

Abstract
Helicobacter pylori is the strongest identified risk factor for gastric adenocarcinoma. One H. pylori virulence constituent that augments cancer risk is the cag secretion system, which translocates CagA and peptidoglycan into host cells, eventuating in activation of signal transduction pathways. AKT is a target of phosphatidylinositol 3-phosphate kinase (PI3K) and is activated in gastric cancer, but the relationship between PI3K-AKT and H. pylori-induced cellular responses with carcinogenic potential remains unclear. We defined the molecular pathways mediating H. pylori-stimulated AKT activation and the biological consequences of these events in gastric epithelial cells. H. pylori enhanced PI3K-AKT signaling in a Src and EGFR-dependent manner, which was also mediated by a functional cag secretion system, and peptidoglycan. PI3K activation attenuated apoptosis in response to infection and was required for H. pylori-induced cell migration. These results indicate that PI3K-AKT signaling regulates pathophysiologic responses to H. pylori that may lower the threshold for carcinogenesis.
Keywords: PI3K, AKT, Helicobacter, cancer, migration, apoptosis
Introduction
Chronic gastritis induced by Helicobacter pylori persists for decades and increases the risk of gastric adenocarcinoma [1]. Although H. pylori-induced gastritis is the strongest known risk factor for gastric cancer, only a fraction of colonized individuals ever develop neoplasia, and enhanced cancer risk is mediated by strain-specific bacterial factors and/or inflammatory responses governed by host genetic diversity. The cag pathogenicity island (cag PAI) is a virulence locus present in approximately 60% of U.S. H. pylori strains [1], and strains that harbor the cag PAI (cag+) significantly augment the risk for distal gastric cancer compared to strains that lack the cag island (cag-) [1].
Several cag genes, such as cagE, encode components of a type IV secretion system that exports bacterial proteins into host cells. The terminal product of the cag island, CagA, is translocated into gastric epithelial cells following bacterial attachment [2, 3]. CagA subsequently undergoes tyrosine phosphorylation by Src and Abl kinases, and phospho-CagA alters gastric cell morphology and aberrantly activates signaling molecules such as SHP-2 [4, 5]. Unphosphorylated CagA can also exert effects within host cells such as alteration of cell polarity and activation of β-catenin, responses that have been implicated in carcinogenesis [6, 7]. In addition to CagA, components of peptidoglycan can be translocated into host cells by the cag secretion system where they are sensed by the intracellular pattern recognition receptor Nod1, which activates NF-κB and induces production of pro-inflammatory cytokines such as IL-8 [8].
Signal transduction pathways activated in response to bacterial contact play an important role in H. pylori pathogenesis. Phosphatidylinositol 3-kinase (PI3K) is an integral component of a signal transduction pathway that regulates host cellular responses altered in tumorigenesis. PI3K signaling can be activated by ligand-dependent activation of receptor tyrosine kinases such as EGFR [9]. Src kinases, acting both downstream and upstream of EGFR, can also activate PI3K signaling [10]. PI3K activation results in stimulation of phosphatidylinositol-dependent kinase 1 (PDK1), a kinase that phosphorylates and activates AKT [11]. AKT mediates the downstream effects of PI3K by phosphorylating multiple targets that regulate diverse cellular functions including proliferation and survival. PI3K-AKT signaling is increased in gastric cancer specimens and enhanced levels of AKT phosphorylation correlate with advanced stages of disease [12]. Thus, PI3K is well positioned to regulate epithelial responses that may predispose to malignancies.
Cellular migration plays an important role in the invasive potential and metastatic growth of cancers and H. pylori can increase gastric epithelial cell migration, although the mechanisms required for this response are not clearly defined [13, 14]. Of note, EGFR transactivation increases intestinal epithelial cell motility in a PI3K- and Src-dependent manner [15]. Cell survival is another response that is regulated by PI3K and AKT activation [16]. AKT-dependent phosphorylation of pro-apoptotic Bcl-2 homology domain 3 (BH3)-only proteins [17] and pro-caspase 9 [18] attenuates apoptosis, thereby promoting cell survival and enhancing the susceptibility of cells to mutagenesis. Since H. pylori increases cell proliferation and attenuates apoptosis in humans and in rodent models of infection [19, 20], we determined the ability of H. pylori to activate PI3K-AKT signaling in gastric epithelial cells and investigated the molecular pathways mediating these events to define potential tumor-promoting responses toward this pathogen.
Methods
Cell Culture and Reagents
AGS or MKN28 human gastric epithelial cells were grown in RPMI medium 1640 (GIBCO/BRL) with 10% FBS (Sigma) and 20 μg/ml gentamicin (GIBCO/BRL) under 5% CO2 air at 37°C. Pharmacological inhibitors LY294002 (Cell Signaling Technology), AG1478 (Calbiochem), PP2 (Calbiochem), SU6656 (Calbiochem), AG1295 (Calbiochem), and STI-571 (LC Laboratories) were used at concentrations of 50 μM, 600 nM, 10 μM, 2μM, 50μM and 10μM, respectively. For Western immunoblot and flow cytometry analysis, AGS cells were plated at 5 × 105 cells/well in 6-well plates in 2 mL culture medium. For cell migration assays, 5 × 105 cells were plated in 35 mm culture dishes in 2 mL medium.
