 261 and 365.
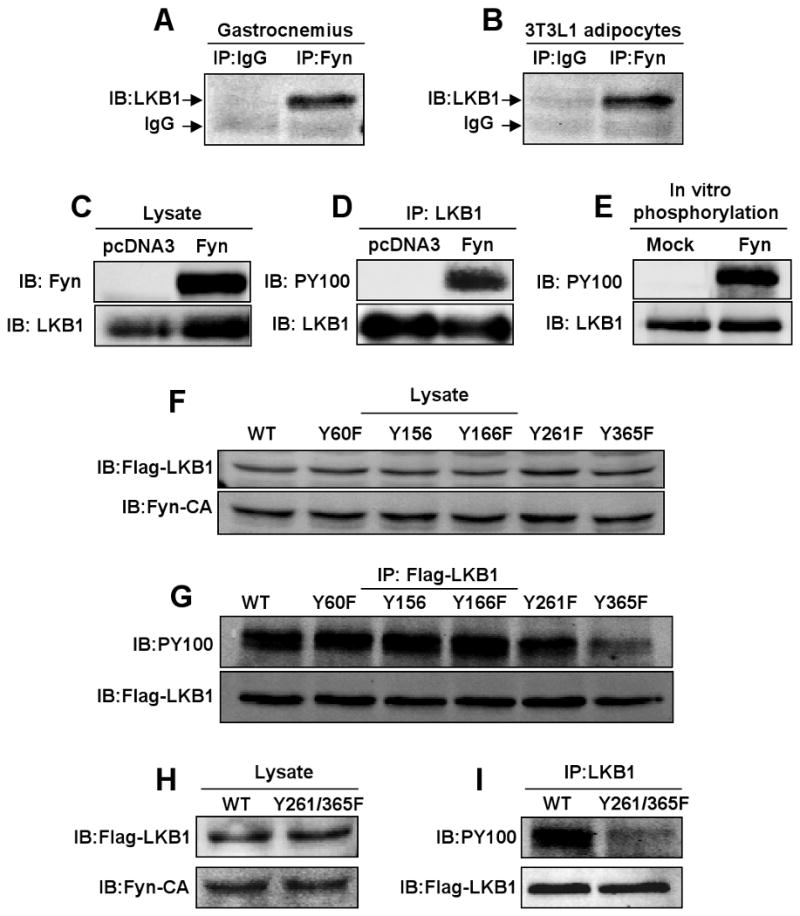
A) gastrocnemius muscle and (B) differentiated 3T3L1 adipocyte extracts were immunoprecipitated with IgG or the Fyn rabbit polyclonal antibody and immunoblotted with the monoclonal LKB1 antibody. 3T3L1 adipocyte were transfected with the pcDNA3 empty vector or pcDNA3-Fyn construct. C) Cell extracts (lysates) were immunoblotted for Fyn and LKB1. D) Cell extracts were immunoprecipitated with the LKB1 monoclonal antibody and immunoblotted with the phosphotyrosine antibody (PY100) or LKB1 antibody. E) Purified His-tagged LKB1 was incubated with ATP in the absence and presence of purified Fyn protein. The samples were then immunoblotted with the LKB1 antibody and the phosphotyrosine antibody PY100. F) pcDNA3-Flag-LKB1 mutant cDNAs and the pcDNA3-Fyn-CA constructs were co-expressed in 3T3L1 adipocytes and levels of expression were determined in whole cell extracts. G) Levels of tyrosine phosphorylation of each LKB1 construct were determined in Flag-immunoprecipitates subjected to immunoblotting with PY100. pcDNA3-Flag-LKB1-Y261/365F double mutant and pcDNA3-Fyn-CA constructs were co-expressed in 3T3L1 adipocytes. H) The levels of LKB1 expression were determined by immunobloting cell extracts with the Flag and Fyn antibodies, respectively. I) LKB1 tyrosine phosphorylation was determined by LKB1 immunoprecipitation followed by immunoblotting with the PY100 and Flag antibodies. (see also Figure S6).
LKB1 is a substrate for Fyn tyrosine kinase
Since Fyn-KD but not Fyn-CA induced the redistribution of LKB1 into the cytoplasm, we next examined whether LKB1 can interact and/or is a substrate for the Fyn tyrosine kinase catalytic activity. First, we observed that endogenous LKB1 co-immunoprecipitated with Fyn kinase in skeletal muscle and in 3T3L1 adipocytes (Fig. 5A and B). We next co-expressed Fyn with LKB1 in 3T3L1 adipocytes. Immunoblotting of LKB1 immunoprecipitates with the PY100 phosphotyrosine antibody demonstrated Fyn-induced tyrosine phosphorylation of LKB1 (Fig. 5C and D). LKB1 is a direct substrate target of Fyn as purified His-LKB1 tagged fusion protein was tyrosine phosphorylated by purified recombinant Fyn kinase in vitro (Fig. 5E). To identify the LKB1 tyrosine sites phosphorylated by Fyn, we utilized Phosphosite Detector from JPT peptide technology. As shown in Figure S6, 141 overlapping peptides (12-15 residues in length) corresponding to the LKB1 sequence were subjected to in vitro phosphorylation utilizing purified recombinant Fyn tyrosine kinase. This analysis identified five tyrosines (Y60, Y156, Y166, Y261 and Y365) as potential LKB1 phosphorylation acceptor sites for the Fyn kinase. To identify the phosphorylation sites in vivo, we next generated single point mutants where each tyrosine site was substituted by a phenylalanine residue. Co-expression of Fyn-CA with wild type LKB1 and each individual mutant demonstrated equal protein expression levels for all LKB1 mutants (Fig. 5F). Tyrosine phosphorylation levels were decreased with LKB1-Y261F mutant and a substantially greater reduction was obtained with LKB1-Y365F (Fig. 5G). In addition, while both single mutants (Y261F and Y365F) partially reduced LKB1 phosphorylation, the double mutation Y261/365F had a more pronounced decrease in LKB1 tyrosine phosphorylation (Fig. 5H and I).
