

Abstract
Major depressive disorder (MDD) constitutes a major public health problem worldwide and affects women twice as frequently as men. Previous genetic studies have revealed significant evidence of linkage of the CREB1 region (2q33–35) to Mood Disorders among women from families with recurrent, early-onset MDD (RE-MDD), a severe and familial subtype of MDD. A rare G to A transition at position −656 in the CREB1 promoter cosegregates with Mood Disorders in women from these families, implicating CREB1 as a sex-related susceptibility gene for unipolar Mood Disorders. In the current study, the functional significance of the CREB1 promoter variant was determined using transfection experiments that employed plasmid constructs containing the wild-type or variant CREB1 promoters coupled to a reporter gene. The results support the hypothesis that the A−656 allele contributes to the development of MDD in women through selective alteration of CREB1 promoter activity by female gonadal steroids in noradrenergic neuronal cells. Furthermore, the exaggeration of these effects during a simulated stress condition may be relevant to reported gene-environment interactions that contribute to the emergence of MDD in clinical populations.
Keywords: CREB1, Genetics, Gonadal Steroids, Noradrenergic Neurons, Sex, Depression, Mood
INTRODUCTION
Major Depressive Disorder (MDD) constitutes a major public health problem worldwide and affects women twice as frequently as men.1 Families identified by individuals with Recurrent, Early-Onset MDD (RE-MDD), a severe and strongly familial form of MDD, have provided an important resource in efforts to identify and characterize genes that contribute to the risk of developing MDD and related conditions.1,2
Model-free linkage analysis of a region of chromosome 2q33–35, highlighted by previous case-control studies3,4 and supported by within-family analyses employing the transmission disequilibrium test,5 has revealed evidence of sex-specific linkage to unipolar Mood Disorders extending over 15 cM in our 81 RE-MDD families.6,7 Peak multipoint LOD scores of 6.33 and 6.87 occurred at D2S2321 and D2S2208, respectively. This finding resulted from linkage of the 2q33–35 region to unipolar Mood Disorders among the women in these 81 RE-MDD families; no evidence of linkage of the 2q33–35 region to Mood Disorders was detected among the male family members (peak LOD score 0.00). The 451Kb region between the adjacent SSTRPs D2S2321 and D2S2208 includes an attractive candidate gene, CREB1(2q33.2-34), which encodes the cAMP response element binding protein.8–10
The CREB1 gene encodes a 43 kDa cAMP-responsive element-binding protein (CREB) consisting of 341 amino acids that is a member of the basic leucine zipper family of transcription factors.11 In addition to the full-length, 341 aa CREB protein (isoform B), additional isoforms are synthesized by alternative mRNA splicing that are tissue-specific and differentially-expressed during development. In response to elevated cAMP levels produced by the activation of g-protein coupled receptors, catalytic subunits of protein kinase A diffuse into the nucleus and induce cellular gene expression by phosphorylating CREB at Ser133. Phosphorylated CREB molecules induce the transcription of genes whose promoters include a cAMP-responsive element (CRE), a conserved palindromic 8-nucleotide sequence (TGACGTCA) or single CGTCA motif. In addition to the cAMP signaling system, several growth factor and stress signals stimulate CREB-mediated transcription by promoting the phosphorylation of CREB at Ser133 by various cellular kinases.11 CREB1 is ubiquitously expressed in human tissues and its target genes encode biosynthetic enzymes and receptors for neurotransmitters, neuropeptides, neuronal and non-neuronal growth factors; transcription factors and proteins that regulate the cell cycle/cell survival/DNA repair, reproduction/development, and intercellular signaling and transport; and a range of metabolic/catabolic enzymes and structural proteins. Promoter sequences in addition to CRE and the participation of co-activation factors impose specificity on the particular programs of gene expression that are initiated by CREB.
CREB1 is an excellent candidate for a susceptibility gene, alleles of which might alter CREB1 gene expression or encode CREB protein variants that influence the risk of developing unipolar Mood Disorders. Alterations in CREB1 gene expression and CREB phosphorylation have been reported in clinicopathologic studies of temporal cortex from patients with MDD, in the hippocampus and nucleus accumbens of animal models of MDD and related disorders, and in the brains of rodents exposed to chronic treatment with antidepressant drugs.8–10 CREB has also been implicated in neuronal plasticity, cognition, and long term memory,12 abnormalities of which commonly occur in patients with MDD, may predispose patients to the onset or recurrence of MDD, and may be related to the eventual development of irreversible dementia in some patients.1,13 Finally, reports of synergistic interactions of CREB with nuclear estrogen receptors14–16 may provide a mechanism by which CREB facilitates sex-specific patterns of gene expression that manifest themselves in the sex-specific effects of risk alleles for unipolar Mood Disorders.
Sequence variants in the CREB1 promoter have been detected that cosegregate with depressive disorders in women from RE-MDD families, providing support for CREB1 as a sex-limited susceptibility gene for unipolar Mood Disorders and related conditions in these families.17 A rare G to A transition at position −656 in the CREB1 promoter (A−656) appeared to confer unipolar Mood Disorders with high penetrance among women in two (2.5%) of the 81 RE-MDD families. By comparison, no A−656 carriers were identified among 149 unrelated women who had no history of psychiatric disorders (allele frequency < 0.003). Based on these observations, we hypothesized that the A variant at position −656 alters the activity of the CREB1 promoter, an effect that is dependent upon or enhanced by the presence of female gonadal steroids.