H. pylori strains
The H. pylori cag+ rodent-adapted strain 7.13, the cag+ clinical strain J166, or the cag- clinical isolate J68, were grown in Brucella broth with 5% FBS for 18 hours, harvested by centrifugation, and were added to gastric cells at a bacteria-to-cell ratio of 100:1. Isogenic cagA-, cagE-, and slt- null mutants were constructed within strain 7.13 by insertional mutagenesis using aphA and were selected with kanamycin (25 μg/ml) as described previously [21]. H. pylori were heat-killed by boiling at 100°C for 10 minutes, while H. pylori filtrates were prepared by passing broth supernatants through a 0.2 μM pore-size filter (Corning).
Western Blot Analysis
Gastric cell lysates were harvested in lysis buffer (50 mM Tris pH 7.2, 150 mM NaCl, 1% Triton X-100, 0.1% SDS) including protease and phosphatase inhibitors (Sigma). Proteins (30 μg) were separated by 10% SDS-PAGE and transferred to poly-vinylidene difluoride membranes (Pall). Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline with 0.05% Tween 20 (TBST), incubated for 24 hours with a purified rabbit polyclonal anti-phospho-AKT antibody (1:1000 dilution; Cell Signaling), a rabbit polyclonal anti-total AKT antibody (1:1000 dilution; Cell Signaling), a monoclonal mouse anti-GAPDH antibody (1:2000 dilution; Santa Cruz Biotech), a mouse monoclonal anti-phospho-tyrosine-99 antibody (1:300 dilution; Santa Cruz Biotech), a rabbit polyclonal anti-CagA antibody (1:5000 dilution; Austral Biologicals), a rabbit polyclonal anti-phospho-Src family antibody (1:1000 dilution; Cell Signaling), a rabbit polyclonal anti-Src antibody (1:1000 dilution; Cell Signaling), a mouse monoclonal anti-phospho-EGFR (Tyr1068) antibody (1:1000; Millipore), a rabbit polyclonal anti-EGFR antibody (1:3000; Millipore), a rabbit polyclonal anti-phospho-Gab1 antibody (1:1000; Cell Signaling), or a rabbit polyclonal anti-Gab1 antibody (1:1000 dilution; Cell Signaling). Goat anti-rabbit (1:5000 dilution; Santa Cruz Biotech) or goat anti-mouse (1:5000 dilution; Santa Cruz Biotech) horseradish peroxidase-conjugated secondary antibodies were used followed by enhanced chemiluminescence detection following the manufacturer’s instructions (Perkin Elmer). Immunoblots were quantified with the GeneTools Software (Syngene).
Flow Cytometry Analysis
AGS cells co-cultured with H. pylori were washed with PBS and harvested using 0.25% Trypsin/EDTA (GIBCO/BRL). Cells were collected by centrifugation and resuspended in binding buffer (10x: 0.1 M HEPES pH 7.4, 1.4 M NaCl, 25 mM CaCl2) at a concentration of 5 × 105 cells/mL. Cells were stained with Annexin V-APC (BD Bioscience) and Propidium iodide/RNAse (BD Bioscience) and were analyzed by quantitative flow cytometry.
Apoptotic Resistance Assays
AGS cells were infected with H. pylori for 3 hours followed by incubation with 50 μM LY294002 for 1 hour. After a four hour exposure to 1 μM Staurosporine (Sigma-Aldrich), cells were harvested for Annexin V-flow cytometry analysis as described [22].
Cell Migration Analysis
Confluent AGS cell monolayers in plates coated with 2.5 μg human fibronectin (BD Bioscience) were pre-incubated with pharmacological inhibitors for one hour. Eight circular wounds were generated in each plate using a rotating silicon tip [23]. H. pylori was then added to the cells and wound images were taken at zero, six and sixteen hours post infection using Q-Capture Imaging Software. Areas were measured using Image J software (NIH).
Transient transfection of siRNA
AGS cells (2.5 × 105) in 6-well plates were transiently transfected using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, transfection reagent (5.0 μl/well) was mixed with siRNA oligos (10 μl of 10 μM solution/well) in 500 μl Opti-MEM (Life Technologies). Cells were incubated with the transfection mixture for 24 hours, fresh medium was added, and bacterial co-cultures were performed 24 hours later.
Statistical Analysis
All experiments were performed on at least three independent occasions. Statistical analysis was performed by Student’s t test and ANOVA using Prism Graph Pad. A P-value < 0.05 was defined as statistically significant.
Results
H. pylori activates PI3K-AKT signaling in gastric epithelial cells
We used a cag+ H. pylori strain, 7.13, that reproducibly causes gastric cancer in rodent models, to determine whether H. pylori alters AKT activity. AGS cells were infected with strain 7.13 at a multiplicity of infection (MOI) of 100 or were exposed to medium alone. H. pylori strain 7.13 increased AKT phosphorylation compared to uninfected controls at each time point (Figure 1). Similar patterns of AKT activation were observed following infection of AGS cells with the cag+ human clinical isolate J166 or infection of MKN28 human gastric epithelial cells with strains 7.13 or J166 (data not shown).
Figure 1. H. pylori induces AKT activation in vitro in a time-dependent manner.