LKB1 subcellular distribution is regulated by tyrosine phosphorylation
Since inhibition of Fyn kinase activity by SU6656 treatment or expression of a Fyn kinase deficient mutant (dominant-negative) resulted in redistribution of LKB1 out of the nucleus, we examined the subcellular localization of the phosphorylation defective mutants of LKB1 (Fig. 6). As previously observed, in adipocytes wild type LKB1 was predominantly nuclear localized as well as the LKB1-Y60F mutant (Fig. 6A, panels a-f). Similarly, both LKB1 mutants Y156F and Y166F were also nuclear localized (data not shown). In contrast, both the LKB1 Y261F and Y365F mutants displayed a greater cytosolic distribution, similar to that observed for the SU6656-treated and Fyn-KD transfected cells (Fig. 6B, panels a-f). Similarly, the LKB1 double mutation Y261/365F displayed a predominant cytoplasmic distribution (Fig. 6C, panels a-c). To determine if the Fyn-dependent tyrosine phosphorylation at Y261 and Y365 was directly responsible for LKB1 nuclear localization, we co-expressed constitutively active Fyn (Fyn-CA) with the LKB1-Y261/365F double mutant. In this case, LKB1-Y261/365F remained cytosolic and Fyn-CA was ineffective in redistributing this mutant into the nucleus (Fig. 6D, panels a-d). Quantification of these data are presented in Figure 6E with approximately 20% of LKB1-WT and LKB1-Y60F displayed a cytoplasmic localization whereas 55-60% of LKB1-Y261F and LKB1-Y365F were in the cytoplasm. Moreover, nearly 90% of the LKB1-Y261/365F double mutant was found in the cytoplasm and co-expression of Fyn-CA was ineffective in altering the subcellular localization of the LKB1-Y261/365F double mutant.
Figure 6. LKB1 tyrosine phosphorylation regulates its subcellular distribution.
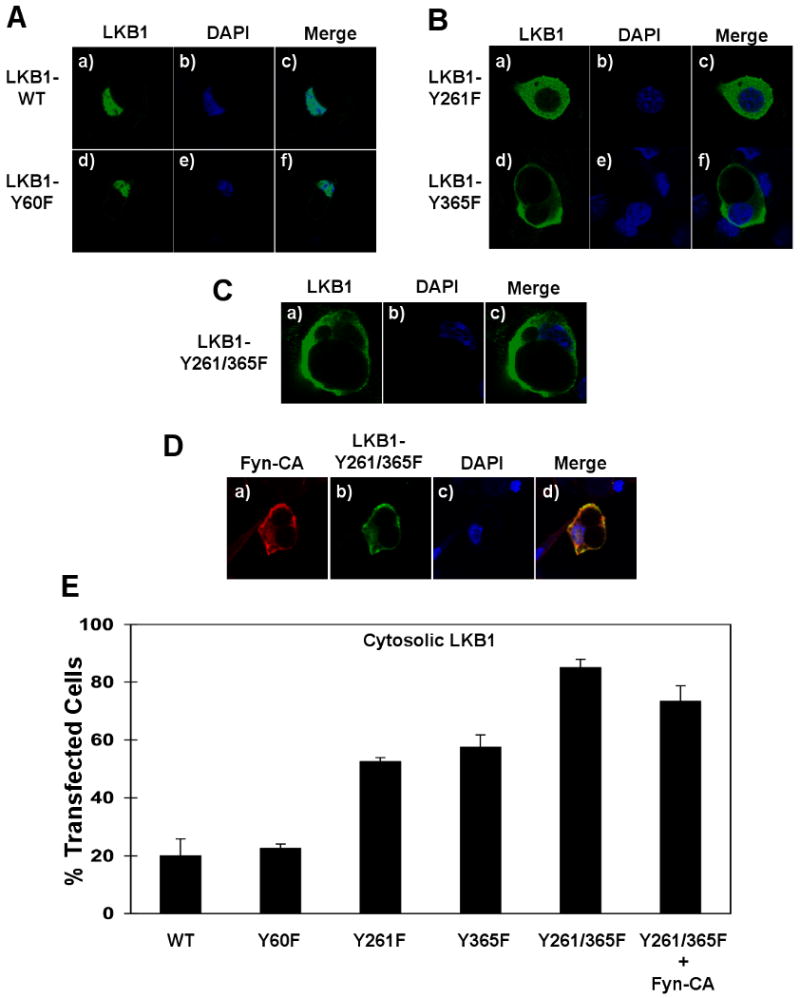
A) 3T3L1 adipocytes were transfected with pcDNA-Flag-LKB1-WT or the pcDNA-Flag-LKB1-Y60F mutant cDNAs. B) 3T3L1 adipocytes were transfected with the pcDNA-Flag-LKB1-Y261F and pcDNA-Flag-LKB1-Y365F cDNAs. C) 3T3L1 adipocytes were transfected with pcDNA-Flag-LKB1-Y261/365F double mutant cDNA. Cells were fixed and subjected to immunofluorescence for the localization of LKB1 (Flag antibody) and DAPI labeling for nuclei identification. D) 3T3L1 adipocytes were transfected with pcDNA-Fyn-CA and the pcDNA-Flag-LKB1-Y261/365F double mutant cDNAs. Cells were fixed and subjected to immunofluorescence for Fyn-CA expression (red), LKB1-Y261/365F double mutant localization (green) and nuclei (blue). E) Percentage of 3T3L1 cells with LKB1 signal detected in the cytoplasm. Data are representative of n = 3 experiments.