We determined the effects of gonadal steroid hormones (17 β-estradiol, progesterone, testosterone) on the activity of the wild-type (wt) human CREB1 promoter and assessed the functional significance of the CREB1 promoter variant using transfection experiments that employed constructs containing the wt or variant CREB1 promoters coupled to a reporter gene, chloramphenicol acetyltransferase (CAT). Transfection was performed using noradrenergic CATH.a cells derived from the locus ceruleus because a substantial body of evidence has implicated alterations of noradrenenergic neurons in the pathogenesis of mood disorders, as well as in the mechanism of action of antidepressant drugs.18,19 Expression was assessed in CATH.a cells grown in the absence and presence of physiologically-relevant concentrations (100nM) of gonadal steroid hormones, at baseline and during activation of the cyclic-adenosine monophosphate (cAMP) signaling pathway. The latter condition simulated stress-induced activation of g protein-coupled neurotransmitter/growth factor receptors on brain cells grown in the presence of different gonadal hormones. Since stressful life events have been reported to contribute to the emergence of major depressive episodes among individuals who carry risk alleles for MDD,20 we hypothesized that the activation of the cAMP signaling pathway augments the effect of the A−656 sequence variant on CREB1 promoter function.
MATERIALS AND METHODS
Source and Growth Conditions for CATH.a Cells
The CATH.a cell line was developed from a brain stem tumor of a transgenic mouse expressing the SV40 T antigen under the control of the tyrosine hydroxylase promoter, exhibits a neural, noradrenergic phenotype, and resembles locus ceruleus neurons in their signal transduction profile.21,22 This cell line was obtained from the American Type Culture Collection (Manassas, VA) and grown in RPMI 1640 medium (Gibco, Grand Island, NY) supplemented with 10 mM HEPES, 4.5 g/L glucose, 1 mM sodium pyruvate, 2 g/L sodium bicarbonate, 8% horse serum, and 4% fetal bovine serum, at 37°C, 5% CO2, 100% humidity. Cells were subcultured at a ratio of 1:2 to 1:4.
Construction of Promoter - CAT Expression Plasmids
The 5’ regulatory region of the CREB1 gene has been intensively studied, and it exhibits high nucleotide sequence homology across mouse, rat, and man.22–28 The human CREB1 promoter includes most of the untranslated exon 1 (bps 1 to 130) and extends 1080 bps from the major transcriptional start site in the 5’ direction. This −1080 to 130 bp sequence is identical to the −1264 to −51 bp promoter region described by Meyer and coworkers that was originally numbered relative to the invariant translational start site of the cloned cDNA sequence.23 This 5’ regulatory region includes 1210 bps with restriction sites for Sau 3AI at both termini.
The wild-type CREB1 promoter and variant promoter containing the G to A transition located at position −656 were cloned using genomic DNA prepared from an individual who was heterozygous for these alleles. To achieve this, a 1580 bp region containing the 1210 bp Sau 3AI fragment was amplified using the primers 5’-CCAGAATCGAACCCTCTCTGCTTCC-3’ and 5’-CCTCCTCCTGCTCCTC TTACCG-3’, and GeneAmp® High Fidelity Enzyme Mix (Applied Biosystems, Foster City, CA). The Sau 3AI fragment containing the CREB1 promoter was excised from the PCR product, purified by phenol extraction and ethanol precipitation, and ligated into the Bgl II site of the pCAT®3-Basic Vector (Promega, Madison, WI). The cloning product was transformed into One Shot® TOP10 Chemically Competent E. coli (Invitrogen, Carlsbad, CA) and plated on selective plates containing ampicillin. Colonies were selected and inoculated into LB medium containing ampicillin, and grown overnight. Plasmid DNA was isolated using the Wizard® Plus Minipreps DNA Purification System (Promega Corporation, Madison, WI). Plasmid insert orientation was determined by digestion with restriction endonuclease Fsp I, followed by agarose gel electrophoresis and staining with ethidium bromide. Distinguishing between wild-type or variant promoter inserts was accomplished by PCR and RFLP analysis that detected the presence/absence of an Msp I restriction site that was eliminated by the G to A transition in the variant promoter.17 Large scale preparation of the plasmids from cultures was performed using Wizard® Plus Maxipreps DNA Purification System (Promega Corporation, Madison, WI). The base sequences of the cloned CREB promoters in the two final plasmid preparations to be used in the transfection experiments were confirmed in their entirety by automated DNA sequencing, to ensure that they differed from one another only by the SNP at position −656 and were devoid of PCR or cloning artifacts.
The female research subject with RE-MDD whose genomic DNA was used to clone the wt and variant CREB1 promoters had experienced two major depressive episodes beginning at age 23. She provided written informed consent to participate in a research project on the molecular genetics of affective disorders that was approved by the Institutional Review Board of the University of Pittsburgh.
Transfection of CATH.a Cells
Approximately 18 hrs prior to transfection, CATH.a cells were seeded in 60 mm cell culture dishes (Corning Inc., Corning, NY) at a density of 0.8 to 1.0 × 106 cells/ dish using medium that lacked or contained physiologically-relevant concentrations (100nM) of a gonadal steroid hormone (17 β-estradiol, E; progesterone, P; or testosterone, T; Sigma, St Louis, MO). This concentration of gonadal steroids is in the midrange of those used in cell culture experiments reported in the literature. In addition, 100nM is in the midrange of circulating concentrations of progesterone achieved during the estrus cycle of the female rat, and is similar to the circulating testosterone levels reported for the male rat. While circulating levels of 17 β-estradiol in the female rat are lower, the synthesis of this hormone in brain is likely to produce substantially higher local concentrations of this gonadal steroid in brain regions. The 100nM concentration of 17 β-estradiol is sufficient to induce both slow, long-lasting genomic effects, as well as more rapid, transient actions through non-genomic mechanisms.29
Cells were transfected with the CREB1 promoter-CAT reporter constructs using methods employing Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) that were optimized for CATH.a cells. The transfection cocktail was formed by diluting 15 µl of Lipofectamine™ 2000 reagent in 500 µl of serum free culture medium, and then adding 3 µg of plasmid DNA followed by gentle mixing. The 3 µg of plasmid DNA included in each trasfection cocktail consisted of an equimolar mixture of a CREB1 promoter-CAT reporter construct and the pSV-β-Galactosidase Control Vector (Promega, Madison, WI), or 3 µg of the native pCAT®3-Basic Vector that served as a sham control. The DNA/Lipofectamine™ reagent complex developed at room temperature for 20 min. The cocktail was then added dropwise to the cell culture dish, which was rocked gently to distribute the complex. Cells were grown overnight for approximately 20 hrs before further manipulation.