(A) AGS cells were co-cultured with the H. pylori cag+ strain 7.13 at a bacteria/cell ratio of 100:1. One through twenty-four hours after incubation, whole cell lysates were harvested and subjected to Western blot analysis using an anti-phospho-AKT antibody. (-), cells incubated with medium alone. A representative blot is shown. Western blots for total AKT served as normalization controls for AGS cell viability under different experimental conditions and Western blots for GAPDH served as loading controls. (B) Densitometric analysis of multiple Western blot repetitions performed on at least 3 occasions. Levels of phospho-AKT were normalized to total AKT and levels were expressed as fold-induction of infected cells compared with uninfected cells at each time point. Error bars = SEM. *P < 0.01 vs. uninfected control.
Inactivation of cagE abolishes AKT activation by H. pylori
We next investigated the role of bacterial factors in PI3K-AKT signaling events. Neither heat-killed bacteria nor soluble factors contained in H. pylori filtrates stimulated phosphorylation of AKT, indicating that viable H. pylori are required for AKT activation (Figure 2A, 2B). The cag secretion system encodes several proteins that affect cellular signaling after live H. pylori have bound host cells. To define the role of cag components in AKT activation, AGS cells were incubated with the H. pylori cag+ strain 7.13 or its isogenic cagA- or cagE- null mutant derivatives. AKT activation was significantly decreased in cells incubated with the 7.13 cagE-, but not the cagA-, mutant versus the wild-type strain (Figure 2C, 2D). Similarly, the cag- clinical isolate J68 failed to induce AKT phosphorylation (data not shown). These findings indicate that a functional cag secretion system, but not cagA, is required for induction of PI3K-AKT signaling.
Figure 2. AKT phosphorylation by H. pylori is dependent on specific genes within the cag pathogenicity island.
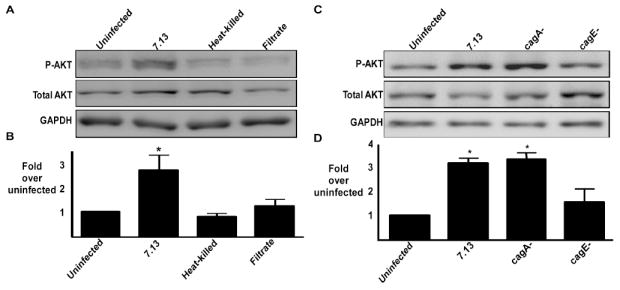
(A) AGS cells were incubated in the absence or presence of live H. pylori strain 7.13 at a bacteria/cell ratio of 100:1, heat-killed H. pylori, or H. pylori 7.13 filtrate for two hours. Whole cell lysates were subjected to Western blot analysis using an anti-phospho AKT antibody. Anti-total AKT blots served as normalization controls for AGS cell viability under different experimental conditions and anti-GAPDH blots served as loading controls. (B) Densitometric analysis of Western blots performed on 3 occasions. Error bars = SEM. *P <0.04 vs. AGS cells alone. (C) AGS cells were cultured in the absence or presence of the H. pylori cag+ strain 7.13 or its isogenic cagA- or cagE- null mutant derivatives at bacteria/cell ratios of 100:1. Two hours post infection, whole cell lysates were subjected to Western blot analysis using an anti-phospho-AKT antibody. A representative blot is shown. Western blots for total AKT served as normalization controls and Western blots for GAPDH served as loading controls. (D) Densitometric analysis of multiple Western blot repetitions performed on at least 5 occasions. Error bars = SEM. *P < 0.002 vs. AGS cells alone.
Peptidoglycan is required for activation of AKT by H. pylori
In addition to CagA, peptidoglycan can be translocated by the cag secretion system, and can alter host signaling. Therefore, we examined the role of peptidoglycan in AKT activation using a 7.13 isogenic slt mutant. The slt mutant lacks the soluble lytic transglycosylase (slt) required for peptidoglycan turnover and release. We first established that inactivation of slt in strain 7.13 does not alter CagA translocation into host cells (Figure 3A). We then co-cultured AGS cells with wild-type strain 7.13 or the isogenic slt- mutant. Cells co-cultured with the slt- mutant contained significantly lower levels of phospho-AKT compared to cells infected by wild-type 7.13 (Figure 3B, 3C). These results indicate that peptidoglycan, in conjunction with a functional cag secretion system, is required for maximal AKT stimulation by H. pylori.
Figure 3. AKT phosphorylation by H. pylori is mediated by peptidoglycan.

(A) AGS cells were cultured in the absence or presence of wild-type H. pylori strain 7.13 or its isogenic cagA- or slt- null mutant at a bacteria/cell ratio of 100:1. Two hours post infection, whole-cell lysates were subjected to Western blot analysis using an anti-phospho-tyrosine 99 antibody or an anti-CagA antibody. A representative blot is shown. Western blots for GAPDH served as loading controls. (B) H. pylori strain 7.13 or its isogenic slt null mutant derivative, were added to AGS cells at a bacteria/cell ratio of 100:1. Two hours after incubation, whole-cell lysates were subjected to Western blot analysis using an anti-phospho-AKT antibody. A representative blot is shown. Western blots for total AKT served as normalization controls for AGS cell viability under different experimental conditions and Western blots for GAPDH served as loading controls. (C) Densitometric analysis of multiple Western blot repetitions performed on at least 3 occasions. Error bars = SEM. *P < 0.04 vs AGS cells alone; **P < 0.009 vs AGS cells incubated with wild-type H. pylori.