To confirm these results in vivo, we took advantage of skeletal muscle transfection by electroporation as described previously (Prud'homme et al., 2006). The expressed Flag-LKB1-WT is predominantly detected in the muscle syncytia that parallel the sarcolemma (Fig. 7A, panels a-c). In contrast, expression of the Flag-LKB1-Y261/365F double mutant resulted in a more cytoplasmic localization (Fig. 7A, panels d-f). The low level of non-specific background labeling is shown in Figure 7A (panels g-i) and a larger magnification of LKB1 localization is provided in Figure 7B. Although it is very difficult to detect the endogenous LKB1 in vivo by immunofluorescence due to the quality of the currently available antibodies, we also observed an increased LKB1 signal in the cytoplasm of muscle cells of wild type mice treated with SU6656 (data not shown).
Figure 7. Subcellular localization of LKB1 in skeletal muscle in vivo is regulated by tyrosine phosphorylation.
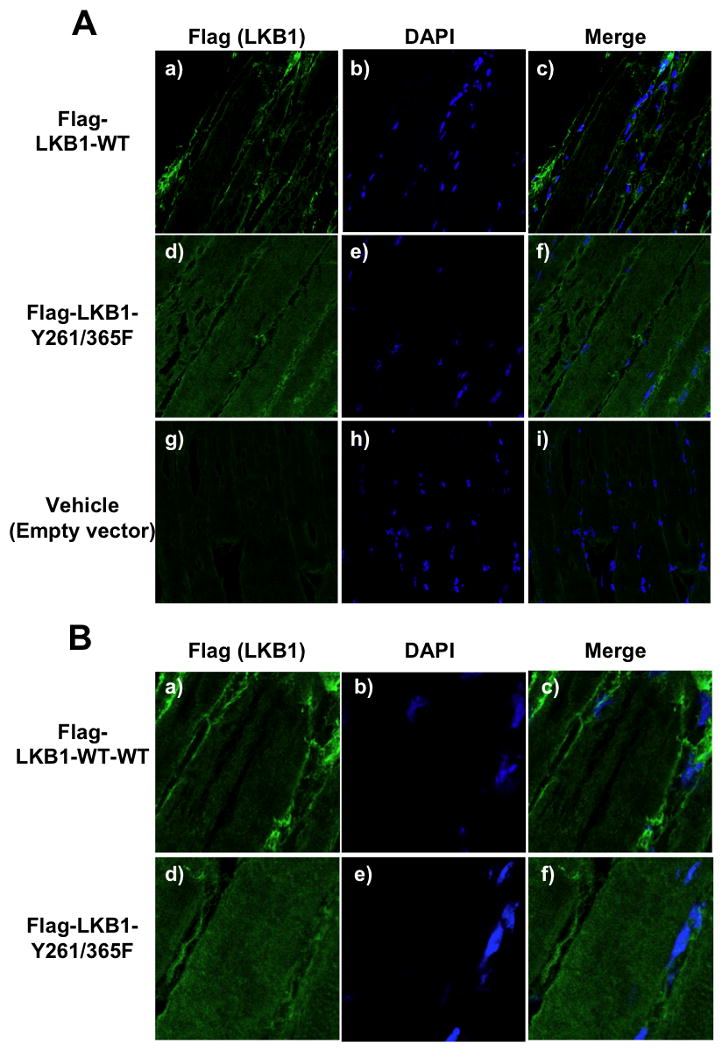
A) Tibialis anterior was transfected with pcDNA-Flag-LKB1-WT (panels a-c), the pcDNA-Flag-LKB1-Y261/365F double mutant (panels d-f) or the pcDNA empty vector (panels g-i) cDNAs. Immunofluorescence was performed on 10 μm frozen sections for the localization of LKB1 (Flag antibody) and nuclei (DAPI). B) Magnified images of muscles transfected with pcDNA-Flag- LKB1-WT (panels a-c) or the pcDNA-Flag-LKB1-Y261/365F double mutant (panels d-f) is shown to more easily visualize the change in LKB1 localization. (see also Figure S7).
To determine if Fyn kinase also induces the tyrosine phosphorylation of LKB1 in vivo, tibialis anterior muscles were transfected with Fyn-CA and endogenous LKB1 was immunoprecipitated and immunoblotted with PY100 (Figure S7A and B). In the absence of Fyn-CA there was a relatively low level of LKB1 tyrosine phosphorylation that was significantly increased by the expression of Fyn-CA. To demonstrate the functional consequence of LKB1 distribution, we first assessed the effect of the LKB1-Y261/365F double mutant on AMPK phosphorylation by co-expression with the GST-AMPK α subunit in HeLa cells. There was a low level of AMPK α subunit T172 phosphorylation when co-expressed with LKB1-WT that was increased in the presence of the LKB1-Y261/365F double mutant (Fig. S7C). Moreover, there was a marked increase in endogenous AMPK α subunit T172 phosphorylation in tibialis anterior muscle transfected with the LKB1-Y261/365F double mutant compared to LKB1-WT (Fig. S7D).
Discussion
AMP-activated protein kinase (AMPK) is considered as a cellular “energy sensor” directly regulated by the alteration of the intracellular AMP/ATP ratio that occurs during prolonged fasting and re-feeding (Hardie, 2008a; Hardie et al., 2006; Hue and Rider, 2007; Schimmack et al., 2006). During states of low energy, activation of AMPK results in the phosphorylation and inhibition of AcetylCoA Carboxylase (ACC) activity, thereby lowering MalonylCoA levels leading to increased fatty acid oxidation and a reduction in fatty acid synthesis (Brownsey et al., 2006). In contrast, during states of caloric excess AMPK is inactive, resulting in increased ACC activity and inhibition of fatty acid oxidation with a concomitant increase in fatty acid storage. The critical role of AMPK in determining energy balance has been clearly demonstrated in both AMPK knockout and over expression of dominant-interfering AMPK mutants that display insulin resistance, reduced energy expenditure and inability to undergo normal metabolic switching between carbohydrate and fatty acid fuels (Hardie, 2008a); (Hardie, 2008b; Viollet et al., 2003). Thus, mechanisms and factors controlling AMPK activity are key issues in the balance of lipid and glucose metabolism that may provide novel therapeutic opportunities.