Activation of the cAMP Signaling Pathway
At 20 hrs (± 1 hr) post transfection, cAMP pathway activation was achieved within the weakly-adherent CATH.a cells by removing sufficient medium to leave 4.5 ml in the dish, and adding 0.5 ml of medium containing a 10x concentration of cAMP activators, such that the resulting culture medium contained a final concentration 10 µM forskolin and 0.25 mM IBMX (Sigma, St. Louis, MO). Cells were then incubated from 0 (no replacement) to 48 hrs prior to harvest and assay. Maximal CREB1 promoter activity occurred after 48 hrs of activation.
Chloramphenicol Acetyltransferase (CAT) Assay
CAT assays were performed using the CAT Enzyme Assay System With Reporter Lysis Buffer (Promega, Madison, WI). Aliquots of cleared cell lysate were assayed in 125 µl reactions containing 40 µM chloramphenicol with 0.20 µCi of 3H-chloramphenicol (PerkinElmer Life Sciences, Boston, MA) added as tracer and 25 µg n-butyryl CoA, in 20 mM Tris, pH 8 (Sigma, St. Louis, MO). Following incubation at 37°C for 2 hours, the reactions were quenched by the addition of 300 µl of mixed xylenes (Sigma, St. Louis, MO), vortexed, and the phases clarified by centrifugation at maximum speed for 3 min at room temperature. The upper organic phase containing the reaction product (n-butyryl chloramphenicol) was back-extracted twice with 0.25 M Tris, pH 8 (Sigma, St. Louis, MO). A 100 µl volume of xylene phase was combined in a 20 ml glass scintillation vial with 10 ml of Opti-Fluor® liquid scintillation cocktail (PerkinElmer, Boston, MA), and counted in a Beckman Instruments (Fullerton, CA) LS 1801 liquid scintillation counter. Enzyme specific activity was expressed as nmol of n-butyryl chloramphenicol produced/hr/mg lysate protein.
β-galactosidase Assay
β-galactosidase assays were performed using the β-Galactosidase Enzyme Assay System with Reporter Lysis Buffer (Promega, Madison, WI). A 150 µl volume of cell lysate was mixed with 150 µl of Assay 2X Buffer, and incubated at 37°C for 3.5 hrs. The reaction was stopped by the addition of 500 µl of 1M sodium carbonate. The hydrolysis of the chromogenic substrate ONPG (o-nitrophenyl-β-D-galactopyranoside) was determined by measuring the absorbance of the reaction product o-nitrophenol at 420 nm using a Beckman DU-640 spectrophotometer (Fullerton, CA). Enzyme specific activity was expressed as pmole of o-nitrophenol produced/min/µg of lysate protein.
Protein Assay
Protein concentrations of cell lysates were determined using the BCA™ Protein Assay (Pierce, Rockford, IL), which is insensitive to the detergent included in the Lysis Buffer. Twenty-five µl volumes of clarified cell lysate were combined with 75 µl of 1 × Reporter Lysis Buffer and 2 ml of prepared assay reagent. Samples were incubated at 37°C for 33 minutes, and then cooled to room temperature for 5 min prior to measuring absorbance at 562 nm. Protein concentrations were determined by comparison to bovine serum albumin, fraction V as a standard.
Statistical Analysis
Statistical analysis was performed using SPSS Version 10 (SPSS, Chicago, IL). Experimental results are presented as means ± SD. The activity of the CREB1 promoter in transfected cells was measured by the ratio of CAT/β-galactosidase specific activity (×1000). The effects of gonadal steroid hormones and promoter genotype on CREB1 promoter activity, during basal conditions or following activation of the cAMP pathway for 48 hrs, were determined using a two-way analysis of variance (ANOVA) with post hoc comparisons. When significant effects of hormone environment were detected by the two-way ANOVA, pairwise comparisons of mean CREB1 promoter activity during different hormonal conditions were made using the Tukey HSD test. When significant effects of CREB1 promoter genotype were detected by the two-way ANOVA, the mean activities of the wild type and variant promoters were compared within each hormone condition using a two-tailed t test. The relationship of basal CREB1 promoter activity to maximal activation during stimulation of the cAMP pathway was explored using linear regression, along with parametric (Pearson r) and nonparametric (Spearman rho) correlation coefficients.
RESULTS
Effects of gonadal steroid hormones on the basal activity of the wild-type and variant CREB1 promoters in CATH.a cells
Cultures of CATH.a cells were grown in medium that lacked or contained physiological (100 nM) concentrations of 17 β-estradiol, progesterone, or testosterone. When the cultures reached a density of approximately 50% confluence, the cells were transfected with an equimolar mixture of a CAT reporter construct containing (a) either the wild-type CREB1 promoter or the G(−656)A variant CREBl promoter, and (b) a pSV-β-galactosidase control vector that constitutively expresses β-galactosidase and was included to adjust for potential differences in transfection efficiency across experiments. Sham transfections employing the native pCAT®3-basic vector were performed to control for any background level of reporter or β-galactosidase activity.