H. pylori-induced AKT activation is dependent on activation of PI3K, Src, and EGFR
AKT activation is regulated by many of the same constituents that are activated by H. pylori (e.g., EGFR, Src) [9, 10, 24, 25]. As a prelude to defining the molecular pathways mediating H. pylori-induced AKT activation, we first confirmed that our prototype strain could activate EGFR and Src, and also established the efficacy of PI3K, EGFR and Src inhibitors. H. pylori strain 7.13 induced phosphorylation of Src and EGFR in AGS cells (Figure 4A). AGS cell lysates were then assessed for phospho-AKT after stimulation with EGF, a potent inducer of AKT activation. Each inhibitor was sufficient to attenuate EGF-stimulated AKT activation (Figure 4B). AGS cells were then co-cultured with strain 7.13 in the absence or presence of the PI3K inhibitor LY294002 or the Src inhibitor PP2. H. pylori alone activated AKT by two hours (Figure 4C, 4D). As predicted, AKT activation in response to H. pylori was completely abolished by PI3K inhibition (Figure 4C, 4D). PI3K-dependent AKT activation was further confirmed using an independent PI3K inhibitor, Wortmannin (200nM) (data not shown).
Figure 4. H. pylori-induced AKT phosphorylation in AGS cells is dependent on activation of PI3K, EGFR, and Src.

(A)_H. pylori strain 7.13 was added to AGS cells at a bacteria/cell concentration of 100:1. Two hours post-infection, whole cell lysates were subjected to Western blot analysis using an anti-phospho-Src or an anti-phospho-EGFR antibody. (-), cells incubated with medium alone. A representative blot is shown. Western blots for total Src or EGFR served as normalization controls for AGS cell viability under different experimental conditions. (B) AGS cells were incubated with the PI3K inhibitor LY294002 (50 μmol/L), EGFR kinase inhibitor AG1478 (600 nmol/L), or Src inhibitor PP2 (10 μmol/L) for one hour prior to EGF exposure for 15 minutes. Levels of phospho- and total AKT were determined by Western blot analysis of whole cell lysates. (C) H. pylori strain 7.13 was added to AGS cells at a bacteria/cell concentration of 100:1 in the absence or presence of vehicle alone (DMSO), or 50 μmol/L LY294002, 600 nmol/L AG1478, or 10 μmol/L PP2. Two hours post-infection, whole cell lysates were subjected to Western blot analysis using an anti-phospho-AKT antibody. (-), cells incubated with medium alone. A representative blot is shown. Western blots for total AKT served as normalization controls for AGS cell viability under different experimental conditions and Western blots for GAPDH served as loading controls. (D) Densitometric analysis of multiple Western blot repetitions performed on at least 3 occasions. Error bars = SEM. *P < 0.0001 vs. AGS cells alone. (E) H. pylori strain 7.13 was added to AGS cells at a bacteria/cell concentration of 100:1 in the absence or presence of vehicle alone (DMSO), or 2 μmol/L SU6656, 50 μmol/L AG1295, or 10 μmol/L STI-571. Two hours post-infection, whole cell lysates were subjected to Western blot analysis using an anti-phospho-AKT antibody. (-), cells incubated with medium alone. A representative blot is shown. Western blots for total AKT served as normalization controls for AGS cell viability under different experimental conditions and Western blots for GAPDH served as loading controls. (F) Densitometric analysis of multiple Western blot repetitions performed on at least 3 occasions. Error bars = SEM. *P < 0.04 vs. AGS cells alone. (G) AGS cells were cultured in the absence or presence of the H. pylori cag+ strain 7.13 or its isogenic cagA-, cagE-, or slt- null mutant derivatives at bacteria/cell ratios of 100:1. Two hours post infection, whole cell lysates were subjected to Western blot analysis using an anti-phospho-Gab1 antibody. EGF was used as a positive control for Gab1 phosphorylation and was added for 15 minutes. A representative blot is shown. Western blots for total Gab1 served as normalization controls and Western blots for GAPDH served as loading controls. Densitometric analysis of multiple Western blot repetitions performed on at least 3 occasions is shown below representative Western blot. Error bars = SEM. *P < 0.02 vs. AGS cells alone.
H. pylori-induced activation of AKT was also dependent on Src, as treatment with the Src inhibitor PP2 blocked AKT activation (Figure 4C, 4D). The inhibitor PP2, however, can also exert activity against platelet-derived growth factor receptor (PDGFR) signaling as well as signaling initiated by c-Abl and c-kit [26]. Therefore, we repeated co-culture experiments in the presence or absence of specific inhibitors of these pathways. H. pylori-induced AKT activation was unchanged in the presence of PDGFR or c-Abl/c-kit inhibitors (Figure 4E, 4F), indicating that Src plays a role in microbial-induced activation of AKT.