Previously, we demonstrated that Fyn functions as a negative regulator of fatty acid oxidation through the inhibition of AMPK in skeletal muscle and adipose tissue (Bastie et al., 2007). This was based upon the observation that conventional Fyn null mice displayed enhanced fatty acid oxidation in adipose tissue and skeletal muscle, increased AMPK activity, increased energy expenditure and insulin sensitivity. However, due to the constitutive loss of Fyn expression these data could neither address potential developmental tissue adaptations that could be responsible for these metabolic alterations nor distinguish whether this resulted from a loss of Fyn kinase activity or protein interaction functions. Therefore, the mechanism responsible for AMPK activation in the Fyn null mice remained enigmatic.
To address these issues, we first took advantage of a selective pharmacological approach to acutely inhibit Src family kinase activity that recapitulated the metabolic phenotype observed in the Fyn null mice, including increased fatty acid oxidation, energy expenditure and AMPK active site phosphorylation. The metabolic effect of the SU6656 inhibitor was likely specific for Fyn as this agent had no effect in Fyn null mice. These changes in whole body metabolism occurred relatively rapidly (2-3h) upon acute treatment with SU6656 consistent with inhibition of Fyn kinase activity rather than developmental compensation or other Fyn-protein interactions being responsible for the observed phenotype. Moreover, the phenotypic characteristics of Fyn inhibition are essentially the same as those reported for other animal models with increased AMPK activity (Viollet et al., 2009). Taken together these data strongly indicate that Fyn kinase activity per se serves to functionally inhibit AMPK activity. Importantly, the acute inhibition of Fyn resulted in significant weight loss due to decreased adipose tissue mass without any significant change in lean mass, supporting a potential therapeutic approach for modulating Fyn function.
In this regard, the primary upstream kinase activator of AMPK in peripheral tissues is LKB1 and we speculated that this was a potential Fyn target responsible for the regulation of AMPK. Several studies have demonstrated that LKB1 activity depends on its subcellular localization, LKB1 being active in the cytoplasm and inactive when restricted to the nucleus of cultured cells (Alessi et al., 2006; Baas et al., 2003). Since AMPK is predominantly localized in the cytoplasm, nuclear export of LKB1 would be required for LKB1-dependent phosphorylation of AMPK. Our data demonstrate that activated Fyn kinase activity maintains LKB1 predominantly in the nucleus and that inhibition of Fyn kinase activity results in LKB1 redistribution into the cytoplasm of cultured cells. Moreover, skeletal muscle nuclei are peripherally organized adjacent to the sarcolemma and LKB1 is also peripherally localized in skeletal muscle in vivo. Although, there appears to be partial overlap with nuclei, inhibition of Fyn kinase activity also results in LKB1 redistribution into the cytoplasm in a manner similar to the cultured cells.
Mechanistically, our data indicate that Fyn-dependent regulation of LKB1 localization occurs through the tyrosine phosphorylation of two critical sites on LKB1 (Y261 and Y365). Mutational analyses demonstrated that the loss of these phosphorylation sites resulted in LKB1 cytoplasmic distribution and prevented constitutively active Fyn from inducing LKB1 nuclear import. The tyrosine phosphorylation regulation of LKB1 localization was dominant over MO25/STRADα, as in the presence of STRADα Fyn-CA was still able to drive nuclear import of LKB1. Functionally, treatment with SU6656 or expression of the LKB1-Y261/365F mutant that results in LKB1 cytoplasmic localization also results in increased AMPK phosphorylation on T172 both in skeletal muscle in vivo and in cultured cells.
We therefore propose a working hypothesis in which, similar to other regulated proteins that undergo nuclear import/export (ie: Foxo1), LKB1 continually undergoes nuclear import/export such that under basal steady-state levels, the equilibrium favors nuclear localization. However, this equilibrium shifts to greater cytoplasmic localization by either increasing nuclear export and/or reducing nuclear import through various regulatory events. For example, previous studies have shown that these include binding to STRADα/MO25 and LKB1 deacetylation (Boudeau et al., 2003; Lan et al., 2008). Since Fyn is predominantly a non-nuclear protein being distributed throughout the cell including the plasma membrane, endomembranes and the cytoplasm (Alland et al., 1994); (Davy et al., 1999; Filipp and Julius, 2004; Filipp et al., 2003; Parravicini et al., 2002), we hypothesize that Fyn interacts and tyrosine phosphorylates the nuclear exported LKB1 that, in turn, increases its rate of nuclear import, resulting in a high steady-state level of nuclear LKB1. Inhibition of Fyn kinase prevents LKB1 tyrosine phosphorylation on Y261 and Y365, reducing the rate of nuclear import that now results in a greater steady-state level of LKB1 in the cytoplasm. One appealing model that can account for these observations is that Fyn-dependent LKB1 tyrosine phosphorylation prevents the assembly of LKB1 into the LKB1/STRADα/MO25 ternary complex, thereby increasing LKB1 nuclear localization. Tyrosine dephosphorylation would then allow for the formation of the ternary complex and promote cytosolic LKB1 localization and kinase activation.
In summary, we have demonstrated that LKB1 is a direct substrate for Fyn tyrosine kinase, LKB1 subcellular distribution is regulated by tyrosine phosphorylation on Y261 and Y365, and that Fyn-dependent redistribution of LKB1 into the cytoplasm results in increased phosphorylation/activation of AMPK. Importantly, the positive metabolic effects observed in Fyn null mice (decreased adiposity and increased energy expenditure) are reproduced by the acute pharmacological inhibition of Fyn activity resulting in weight loss via decreased adiposity without affecting lean mass. Since the deregulation of the whole body energy homeostasis is one of the main events leading to the development of obesity, insulin resistance and diabetes, these data highlight the therapeutic potential of inhibiting Fyn kinase signaling.