At 20 hrs (± 1 hr) post-transfection, cells were harvested, washed in PBS, lysed, and assayed for CAT, β-galactosidase activity, and protein concentration. Similar β-galactosidase specific activities were observed across experiments, reflecting the reproducible transfection efficiency of CATH.a cells under the conditions employed. Exposure of transfected cells to 100 nM concentrations of 17 β-estradiol, progesterone, or testosterone had negligible effects on β-galactosidase specific activity, confirming the appropriateness of the pSV-β-galactosidase control vector for use under these experimental conditions. Negligible CAT specific activity was found in cells that lacked the CREB1 promoter-CAT reporter construct. CREB1 promoter activity was expressed as the ratio of CAT/β-galactosidase specific activity (×1000). Each experiment was performed six times and the results were calculated as mean ± standard deviation (SD).
As shown in Figure 1, the hormonal environment had a significant effect on the basal activity of the wild-type and variant CREB1 promoters (two-way ANOVA hormone effect; F = 107.11, df = 3,40, p < 0.000001). Progesterone was associated with a significant reduction in promoter activity (p < 0.05, post hoc Tukey HSD), while testosterone increased promoter activity compared to the no hormone condition (p < 0.05 for both comparisons, post hoc Tukey HSD). The G to A transition at position −656 also had a significant effect on basal CREB1 promoter activity compared to the wild type promoter (two-way ANOVA genotype effect; F = 11.21, df = 1,40, p < 0.002). A significant hormone-genotype interaction (F = 3.04, df = 3,40, p = 0.04) reflected the observation that the effect of the G(−656)A polymorphism on CREB1 promoter activity was hormone-dependent. Significant differences between the activity of the wild-type and variant CREB1 promoters occurred in absence of gonadal steroids (t=3.14, df=10, p=0.01), and in the presence of the female gonadal steroids 17 β-estradiol (t = 2.76, df = 6.37, p = 0.03) and progesterone (t = 3.14, df = 10, p = 0.01). In both the no hormone and 17 β-estradiol conditions, the activity of the variant promoter exceeded that of the wild-type promoter. In the presence of progesterone, the variant promoter exhibited a small reduction in activity compare to the wild-type promoter. The G(−656)A polymorphism had no effect on the activity of the CREB1 promoter when CATH.a cells were grown in the presence of testosterone.
Figure 1.
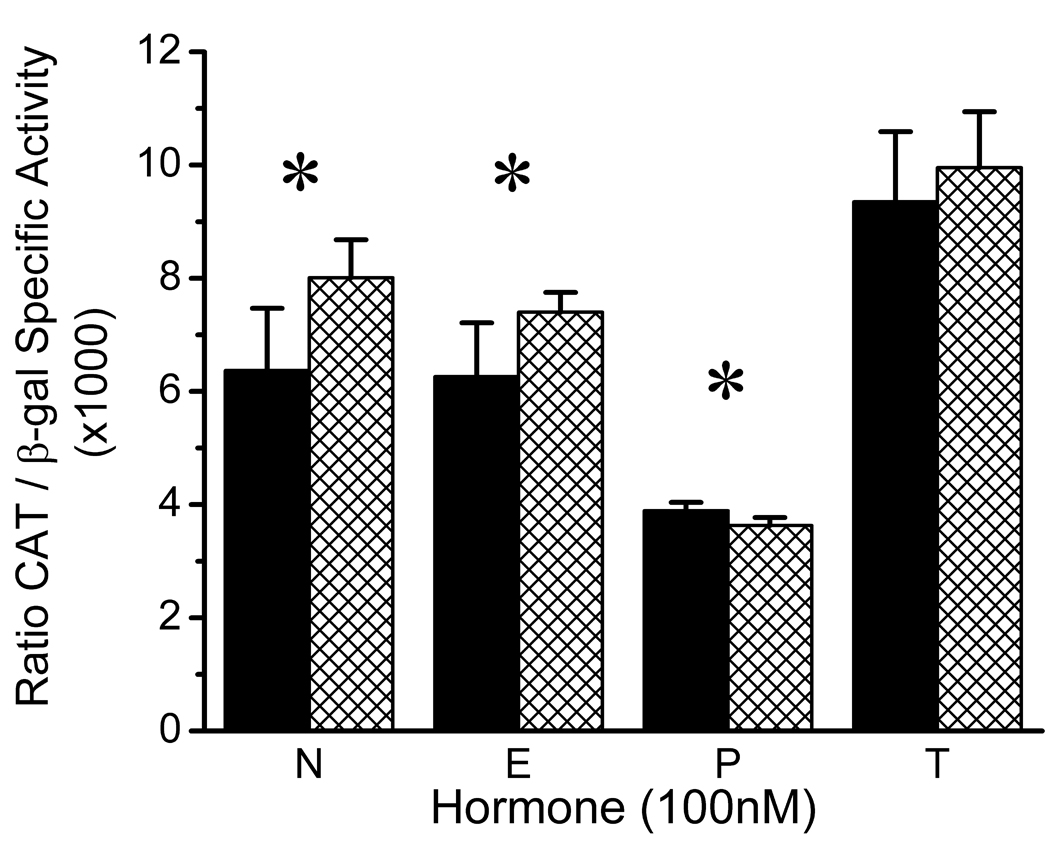
Effects of Gonadal Steroid Hormones on the Basal Activity of the Wild-Type and Variant CREB1 Promoters in CATH.a Neuronal Cells. Wild-type promoter, solid bars. Variant promoter, hatched bars. Corresponding means (±SD) for wild-type and variant promoter activity were: No Hormone (N), 6.37 (1.10) and 8.01 (0.67); 17 β-Estradiol (E), 6.26 (0.95) and 7.40 (0.35); Progesterone (P), 3.89 (0.15) and 3.63 (0.14); and Testosterone (T), 9.35 (1.24) and 9.95 (0.99). Results of two-way ANOVA: Hormone effect, F = 107.11; df = 3,40; p < 0.000001; Genotype effect, F = 11.21; df = 1,40; p < 0.002; Hormone×Genotype interaction, F = 3.04, df = 3,40; p = 0.04. All pairwise post hoc comparisons of hormone effects, p < 0.05, Tukey HSD, except for N vs. E (p = 0.69). Significant pairwise comparisons of G(−656)A genotypes within hormone conditions are indicated by an asterisk: N, t = 3.14, df = 10, p = 0.01; E, t = 2.76, df = 6.37, p = 0.03; P, t = 3.14, df = 10, p = 0.01.