EGFR transactivation can mediate PI3K and Src activation and this receptor can be transactivated by H. pylori (Figure 4A) [24]; therefore we next determined the role of EGFR in H. pylori-induced AKT activation. Co-culture of AGS cells with H. pylori in the presence of the EGFR kinase inhibitor AG1478 significantly reduced AKT phosphorylation to levels seen in uninfected controls (Figure 4C, 4D). However, the EGFR inhibitor AG1478 also inhibits FAK, a component of another PI3K-dependent pathway. Therefore, to rule out involvement of FAK, we determined whether H. pylori infection could stimulate Gab1 phosphorylation, an event dependent on EGFR transactivation. As shown in Figure 4G, co-culture with wild-type strain 7.13 induced Gab1 phosphorylation. The pattern of Gab1 phosphorylation mirrored AKT activation as H. pylori mutant strains that lacked cagE or slt failed to induce phosphorylation of Gab1 (Figure 4G). Finally, to more firmly implicate EGFR and Src signaling in these events, we co-cultured H. pylori strain 7.13 with AGS cells in the presence or absence of a Src family kinase inhibitor that does not activate PDGFR (SU6656) [27]. As demonstrated in Figures 4E and 4F, pre-incubation with SU6656 attenuated the ability of H. pylori to activate AKT. Collectively, these results indicate that transactivation of EGFR and Src activation are likely required for H. pylori-induced AKT activation.
PI3K signaling is required for cell migration in response to H. pylori
Colonic epithelial migration is dependent on PI3K and Src activation [28]. To determine if H. pylori promotes cell migration in a PI3K-dependent manner, AGS cells were treated with the PI3K inhibitor LY294002 and infected with H. pylori. Wounds were then induced and measured over time using time-lapse microscopy. Inhibition of PI3K did not significantly alter cell motility in uninfected cells (Figure 5A, 5B). H. pylori significantly increased wound healing compared to uninfected cells, but this was abolished by inhibition of PI3K (Figure 5A, 5B). Treatment of cells with EGFR (AG1478) and Src (PP2) inhibitors also blocked migration in response to H. pylori (Figure 5C), which mirrored results investigating the effects of H. pylori on AKT activation (Figure 4). Since PP2 can also inhibit PDGFR signaling, we repeated migration assays in the presence or absence of the specific PDGFR inhibitor AG1295. H. pylori-induced cell migration was unchanged in the presence of the PDGFR inhibitor (Figure 5C), indicating that Src likely plays a role in cell motility that is induced by H. pylori.
Figure 5. Activation of PI3K-AKT and Rac mediates H. pylori-induced cell migration.

(A) AGS cells were grown to confluency and incubated with the PI3K inhibitor LY294002 (50 μM) or vehicle alone (DMSO) for one hour. A wound was then introduced into the cell monolayer and medium or H. pylori strain 7.13 was added. Wound areas were measured at zero, six and sixteen hours post-infection. (B) Quantification of wound closure for each treatment group in experiments performed on at least 5 independent occasions. (-), cells incubated without H. pylori. Error bars = SEM. * P < 0.04 vs AGS cells infected with H. pylori strain 7.13 in the presence of the PI3K inhibitor LY294002 at both six and 16 hours. (C) AGS cells were grown to confluency and incubated with 600 nmol/L AG1478, 10 μmol/L PP2, or 50 μmol/L AG1295, or vehicle alone (DMSO) for one hour. A wound was then introduced into the cell monolayer and medium or H. pylori strain 7.13 was added. Wound areas were measured at zero and six hours post-infection. Quantification of wound closure for each treatment group in experiments performed on at least 3 independent occasions is shown. (-), cells incubated without H. pylori. Error bars = SEM. * P < 0.005 vs uninfected AGS cells alone or in the presence of AG1295. (D) AGS cells were grown to confluency and incubated with the Rac1 inhibitor NSC23766 (50 μM) or vehicle alone (water) for one hour. A wound was then introduced into the cell monolayer and medium or H. pylori strain 7.13 was added. Wound areas were measured at zero, six and sixteen hours post-infection. (E) Quantification of wound closure for each treatment group in experiments performed on at least 5 independent occasions. (-), cells incubated without H. pylori. Error bars = SEM. ** P < 0.01 vs AGS cells infected with H. pylori strain 7.13 in the presence of the Rac inhibitor NSC23766 at both six and 16 hours.
H. pylori and PI3K can also activate the small GTPase Rac [29, 30], an important regulator of the migratory phenotype of cancer cells. Therefore, we next investigated whether Rac activation influenced H. pylori-induced wound closure by repeating wound-healing assays in the presence of a specific Rac1 inhibitor (NSC23766). Inhibition of Rac did not significantly alter cell motility in uninfected cells (Figure 5D, 5E). Similar to results seen with PI3K inhibition, H. pylori-induced cell migration was completely abolished in the presence of Rac inhibition (Figure 5D, 5E). These results indicate that H. pylori promotes gastric epithelial cell migration via a PI3K, Src and Rac-dependent pathway, likely transduced by upstream signaling from EGFR transactivation.
H. pylori-induced cell migration is dependent on cagE and peptidoglycan
Having demonstrated that cagE and peptidoglycan are required for H. pylori-induced AKT activation, we next determined the role of these bacterial factors in cell migration. Similar to the patterns observed for AKT activation (Figure 2), cell migration was significantly decreased following infection with the cagE- or slt- mutants compared to wild-type 7.13 (Figure 6). These results indicate that PI3K signaling and cag-mediated peptidoglycan translocation mediate H. pylori-induced cell migration.