Experimental Procedures
Animals
Eight to ten week old males C57BLK6/J, pp59fyn null mice and their controls were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed in a facility equipped with a 12 h light/dark cycle. Animals were fed ad libitum a standard chow diet (Research Diets, New Brunswick, NJ) containing 75.9% (Kcal) carbohydrates, 14.7% protein, and 9.4% fat. All studies were approved by and performed in compliance with the guidelines of the Yeshiva University Institutional Animal Care and Use Committee (IACUC).
Whole body indirect calorimetry
Oxygen and carbon dioxide consumption were simultaneously determined by Oxymax open-circuit indirect calorimetry system (eight-cage system) (Columbus Instruments). Animals were allowed to acclimatize for two complete light and dark cycles (48 h) and SU6656 injections were performed at the beginning of the light cycle the following day. Measurements were subsequently taken 12 h following the injection. Data were analyzed as the average of 1 h measurements for each mouse. Instrument settings were as follow: gas flow rate= 0.6 l/min, sample flow rate=0.5 l/min, settle time =120 s, measure time =60 s.
Total Body Mass and Magnetic Resonance Imaging
Total body mass (g) was recorded before (T= 0) and 12 h (T= 12 h) after the SU6656 injection. To determine fat and lean mass, animals were placed in a clear plastic holder without anesthesia or sedation and inserted into the EchoMRI-3-in-1™ System from Echo Medical Systems (Houston, TX, USA). Total body fat and lean mass were measured before (T= 0) and after (T=12 h) after the injection.
Western Blot Analysis
Animals were injected with vehicle or SU6656 (4mg/kg) at the beginning of the light cycle. They were anesthetized and sacrificed by cervical dislocation 3 h after the injection. Tissues were rapidly freeze-clamped in liquid nitrogen and stored at -80°C. Protein preparation and blotting were performed as described below. Tissues were ground in liquid nitrogen and homogenized in a buffer containing 50mM Tris (pH 7.4), 1% glycerol and 1% Triton X-100 supplemented with protease inhibitor (Complete mini, Roche Pharmaceuticals, Nutley, NJ). Homogenates were centrifuged for 30 min at 14,000 rpm at 4°C, and supernatants were collected. Protein concentration was determined with the BCA™ Protein Assay (Thermo Scientific, Rockford, IL).
Protein samples (40 μg) were separated on 8 or 10% reducing polyacrylamide gels and trasnferred onto Immobilon-P polyvinylidene difluoride membranes. Immunoblots were blocked with 2% milk and 3% BSA in Tris-buffered saline for 60 min at room temperature and incubated overnight at 4°C with the indicated antibodies (Cell Signalling, Upstate, and Alpha Diagnostic international) in Tris-buffered saline and 0.05% Tween 20 (TBST) containing 1% BSA. Blots were washed in TBST and incubated with horseradish peroxidase-conjugated secondary antibodies (1:30,000) for 30 min at room temperature. Membranes were washed in TBST, and antigen-antibody complexes were visualized by chemiluminescence using an ECL kit (Pierce). Alternatively, immunoblots were incubated with IRDye800CW Goat Anti Mouse (H+L) or IRDye680 Goat Anti Rabbit (H+L) secondary antibodies and signal was detected with the Odyssey® Infrared Imaging System (Li-COR Biotechnology, Lincoln, NE).
Fatty acid oxidation in isolated muscles
Animals received a single intra-peritoneal injection of SU6656 or vehicle in the morning. Mice were sacrificed by cervical dislocation 3 h after the injection. Skeletal muscles (red soleus and white gastrocnemius) were rapidly removed and pre-incubated for 10 min in oxygenated (95% O2, 5% CO2) Earle's Solution (Invitrogen, Carlsbad, CA) supplemented with 5mM D-Glucose, 250μM palmitate and 0.5% BSA plus either 10μM SU6656 or an equal volume of DMSO. Tissues were incubated for 45 min in the same buffer containing 250μM palmitate containing 1μCi/ml [1-14C] palmitate tracer (Amersham, Piscataway, NJ) bound to 0.5% BSA. Incubations were carried out under an atmosphere of 95% O2/5% CO2 at 30°C in glass vials (Kontes, Vineland, NJ) equipped with a center well filled with 200μl of 2N NaOH (trapping agent). At the end of the incubation, perchloric acid was added through the cap to a concentration of 0.6 mM and vials were incubated for 3 h at 30°C with moderate shaking. The 14CO2 produced was determined by scintillation counting of NaOH using the UniScintBD scintillation liquid (National Diagnostics, Atlanta, GO).
Cell culture
C2C12 myoblasts were grown in Dulbecco's Modified Essential Medium (DMEM, Invitrogen, Carlsbad, CA) with 10% fetal bovine serum. Differentiation into myotubes was initiated by switching the myoblasts to DMEM complemented with 2% horse serum for 4-6 days as described previously (Yaffe and Saxel, 1977a, b). 3T3L1 preadipocytes were cultured in DMEM supplemented with 10% calf serum at 37°C. Confluent cultures were induced to differentiate into adipocytes as described previously (Min et al., 1999).