Effects of the cAMP signaling pathway on wild-type and variant CREB1 promoter activity in CATH.a cells grown in the absence/presence of gonadal steroid hormones
CATH.a cells were grown in the absence or presence of gonadal steroids and transfected with equimolar amounts of either of the CREB1 promoter-CAT reporter constructs and pSV-β-galactosidase control vector, as described in the previous section. At 20 hrs (± 1 hr) post transfection, cAMP pathway activation was achieved by removing sufficient medium to leave 4.5 ml in the dish, and adding 0.5 ml of medium (± steroids) containing a 10x concentration of cAMP activators, such that the resulting culture medium contained a final concentration 10 µM forskolin and 0.25 mM IBMX. Forskolin increases intracellular cAMP levels by direct stimulation of adenylate cyclase, while IBMX inhibits the breakdown of cAMP by inhibition of phosphodiesterase. This condition simulates the activation of g protein-coupled neurotransmitter/growth factor receptors on brain cells grown in the presence of different gonadal hormones. Cells were harvested at intervals after activation, lysed, and assayed for CAT, β-galactosidase, and protein concentration. Each experiment was performed six times and the results expressed as mean ± SD.
Maximal CREB1 promoter activity occurred 48 hours after activation of the cAMP signaling pathway and the results at this time point are presented in Figure 2. As observed for the basal condition, both hormonal environment and promoter genotype had significant effects on maximal CREB1 promoter activity (two-way ANOVA hormone effect; F = 295.03; df = 3,40; p < 0.000001 and genotype effect, F = 35.40; df = 1,40; p = 0.000001, respectively). A significant hormone-genotype interaction (F = 7.69, df = 3,40; p = 0.0004) reflected the observation that the effect of the G(−656)A polymorphism on CREB1 promoter activity was hormone-dependent. Significant differences between the activity of the wild-type and variant CREB1 promoters occurred in absence of gonadal steroids (t = 12.24, df = 10, p < 0.001), and in the presence of the female gonadal steroids 17 β-estradiol (3.43, df = 10, p = 0.006) and progesterone (t = 3.74, df = 10, p = 0.004). In both the no hormone and 17 β-estradiol conditions, the activity of the variant promoter exceeded that of the wild-type promoter. In the presence of progesterone, the variant promoter exhibited a small reduction in activity compared to the wild-type promoter. The G(−656)A polymorphism had no effect on the activity of the CREB1 promoter when CATH.a cells were grown in the presence of testosterone.
Figure 2.
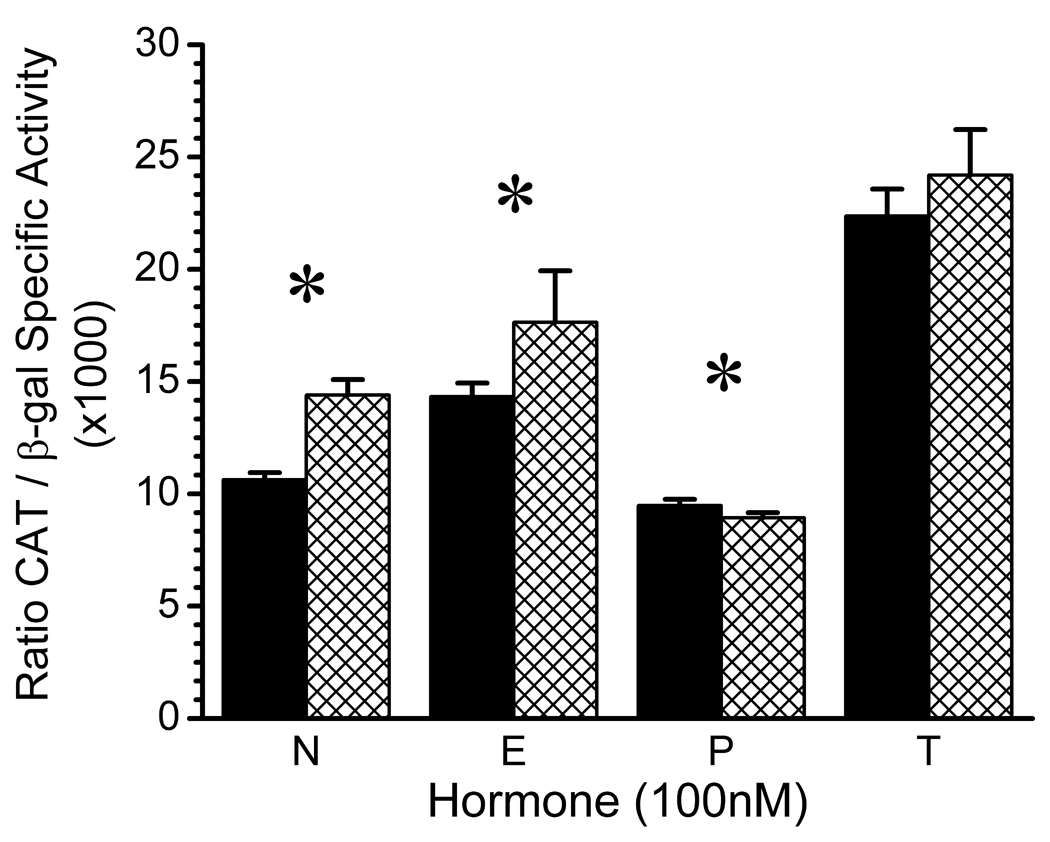
Effects of the cAMP Signaling Pathway on Wild-Type and Variant CREB1 Promoter Activity in CATH.a Neuronal Cells Grown in the Absence/ Presence of Gonadal Steroid Hormones. Wild-type promoter, solid bars. Variant promoter, hatched bars. Corresponding means (±SD) for wild-type and variant promoter activity were: No Hormone (N), 10.63 (0.31) and 14.40 (0.69); 17 β-Estradiol (E), 14.33 (0.60) and 17.64 (2.29); Progesterone (P), 9.48 (0.28) and 8.93 (0.23); and Testosterone (T), 22.37 (1.20) and 24.19 (2.03). Results of two-way ANOVA: Hormone effect, F = 295.03; df = 3,40; p < 0.000001; Genotype effect, F = 35.40; df = 1,40; p = 0.000001; Hormone × Genotype interaction, F = 7.69, df = 3,40; p = 0.0004. All pairwise post hoc comparisons of hormone effects, p < 0.05, Tukey HSD. Significant pairwise comparisons of G(−656)A genotypes within hormone conditions are indicated by an asterisk: N, t = 12.24, df = 10, p < 0.001; E, t = 3.43, df = 10, p = 0.006; P, t = 3.74, df = 10, p = 0.004.
As shown in Figure 2, stimulating the cAMP signaling pathway significantly increased the activity of the CREB1 promoter in CATH.a cells in ways that were dependent on hormonal environment and promoter genotype. However, the relative effects of hormonal environment and genotype resembled those observed for the unstimulated, basal conditions described in Figure 1. The relationship between the basal and maximally-stimulated activity of the CREB1 promoter in transfected CATH.a cells was evaluated using linear regression, which revealed a strong positive correlation between these variables (Figure 3; Pearson r = 0.925, p = 0.001; Spearman rho = 0.952, p = 0.0003). Upon activation of the cAMP signaling pathway, the effects of the A−656 variant on CREB1 promoter activity were enhanced compared to the unstimulated, basal conditions.