Figure 6. H. pylori-induced cell migration is dependent on the cag pathogenicity island and peptidoglycan.

AGS cells were grown to confluency and a wound was introduced into the monolayer. Medium, H. pylori strain 7.13, or isogenic cagA-, cagE-, or slt- null mutant derivatives were then added at bacteria/cell ratios of 100:1. Wound areas were measured at time zero and six hours post-infection. Quantification of wound closure is shown for each treatment group in experiments performed on at least 3 occasions. Error bars = SEM. * P < 0.007 vs uninfected control; ** P < 0.007 vs AGS cells infected with H. pylori strain 7.13.
Activation of AKT by H. pylori attenuates apoptosis and promotes cell survival
Attenuation of H. pylori-induced cell migration by PI3K inhibition persisted to 16 hours (Figure 5), but this was accompanied by an increase in cellular detachment, indicative of cell death. Because AKT activation by PI3K attenuates apoptosis, we next determined the contribution of PI3K signaling to H. pylori-mediated apoptosis. AGS cells co-cultured with strain 7.13 in the absence or presence of the PI3K inhibitor LY294002 or vehicle control were stained with Annexin V and Propidium iodide for analysis of apoptosis using flow cytometry. As expected, treatment with the PI3K inhibitor alone induced a small population of uninfected cells into early apoptosis. Co-culture of AGS cells with H. pylori increased apoptosis, but this phenotype was significantly enhanced in the presence of the PI3K inhibitor (Figure 7A, 7B).
Figure 7. Activation of AKT by H. pylori promotes cell survival.

(A) AGS cells were co-cultured with H. pylori strain 7.13 at a bacteria/cell concentration of 100:1, in the absence or presence of the PI3K inhibitor LY294002 (50 μM) or vehicle alone (DMSO) for 24 hours. Live cells were stained with Annexin V-APC and PI, and apoptosis was quantified by flow cytometry. The upper right quadrant represents late apoptosis, and the lower right quadrant represents early apoptosis. (B) Combined percentage of early and late apoptotic cells for experiments performed on at least 5 occasions. (-), cells incubated without H. pylori. Error bars = SEM. * P < 0.005 vs AGS cells infected with H. pylori strain 7.13 at MOI of 100:1 in the presence of vehicle alone. (C) AGS cells were transiently transfected with scrambled or AKT-specific siRNA, total protein was extracted and subjected to Western blot analysis using an anti-AKT antibody. (D) AGS cells transiently transfected with control or AKT-specific siRNA were co-cultured with H. pylori strain 7.13 at a bacteria/cell concentration of 100:1 for 24 hours. Live cells were stained with Annexin V-APC and PI, and apoptosis was quantified by flow cytometry. The upper right quadrant represents late apoptosis, and the lower right quadrant represents early apoptosis. (E) Combined percentage of early and late apoptotic cells for experiments performed on at least 3 occasions. (-), cells incubated without H. pylori. Error bars = SEM. * P < 0.05 vs AKT siRNA-treated AGS cells infected with H. pylori strain 7.13. (F) AGS cells were co-cultured with or without H. pylori strain 7.13 at a bacterial/cell concentration of 100:1, treated with LY294002 (50μM) or medium alone, and then exposed to Staurosporine (Stsp). Cells were then stained with Annexin V-APC and PI, and subjected to flow cytometry. (-), cells incubated without H. pylori. ** P< 0.05 vs AGS cells infected with H. pylori strain 7.13 in the presence of Stsp alone.
To more robustly demonstrate that PI3K-AKT signaling regulates H. pylori-induced cell survival, we transiently transfected AGS cells with scrambled or AKT-specific siRNA. Western blot analysis indicated that AKT expression was significantly reduced using AKT-specific, but not scrambled, siRNA (Figure 7C). H. pylori strain 7.13 was then co-cultured with AKT-deficient or wild-type control AGS cells and apoptosis was assessed using flow cytometry. Similar to results obtained using a chemical inhibitor of PI3K, inhibition of AKT significantly augmented the ability of H. pylori to induce apoptosis (Figures 7D, 7E). These data indicate that activation of PI3K-AKT promotes gastric cell survival in the presence of H. pylori.
A recent study demonstrated that H. pylori can not only induce apoptosis, but can also promote resistance to this phenotype in response to a known apoptosis-inducing agent, Staurosporine (Stsp) [22]. To extend our data implicating PI3K-AKT in cell survival, we evaluated the ability of PI3K to promote apoptotic resistance in H. pylori-infected AGS cells. Cells were infected with H. pylori strain 7.13 and then treated with Stsp to induce apoptosis. Inhibition of PI3K did not significantly alter apoptosis in uninfected cells exposed to Stsp (Figure 7F). AGS cells infected with H. pylori were more resistant to Stsp-induced apoptosis than cells pre-treated with medium alone (Figure 7F). However, inhibition of PI3K attenuated the anti-apoptotic activity of H. pylori, further supporting a role for H. pylori-induced PI3K signaling in promoting cell survival.