cDNA constructs
pcDNA3.1-Fyn-V5 was generated by RT-PCR performed on spleen total RNA using the SuperScript First-Strand Synthesis System (Invitrogen, Carlsbad, CA) with a pair of oligonucleotides: 5′-CACCATGGGCTGTGTGCAATGTAAGG-3′ and 5′-CAGGTTTTCACCGGGCTGAT-3′. The PCR product was separated on 2% agarose gel, and the specific single band was extracted using the QIAquick PCR purification kit (Qiagen). The purified PCR product was cloned into the pcDNA3.1D/V5-His-TOPO using the pcDNA3.1 Directional TOPO Expression Kit (Invitrogen, Carlsbad, CA). pcDNA3.1-Fyn-CA(Y527F)-V5 was obtained using the oligonucleotides: 5′-CACCATGGGCTGTGTGCAATGTAAGG-3′ and 5′-CAGGTTTTCACCGGGCTGAAACTGGGGCTCT-3′ and following by the same protocol. pcDNA3.1-Fyn-KD(K299M)-V5 was constructed by overlapping extension PCR. The gene encoding Fyn was amplified with the pair of oligonucleotides: 5′-CACCATGGGCTGTGTGCAATGTAAGG-3′ and 5′-CTGGCTTAAGGGTCATTATGGCTACTTTT-3′ and the pair of oligonucleotides: 5′-AAAAGTAGCCATAATGACCCTTAAGCCAG-3′ and 5′-CAGGTTTTCACCGGGCTGAT-3′. PCR products were extracted and purified. Each product was mixed and a second PCR was performed using the oligonucleotides: 5′-CACCATGGGCTGTGTGCAATGTAAGG-3′ and 5′-CAGGTTTTCACCGGGCTGAT-3′. Products were cloned into the pcDNA3.1D/V5-His-TOPO. The pYX-LKB1 construct was obtained from Open biosystems (Rockford, IL) and used to generate the pcDNA3.1-Flag-LKB1 construct. The gene encoding LKB1 was amplified with the oligonucleotides: 5′-ATG GAC TAC AAG GAC GAT GAC GAC AAG ATG GAC GTG GCG GAC CCC-3′ and 5′-TCACTGCTGCTTGCAGGC-3′ and cloned to pcDNA3.1D/V5-His-TOPO. pEGFPC2 and pcDNA3.1-LKB1 were digested by Hind3 and Sac2. Products were purified and ligation was performed using the DNA Ligation Kit (Takara, Shiga, Japan) to obtain the pEGFPC2-LKB1 construct. LKB1 mutants were obtained using an overlapping extension PCR with the following primers: LKB1-Y60F: 5′- AGG GCT CGT TCG GCA AGG TGA -3′, 5′- TCA CCT TGC CGA ACG AGC CCT -3′, LKB1-Y156F: 5′- AGC TCA TGG GTT CTT CCG CCA G -3′, 5′- CTG GCG GAA GAA CCC ATG AGC T -3′, LKB1-Y166F: 5′- GGC CTG GAA TTC CTA CAC AGC -3′, 5′- GCT GTG TAG GAA TTC CAG GCC -3′, LKB1-Y261F: 5′- GGG GAC AAT ATC TTC AAG CTC TTT GAG AAC -3′, 5′- GTT CTC AAA GAG CTT GAA GAT ATT GTC CCC -3′, LKB1-Y365F: 5′- GAC GGC ATT ATC TTC ACC CAG GAC TT -3′, 5′- AAG TCC TGG GTG AAG ATA ATG CCG TC -3′. LKB1-Y261/365F was obtained using primers for LKB1-Y261F and for LKB1-Y261F. The GST-AMPK α subunit and Omni-STRADα cDNAs were kind gifts from Dr. Bin Zheng, Harvard Medical School.
In vitro LKB1 phosphorylation assay
His-LKB1 fusion protein was purified using HisPur Purification kit and Slide-A-Lyzer Dialysis Cassette (Pierce, Rockford, IL). His-LKB1 protein (1μg) was incubated with the recombinant His-FynT kinase (1.8U) (Calbiochem, Gibbstown, NJ) in presence of Src Mg/ATP cocktail (Millipore, Billerica, MA) and kinase reaction was performed for 1 hour at 35°C. Samples were separated on 10% SDS-polyacrylamide gels and immunoblotting was performed with PY100 monoclonal antibody and LKB1 polyclonal antibody. Signals were detected with the Odyssey® Infrared Imaging System (Li-COR Biotechnology, Lincoln, NE).
Transfection of C2C12 myotubes and 3T3L1 adipocytes
C2C12 myotubes and 3T3L1 adipocytes were electroporated as previously described (Waters et al., 1995). A suspension of 3T3L1 adipocytes was electroporated with 500μg of plasmid under low-voltage condition (0.16kV, 950μF). C2C12 myotubes were electroporated with a total of 250μg of plasmid under 0.22kV, 950μF. Adipocytes and myotubes were allowed to adhere onto collagen-coated tissue culture dishes for 30–48 h.
Transfection of skeletal muscle in vivo
Three month old wild type mice were anesthetized with isoflurane and the right tibialis anterior was injected with 125 μg of pcDNA3-Flag-LKB1-WT or pcDNA3-FlagLKB1-(Y261/365F) cDNAs and the left tibialis anterior with the pcDNA empty vector as control. Electroporation (8 shocks) was performed using the S48 SQUARE PULSE STIMULATOR (Grass Technologies, West Warwick, RI) with the following settings: train rate = 1TPS; train duration =500ms; pulse rate =1PPS; duration = 20ms, voltage = 80V. Electroporation was repeated 5 days later. Animals were sacrificed 5 days after the second set of electroporation. The tibialis anterior muscles were rapidly removed and immediately embedded into Optimal Cutting Temperature (O.C.T) compound (Sakura Finetek USA, inc., Torrance, CA). Tissues were frozen in liquid nitrogen. 10 μm frozen sections were prepared and subjected to immunofluorescence labeling as previously described, using anti-Flag Mouse mAb antibody and Alexa Fluor 488 anti-Mouse secondary antibody. Sections were washed 3 times with PBS and mounted with Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA) and were imaged as described above. Settings (Iris (pinhole), laser intensity, gain and offset) were fixed and identical for all samples. Muscle extracts were also used for immunoprecipitation and immunoblotting as described below.