Figure 3.

Relationship of Basal and Maximal CREB1 Promoter Activitity Following Activation of the cAMP Signaling Pathway in CATH.a Neuronal Cells. Wild-type promoter, circle. Variant promoter, triangle. Linear regression: slope = 2.31; y-intercept = −0.61; x-intercept = −0.26; Pearson r = 0.925, p = 0.001; Spearman rho = 0.952, p = 0.0003.
DISCUSSION
These findings revealed that the G to A transition at position −656 had functional consequences for CREB1 promoter activity in CATH.a neuronal cells that were dependent on exposure to gonadal steroid hormones. The A−656 genotype produced an increase in basal promoter activity when cells were grown in the presence of 17 β-estradiol and a small, but statistically-significant, reduction in promoter activity when cells were grown in the presence of progesterone. Activation of the cAMP signaling pathway by forskolin/IBMX, which simulated stress-induced activation of g protein-coupled neurotransmitter/growth factor receptors, augmented the changes in CREB1 promoter activity produced by the A−656 genotype in the presence of 17 β-estradiol and progesterone. In contrast, the A−656 genotype did not have a significant effect on the basal or stimulated activity of the CREB1 promoter when CATH.a cells were grown in the presence of testosterone.
These findings support the hypothesis that the A−656 allele contributes to the development of MDD in women by selectively altering the activity of the CREB1 promoter in noradrenergic neurons exposed to female gonadal steroids. The exaggeration of this functional consequence of the A−656 allele during a simulated stress condition may also provide a molecular model that is relevant to reported gene-environment interactions that contribute to the emergence of MDD in clinical populations. The mechanism and level of expression of CREB1, as well as the splicing of its transcript, are cell-specific characteristics.10,11 Therefore, the manifestation of the functional effects of this pathogenic allele in noradrenergic neurons is consistent with a role of this brain cell type in the pathogenesis of MDD and related disorders.13,19 Although the relationship of CREB1 expression levels to behavior in animal models is complex and region-specific, elevated CREB1 expression in neurons within the nucleus accumbens of rats produces multiple "depression-like" effects in behavioral tests of these rodents.10
In addition to alterations of CREB1 promoter activity during exposure of neuronal cells to static physiological concentrations of female gonadal steroids, the results of these transfection experiments (Figure 1 and Figure 2) suggest that natural fluctuations between 17 β-estradiol and progesterone predominance in women may lead to substantial variations in CREB1 promoter activity in neuronal (and potentially other brain) cells regardless of genotype. This dynamic phenomenon may contribute to the increased lifetime prevalence of MDD in women compared to men, and may be especially relevant to the development of depressive disorders in women at times of fluctuations in gonadal hormones that occur during menarche, menses, pregnancy/childbirth, and menopause. At such times, our findings suggest that the A−656 allele would augment the amplitude of the variations in CREB1 promoter activity and may thereby enhance the risk of an emergent depressive disorder in female carriers. The experimental results also suggest that environmental stresses that impact the cAMP signaling pathway may further exaggerate swings in CREB1 promoter activity along with the risk of developing a depressive disorder. This model is also consistent with the reduction in age-specific prevalence of MDD that occurs in late adulthood in both sexes as circulating levels of gonadal steroids wane.