Discussion
PI3K is a host signaling molecule related to carcinogenesis. Our current experiments have shown that activation of PI3K-AKT can regulate microbially-induced carcinogenic responses by 1) demonstrating that H. pylori can activate AKT in gastric epithelial cells in vitro, 2) capitalizing on an H. pylori isogenic mutant system to demonstrate a requirement for peptidoglycan translocation in AKT activation, 3) defining upstream signaling mediators of H. pylori-induced AKT activation and 4) combining transient inhibitor and gene silencing techniques with studies of epithelial responses that have carcinogenic potential (e.g. cell migration, survival). Collectively, these studies indicate that H. pylori coopts the PI3K-AKT signaling cascade, which, over prolonged periods of time, may lower the threshold for carcinogenesis.
In contrast to extensive literature invoking PI3K and AKT as tumorigenic molecules, few reports have examined the effects of bacterial pathogens on this signaling cascade. Haemophilus influenzae activates PI3K-AKT in epithelial cells, which then leads to a down-regulation of p38-MAPK activation [31]. Salmonella exploits PI3K in intestinal epithelial cells as an anti-inflammatory signal to reduce IL-8 production, which may contribute to the establishment of colonization in the intestine [32]. Our results suggest that induction of PI3K-AKT signaling by H. pylori requires a functional cag secretion apparatus and peptidoglycan, revealing a previously unrecognized effect of this cag island substrate, since the only defined role to date of cag-mediated peptidoglycan delivery is NOD1-dependent induction of IL-8 secretion [8]. In other cell systems, such as eosinophils, peptidoglycan has been shown to activate PI3K signaling and to regulate IL-8 production through Toll-like receptor (TLR) 2 [33]. However, further experiments are required to determine the precise mechanism through which PI3K is activated in H. pylori-infected gastric epithelial cells.
Hyperproliferation has been reproducibly demonstrated in H. pylori-infected tissue [34] and this is accompanied by decreased levels of apoptosis in colonized human and rodent gastric epithelium [19, 20]. Several reports have demonstrated that one role of AKT is to inhibit the function of caspases, which induce apoptosis and cell-cycle arrest. In addition to PI3K activation, however, H. pylori activates other pathways that influence cell survival. For example, MEK/ERK activation in response to H. pylori has been shown to increase Mcl-1 levels, leading to apoptosis resistance [22]. The collective result of activation of these pathways is inhibition of apoptosis and increased cell proliferation, events that favor tumorigenesis.
In macrophages, H. pylori cag+ strains activate PI3K, leading to actin polymerization and delayed phagocytosis [35]. Our current studies focused on epithelial cells demonstrate a dramatic reduction in H. pylori-induced cell migration in the presence of PI3K inhibitors, suggesting that PI3K may also mediate actin dynamics in gastric epithelial cells. We determined that cell migration was not affected by loss of CagA, but did require a functional type IV cag secretion system and peptidoglycan. Our results differ from studies demonstrating that CagA is required for a full motogenic response to H. pylori through its interactions with c-Met and subsequent MEK/ERK signaling [36]. We speculate that these differences may be due in part to the use of different strains as well as different techniques to assess cell migration. However, Al-Ghoul et al. have shown that H. pylori mutants that do not translocate CagA can still stimulate cell motility. These findings suggest that additional factors translocated by the type IV secretion system may affect H. pylori-dependent motility [13], and our results indicating that peptidoglycan is required to promote cell migration are consistent with these findings.
In summary, H. pylori induces PI3K-AKT signaling in gastric epithelial cells, which requires the cag secretion system and peptidoglycan as well as EGFR transactivation and Src activation in host cells. H. pylori-induced PI3K activation mediates cell migration and protection from apoptosis, two phenotypes related to carcinogenesis. Taken together, these data present insights into the pathogenic mechanisms underlying H. pylori infection.
Acknowledgments
The authors would like to thank Lydia Wroblewski, Shannon Allen, Daniel O’Brien, and Seth Ogden for their helpful discussions during the course of this project. We would also like to thank Ivo Boneca, Institut Pasteur, Paris France, for supplying the slt mutant construct.
This work was supported in part by NIH grants DK 53902, CA 77955 and DK 58587 (to RMP).
Footnotes
There are no conflicts of interest for any of the authors in regard to this manuscript.
These data were presented at Digestive Disease Week May, 2007, Washington D.C. and Digestive Disease Week May, 2008, San Diego, CA.