Signal quantification
The ratio of cytosolic and nuclear LKB1 was quantified using the Image J software (National Institutes of Health). Images of 15 representative cells were processed and results represent mean ± s.e from 3 independent experiments
Immunoprecipitation
Cells were homogenized in a NP-40 lysis buffer containing 25mM Hepes, pH 7.4, 10% glycerol, 50mM sodium fluoride, 10mM sodium phosphate, 137mM sodium chloride, 1mM sodium orthovanadate, 1mM PMSF, 10μg/ml aprotinin, 1μg/ml pepstatin and 5μg/ml leupeptin and rocked for 10 min at 4°C. Muscle extracts (100mg) were homogenized in the Bullet Blender (Next Advance, Inc., Averill Park, NY) using zirconium silicate beads (speed 8 for 3 min) in the buffer described above. Homogenates were centrifuged for 10-30 min at 13,000g at 4°C, and supernatants were collected. Protein concentration was determined using the BCA™ Protein assay. Cell lysates (3-4 mg) were incubated with 10μg of antibody for 2 h at 4°C. 50 μl of TrueBlot™ anti-Rabbit Ig IP Beads (eBioscience, Inc., San Diego, CA) was added and samples were rocked for 60 min at 4°C. Samples were washed three times with NP-40 lysis buffer and were resuspended in 100μl of Laemmli Buffer containing 50mM DTT. Samples were heated at 90-100°C for 10 min and centrifuged at 10,000×g for 3 min. Supernatants were collected and loaded on 10% SDS-polyacrylamide gels.
Immunofuorescence
C2C12 myotubes were co-transfected with 50μg pEGFP-C2-LKB1 or pcDNA3-Flag-LKB1 or pcDNA3-Flag-LKB1 mutants and 200μg of the indicated pcDNA3-Fyn constructs. Transfected cells were washed with PBS and fixed for 10 min in PBS containing 4% PFA and 0.2% Triton X-100. Immunofluorescence was performed using a rabbit LKB1 polyclonal antibody, a rabbit Flag specific polyclonal antibody and a mouse Fyn monoclonal antibodies followed by Alexa Fluor 488 anti-rabbit IgG and Alexa Fluor 594 anti-mouse IgG. Samples were mounted on glass slides with Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA). Cells were imaged using a confocal fluorescence microscope (TCS SP5 confocal; Leica microsystems).
Statistics
Results are expressed as mean ± standard error of the mean (SEM). Differences between animals and/or treatments were tested for statistical significance (p<0.05) using Student's unpaired t test
Supplementary Material
Acknowledgments
This work was supported by The Ellison Medical Foundation (New Scholars Award in Aging) (C.C.B), the National Institutes of Health (DK078886, DK020541) (J.E.P), the American Diabetes Association (1-07-RA-142) (J.E.P) and the NY Obesity Research Center NIH (P30DK026687)(G.J.S). The authors wish to thank Dr. Jun Xu (University of California, Los Angeles) for his helpful comments and Dr. Tsugumichi Saito (Dept of Medicine and Molecular Science, Gunma University Graduate School of Medicine) for kindly providing the authors with his technical expertise of in vivo muscle electroporation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- Alland L, Peseckis SM, Atherton RE, Berthiaume L, Resh MD. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J Biol Chem. 1994;269:16701–16705. [PubMed] [Google Scholar]
- Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, Means AR. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J Biol Chem. 1998;273:31880–31889. doi: 10.1074/jbc.273.48.31880. [DOI] [PubMed] [Google Scholar]
- Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastie CC, Zong H, Xu J, Busa B, Judex S, Kurland IJ, Pessin JE. Integrative metabolic regulation of peripheral tissue fatty acid oxidation by the SRC kinase family member Fyn. Cell Metab. 2007;5:371–381. doi: 10.1016/j.cmet.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Scott JW, Resta N, Deak M, Kieloch A, Komander D, Hardie DG, Prescott AR, van Aalten DM, Alessi DR. Analysis of the LKB1-STRAD-MO25 complex. J Cell Sci. 2004;117:6365–6375. doi: 10.1242/jcs.01571. [DOI] [PubMed] [Google Scholar]
- Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM. Regulation of acetyl-CoA carboxylase. Biochem Soc Trans. 2006;34:223–227. doi: 10.1042/BST20060223. [DOI] [PubMed] [Google Scholar]
- Bull HA, Brickell PM, Dowd PM. Src-related protein tyrosine kinases are physically associated with the surface antigen CD36 in human dermal microvascular endothelial cells. FEBS Lett. 1994;351:41–44. doi: 10.1016/0014-5793(94)00814-0. [DOI] [PubMed] [Google Scholar]
- Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J. 2000;346(Pt 3):659–669. [PMC free article] [PubMed] [Google Scholar]
- Choudhury GG, Mahimainathan L, Das F, Venkatesan B, Ghosh-Choudhury N. c-Src couples PI 3 kinase/Akt and MAPK signaling to PDGF-induced DNA synthesis in mesangial cells. Cell Signal. 2006;18:1854–1864. doi: 10.1016/j.cellsig.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Daval M, Foufelle F, Ferre P. Functions of AMP-activated protein kinase in adipose tissue. J Physiol. 2006;574:55–62. doi: 10.1113/jphysiol.2006.111484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davy A, Gale NW, Murray EW, Klinghoffer RA, Soriano P, Feuerstein C, Robbins SM. Compartmentalized signaling by GPI-anchored ephrin-A5 requires the Fyn tyrosine kinase to regulate cellular adhesion. Genes Dev. 1999;13:3125–3135. doi: 10.1101/gad.13.23.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drover VA, Abumrad NA. CD36-dependent fatty acid uptake regulates expression of peroxisome proliferator activated receptors. Biochem Soc Trans. 2005;33:311–315. doi: 10.1042/BST0330311. [DOI] [PubMed] [Google Scholar]