A probable mechanism by which the SNP−656 influences the activity of the CREB1 promoter is by affecting the biological activity of a transcription factor binding site at this location. Previous in silico analysis identified four putative binding sites whose corresponding transcription factors are expressed in human brain.30 Of these four candidates, several lines of evidence suggest that the effects of the SNP−656 may be mediated by CP2 binding. The pathogenic A−656 allele creates a perfect match to the core of the CP2 binding site, reflecting a gain of function that is consistent with the dominant effect (penetrance ≥ 82%) of this variant on the development of depressive disorders among women who are heterozygous carriers.17 Among its target genes, CP2 appears to regulate the expression of glycogen synthase kinase 3β,31 which has been implicated in the pathophysiology of both mood disorders and AD.8,32,33 In addition, a non-coding polymorphism in the 3' untranslated region of the CP2 gene has been reported to affect the risk of MDD34 and Alzheimer's disease,35 both of which aggregate in RE-MDD families.1 Nonetheless, a degree of caution is warranted in the interpretation of the in silico results, which are sensitive to the threshold settings employed to minimize false positive and false negative results, and which do not always reflect the biological activity of a putative binding site.
It is noteworthy that the in silico analysis did not identify an estrogen receptor binding site, even when relaxed similarity score thresholds were employed. This finding suggests that the influence of 17 β-estradiol on the activity of the wt CREB1 promoter, and the interaction of this gonadal steroid with the SNP−656 genotype, were mediated through effects upstream of promoter binding. Potential examples include the involvement of estrogen-induced effects on the expression or kinase-activation of transcription factor(s) that bind to the CREB1 promoter, or by the involvement of a co-transcription factor whose binding to the CREB1 promoter is dependent on a physical interaction with an estrogen receptor.11,15,36
CREB and other transcription factors appear to participate at the top level of the molecular and cellular cascade that controls aspects of neuronal plasticity that regulate mood, cognition, and related phenotypes. The interaction of sex with CREB1 variants that influence the development of psychiatric syndromes, or their clinical features, seems likely to be complex and allele specific. As an example, Perlis and colleagues have recently described associations of non-coding SNPs in the CREB1 region with expressed anger and treatment-emergent suicidal ideation among men with MDD that are less evident or absent among women with this disorder.37,38 Whether these genotypes affect the risk of developing syndromic MDD among men or women cannot be determined from these studies. Since testosterone potentiates aggression/impulsivity, it is tempting to speculate that the association of these CREB1 alleles with anger and suicidal ideation in men with MDD may be related to the effects of testosterone on CREB-mediated gene expression. Molecular consequences of target genes and signaling pathways lower in this regulatory hierarchy may also contribute to sex-related differences in vulnerability to developing MDD and/or modify its clinical presentation.
ACKNOWLEDGMENTS
This work was supported by research project grants MH43261, MH60866, and MH47346 from the National Institute of Mental Health (GSZ).
REFERENCES
- 1.Zubenko GS, Zubenko WN, Spiker DG, Giles DE, Kaplan BB. The malignancy of recurrent, early-onset major depression: A family study. Am J Med Genet (Neuropsychiatr Genet) 2001;105(8):690–699. doi: 10.1002/ajmg.1554. [DOI] [PubMed] [Google Scholar]
- 2.Maher BS, Marazita ML, Zubenko WN, Spiker DG, Giles DE, Kaplan BB, et al. Genetic segregation analysis of recurrent, early-onset major depression: Evidence for single major locus transmission. Am J Med Genet (Neuropsychiatr Genet) 2002;114(2):214–221. doi: 10.1002/ajmg.10158. [DOI] [PubMed] [Google Scholar]
- 3.Zubenko GS, Hughes HB, Stiffler JS, Zubenko WN, Kaplan BB. Genome survey for susceptibility loci for recurrent, early-onset major depression: Results at 10cM resolution. Am J Med Genet (Neuropsychiatr Genet) 2002;114:413–422. doi: 10.1002/ajmg.10381. [DOI] [PubMed] [Google Scholar]
- 4.Philibert R, Caspers K, Langbehn D, Troughton EP, Yucuis R, Sandhu HK, et al. The association of the D2S2944 124 bp allele with recurrent early onset major depressive disorder in women. Am J Med Genet (Neuropsychiatr Genet.) 2003;121(1):39–43. doi: 10.1002/ajmg.b.20062. [DOI] [PubMed] [Google Scholar]
- 5.Zubenko GS, Hughes HB, Stiffler JS, Zubenko WN, Kaplan BB. D2S2944 identifies a likely susceptibility locus for recurrent, early-onset, major depression in women. Mol Psychiatry. 2002;7(5):460–467. doi: 10.1038/sj.mp.4001121. [DOI] [PubMed] [Google Scholar]
- 6.Zubenko GS, Hughes HB, III, Maher BS, Stiffler JS, Zubenko WN, Marazita ML. Genetic linkage of region containing the CREB1 gene to depressive disorders in women from families with recurrent, early-onset, major depression. Am J Med Genet (Neuropsychiatr Genet) 2002;114:980–987. doi: 10.1002/ajmg.b.10933. [DOI] [PubMed] [Google Scholar]