References
- 1.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 2.Backert S, Ziska E, Brinkmann V, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cellular Microbiology. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- 3.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 4.Higashi H, Tsutsumi R, Muto S, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683–6. doi: 10.1126/science.1067147. [DOI] [PubMed] [Google Scholar]
- 5.Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775–6778. doi: 10.1074/jbc.C100754200. [DOI] [PubMed] [Google Scholar]
- 6.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–4. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franco AT, Israel D, Washington MK, et al. Activation of Beta-catenin by carcinogenic Helicobacter pylori. PNAS. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 9.Rodrigues GA, Falasca M, Zhang Z, Ong SH, Schlessinger J. A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol Cell Biol. 2000;20:1448–1459. doi: 10.1128/mcb.20.4.1448-1459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osherov N, Levitzki A. Epidermal-growth-factor-dependent activation of the src-family kinases. Eur J Biochem. 1994;225:1047–1053. doi: 10.1111/j.1432-1033.1994.1047b.x. [DOI] [PubMed] [Google Scholar]
- 11.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi I, Semba S, Matsuda Y, Kuroda Y, Yokozaki H. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology. 2006;73:8–17. doi: 10.1159/000093087. [DOI] [PubMed] [Google Scholar]
- 13.Al-Ghoul L, Wessler S, Hundertmark T, Krüger S, Fischer W, Wunder C, Haas R, Roessner A, Naumann M. Analysis of the type IV secretion system-dependent cell motility of Helicobacter pylori-infected epithelial cells. Biochem and Biophys Res Comm. 2004;322:860–866. doi: 10.1016/j.bbrc.2004.07.199. [DOI] [PubMed] [Google Scholar]
- 14.Moese S, Selbach M, Meyer TF, Backert S. cag+ Helicobacter pylori induces homotypic aggregation of macrophage-like cells by up-regulation and recruitment of intracellular adhesion molecule 1 to the cell surface. Infect Immun. 2002;70:4687–91. doi: 10.1128/IAI.70.8.4687-4691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frey M, Golovin A, Polk DB. Epidermal growth factor-stimulated intestinal epithelial cell migration requires src family kinase-dependent p38 MAPK signaling. J Biol Chem. 2004;279:44513–44521. doi: 10.1074/jbc.M406253200. [DOI] [PubMed] [Google Scholar]
- 16.Marte BM, D J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 17.Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2002;6:41–51. [PubMed] [Google Scholar]
- 18.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 19.Maeda S, Yoshida H, Mitsuno Y, et al. Analysis of apoptotic and antiapoptotic signalling pathways induced by Helicobacter pylori. Mol Pathol. 2002;55:286–93. doi: 10.1136/mp.55.5.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peek RM, Wirth HP, Moss SF, et al. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology. 2000;118:48–59. doi: 10.1016/s0016-5085(00)70413-6. [DOI] [PubMed] [Google Scholar]
- 21.Peek RM, Jr, Blaser MJ, Mays DJ, et al. Helicobacter pylori strain-specific genotypes and modulation of the gastric epithelial cell cycle. Cancer Res. 1999;59:6124–31. [PubMed] [Google Scholar]
- 22.Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, Haas R, Sasakawa C. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–263. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, Whitehead R, Polk DB. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Phys Cell Physiol. 2003;284:C953–C961. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 24.Keates S, Sougioultzis S, Keates AC, et al. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem. 2001;276:48127–34. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 25.Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol. 2002;43:971–80. doi: 10.1046/j.1365-2958.2002.02781.x. [DOI] [PubMed] [Google Scholar]
- 26.Meyn MA, 3, Schreiner SJ, Dumitrescu TP, Nau GJ, S TE. SRC family kinase activity is required for murine embryonic stem cell growth and differentiation. Mol Pharm. 2005;68:1320–1330. doi: 10.1124/mol.104.010231. [DOI] [PubMed] [Google Scholar]
- 27.Blake R, Broome M, Liu X, et al. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates rac activation through src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2008;294:G276–285. doi: 10.1152/ajpgi.00340.2007. [DOI] [PubMed] [Google Scholar]
- 29.Posern G, Saffrich R, Ansorge W, Feller SM. Rapid lamellipodia formation in nerve growth factor-stimulated PC12 cells is dependent on Rac and PI3K activity. J Cell Physiol. 2000;183:416–424. doi: 10.1002/(SICI)1097-4652(200006)183:3<416::AID-JCP15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 30.Churin Y, Kardalinou E, Meyer TF, Naumann M. Pathogenicity island-dependent activation of Rho GTPases Rac1 and Cdc42 in Helicobacter pylori infection. Mol Microbiol. 2001;40:815–823. doi: 10.1046/j.1365-2958.2001.02443.x. [DOI] [PubMed] [Google Scholar]
- 31.Li JD. Exploitation of host epithelial signaling networks by respiratory bacterial pathogens. J Pharmacol Sci. 2003;91:1–7. doi: 10.1254/jphs.91.1. [DOI] [PubMed] [Google Scholar]
- 32.Huang FC, Li Q, Cherayil BJ. A phosphatidyl-inositol-3-kinase-dependent anti-inflammatory pathway activated by Salmonella in epithelial cells. FEMS Microbiol Lett. 2005;243:265–270. doi: 10.1016/j.femsle.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 33.Wong CK, Cheung PF, Ip WK, Lam CW. Intracellular signaling mechanisms regulating toll-like receptor-mediated activation of eosinophils. Am J Respir Cell Mol Biol. 2007;37:85–96. doi: 10.1165/rcmb.2006-0457OC. [DOI] [PubMed] [Google Scholar]
- 34.Fraser AG, Sim R, Sankey EA, Dhillon AP, Pounder RE. Effect of eradication of Helicobacter pylori on gastric epithelial cell proliferation. Aliment Pharmacol Ther. 1994;8:167–73. doi: 10.1111/j.1365-2036.1994.tb00274.x. [DOI] [PubMed] [Google Scholar]
- 35.Allen LA, Allgood JA, Han X, Wittine LM. Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of Helicobacter pylori. Journal of Leukocyte Biology. 2005;78:220–230. doi: 10.1189/jlb.0205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol. 2003;161:249–55. doi: 10.1083/jcb.200208039. [DOI] [PMC free article] [PubMed] [Google Scholar]