- Drover VA, Ajmal M, Nassir F, Davidson NO, Nauli AM, Sahoo D, Tso P, Abumrad NA. CD36 deficiency impairs intestinal lipid secretion and clearance of chylomicrons from the blood. J Clin Invest. 2005;115:1290–1297. doi: 10.1172/JCI21514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipp D, Julius M. Lipid rafts: resolution of the “fyn problem”? Mol Immunol. 2004;41:645–656. doi: 10.1016/j.molimm.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Filipp D, Zhang J, Leung BL, Shaw A, Levin SD, Veillette A, Julius M. Regulation of Fyn through translocation of activated Lck into lipid rafts. J Exp Med. 2003;197:1221–1227. doi: 10.1084/jem.20022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty S, Hardie DG. C-terminal phosphorylation of LKB1 is not required for regulation of AMP-activated protein kinase, BRSK1, BRSK2, or cell cycle arrest. J Biol Chem. 2009;284:77–84. doi: 10.1074/jbc.M806152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008a;32 4:S7–12. doi: 10.1038/ijo.2008.116. [DOI] [PubMed] [Google Scholar]
- Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008b;582:81–89. doi: 10.1016/j.febslet.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase--development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hemminki A, Tomlinson I, Markie D, Jarvinen H, Sistonen P, Bjorkqvist AM, Knuutila S, Salovaara R, Bodmer W, Shibata D, de la Chapelle A, Aaltonen LA. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet. 1997;15:87–90. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- Huang MM, Bolen JB, Barnwell JW, Shattil SJ, Brugge JS. Membrane glycoprotein IV (CD36) is physically associated with the Fyn, Lyn, and Yes protein-tyrosine kinases in human platelets. Proc Natl Acad Sci U S A. 1991;88:7844–7848. doi: 10.1073/pnas.88.17.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue L, Rider MH. The AMP-activated protein kinase: more than an energy sensor. Essays Biochem. 2007;43:121–137. doi: 10.1042/BSE0430121. [DOI] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007;292:E1308–1317. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastick CC, Saltiel AR. Insulin-stimulated tyrosine phosphorylation of caveolin is specific for the differentiated adipocyte phenotype in 3T3-L1 cells. J Biol Chem. 1997;272:20706–20714. doi: 10.1074/jbc.272.33.20706. [DOI] [PubMed] [Google Scholar]
- Meex SJ, van der Kallen CJ, van Greevenbroek MM, Eurlings PM, El Hasnaoui M, Evelo CT, Lindsey PJ, Luiken JJ, Glatz JF, de Bruin TW. Up-regulation of CD36/FAT in preadipocytes in familial combined hyperlipidemia. FASEB J. 2005;19:2063–2065. doi: 10.1096/fj.04-2403fje. [DOI] [PubMed] [Google Scholar]
- Min J, Okada S, Kanzaki M, Elmendorf JS, Coker KJ, Ceresa BP, Syu LJ, Noda Y, Saltiel AR, Pessin JE. Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol Cell. 1999;3:751–760. doi: 10.1016/s1097-2765(01)80007-1. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Zhang Y, Aldaz GA, Grammer T, Glasheen EM, Yenush L, Wang LM, Sun XJ, Blenis J, Pierce JH, White MF. YMXM motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol Cell Biol. 1996;16:4147–4155. doi: 10.1128/mcb.16.8.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezu J, Oku A, Shimane M. Loss of cytoplasmic retention ability of mutant LKB1 found in Peutz-Jeghers syndrome patients. Biochem Biophys Res Commun. 1999;261:750–755. doi: 10.1006/bbrc.1999.1047. [DOI] [PubMed] [Google Scholar]
- Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalez-Espinosa C, Furumoto Y, Saitoh S, Samelson LE, O'Shea JJ, Rivera J. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat Immunol. 2002;3:741–748. doi: 10.1038/ni817. [DOI] [PubMed] [Google Scholar]
- Pravenec M, Landa V, Zidek V, Musilova A, Kazdova L, Qi N, Wang J, St Lezin E, Kurtz TW. Transgenic expression of CD36 in the spontaneously hypertensive rat is associated with amelioration of metabolic disturbances but has no effect on hypertension. Physiol Res. 2003;52:681–688. [PubMed] [Google Scholar]
- Prud'homme GJ, Glinka Y, Khan AS, Draghia-Akli R. Electroporation-enhanced nonviral gene transfer for the prevention or treatment of immunological, endocrine and neoplastic diseases. Curr Gene Ther. 2006;6:243–273. doi: 10.2174/156652306776359504. [DOI] [PubMed] [Google Scholar]
- Ribon V, Printen JA, Hoffman NG, Kay BK, Saltiel AR. A novel, multifuntional c-Cbl binding protein in insulin receptor signaling in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18:872–879. doi: 10.1128/mcb.18.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmack G, Defronzo RA, Musi N. AMP-activated protein kinase: Role in metabolism and therapeutic implications. Diabetes Obes Metab. 2006;8:591–602. doi: 10.1111/j.1463-1326.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae. J Cell Biol. 1994;126:353–363. doi: 10.1083/jcb.126.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Carraway KL, 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, et al. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- Steinberg GR. Inflammation in obesity is the common link between defects in fatty acid metabolism and insulin resistance. Cell Cycle. 2007;6:888–894. doi: 10.4161/cc.6.8.4135. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Pons S, Asano T, Myers MG, Jr, Glasheen E, White MF. The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J Biol Chem. 1996;271:10583–10587. doi: 10.1074/jbc.271.18.10583. [DOI] [PubMed] [Google Scholar]
- Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum Mol Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, Foretz M, Andreelli F, Ventura-Clapier R, Bertrand L. AMPK: Lessons from transgenic and knockout animals. Front Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters SB, Yamauchi K, Pessin JE. Insulin-stimulated disassociation of the SOS-Grb2 complex. Mol Cell Biol. 1995;15:2791–2799. doi: 10.1128/mcb.15.5.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. A myogenic cell line with altered serum requirements for differentiation. Differentiation. 1977a;7:159–166. doi: 10.1111/j.1432-0436.1977.tb01507.x. [DOI] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977b;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.