- 7.Zubenko GS, Maher BS, Hughes HB, III, Zubenko WN, Stiffler JSS, Kaplan BB, et al. Genome-wide linkage survey for genetic loci that influence the development of depressive disorders in families with recurrent, early-onset, major depression. Am J Med Genet (Neuropsychiatr Genet) 2003;123B:1–18. doi: 10.1002/ajmg.b.20073. [DOI] [PubMed] [Google Scholar]
- 8.Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nature Medicine. 2001;7(5):541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 9.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 10.Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends in Neurosciences. 2005;28(8):436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 12.Weeber EJ, Sweatt JD. Molecular biology of human cognition. Neuron. 2002;33:845–848. doi: 10.1016/s0896-6273(02)00634-7. [DOI] [PubMed] [Google Scholar]
- 13.Zubenko GS. Do susceptibility loci contribute to the expression of more than one mental disorder? A view from the genetics of Alzheimer’s disease. Mol Psychiatry. 2000;5:131–136. doi: 10.1038/sj.mp.4000713. [DOI] [PubMed] [Google Scholar]
- 14.Lazennec G, Thomas JA, Katznellenbogen BS. Involvement of cyclic AMP response element binding protein (CREB) and estrogen receptor phosphorylation in the synergistic activation of the estrogen receptor by estradiol and protein kinase activators. J Steroid Biochem Mol Biol. 2001;77:193–203. doi: 10.1016/s0960-0760(01)00060-7. [DOI] [PubMed] [Google Scholar]
- 15.McEwen BS. Estrogens effects on the brain: Multiple sites and molecular mechanisms. J Appl Physiol. 2001;91:2785–2801. doi: 10.1152/jappl.2001.91.6.2785. [DOI] [PubMed] [Google Scholar]
- 16.Tremblay A, Giguere V. Contribution of steroid receptor co-activator-1 and CREB binding protein in ligand-independent activity of estrogen receptor beta. J Steroid Biochem Mol Biol. 2001;77:19–27. doi: 10.1016/s0960-0760(01)00031-0. [DOI] [PubMed] [Google Scholar]
- 17.Zubenko GS, Hughes HB, Stiffler JS, Brechbiel A, Zubenko WN, Maher B, et al. Sequence variations in CREB1 cosegregate with depressive disorders in women. Mol Psychiatry. 2003;8:611–618. doi: 10.1038/sj.mp.4001354. [DOI] [PubMed] [Google Scholar]
- 18.Ressler KJ, Nemeroff CB. Role of norepinephrine in the pathophysiology and treatment of mood disorders. Biol Psychiatry. 1999;46:1219–1233. doi: 10.1016/s0006-3223(99)00127-4. [DOI] [PubMed] [Google Scholar]
- 19.Harro J, Oreland L. Depression as a spreading adjustment dosrder of monoaminergic neurons: a case for primary implication of the locus coeruleus. Brain Research Reviews. 2001;38:79–128. doi: 10.1016/s0165-0173(01)00082-0. [DOI] [PubMed] [Google Scholar]
- 20.Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 21.Suri C, Fung BP, Tischler AS, Chikaraishi DM. Catecholaminergic cell lines from the brain and adrenal glands of tyrosine hydroxylase-SV40 T antigen transgenic mice. J Neurosci. 1993;13:1280–1291. doi: 10.1523/JNEUROSCI.13-03-01280.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Widnell KI, Russell DS, Nestler EJ. Regulation of expression of cAMP response element-binding protein in the locus coeruleus in vivo and in a locus coeruleus-like cell line in vitro. Proc. Natl. Acad. Sci. USA. 1994;91:10947–10951. doi: 10.1073/pnas.91.23.10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer TE, Waeber G, Lin J, Beckmann W, Habener JF. The promoter of the gene encoding 3’,5’-cyclic adenosine monophosphate (cAMP) response element binding protein contains cAMP response elements: Evidence for positive autoregulation of gene transcription. Endocrinology. 1993;132:770–780. doi: 10.1210/endo.132.2.8381074. [DOI] [PubMed] [Google Scholar]
- 24.Walker WH, Fucci L, Habener JF. Expression of the gene encoding transcription factor cyclic adenosine 3’,5’-monophosphate (cAMP) response element binding protein (CREB); Regulation by follicle-stimulating hormone-induced cAMP signaling in primary rat Sertoli cells. Endocrinology. 1995;136:3534–3545. doi: 10.1210/endo.136.8.7628390. [DOI] [PubMed] [Google Scholar]
- 25.Widnell KI, Chen J-S, Iredale PA, Walker WH, Duman RS, Habener JF, et al. Transcriptional regulation of CREB (cyclic AMP response element-binding protein) expression in CATH.a cells. J Neurochem. 1996;66(4):1770–1773. doi: 10.1046/j.1471-4159.1996.66041770.x. [DOI] [PubMed] [Google Scholar]
- 26.Coven E, Ni Y, Widnell KI, Chen J, Walker WH, Habener JF, et al. Cell type-specific regulation of CREB gene expression: Mutational analysis of CREB promoter activity. J Neurochem. 1998;71(5):1865–1874. doi: 10.1046/j.1471-4159.1998.71051865.x. [DOI] [PubMed] [Google Scholar]
- 27.Delfino FJ, Walker WH. NF-κB induces cAMP-response element-binding protein gene transcription in Sertoli cells. J Biol Chem. 1999;274:35607–35613. doi: 10.1074/jbc.274.50.35607. [DOI] [PubMed] [Google Scholar]
- 28.Shell SA, Fix C, Olejniczak D, Gram-Humphrey N, Walker WH. Regulation of cyclic adenosine 3’,5’-monophosphate response element binding protein (CREB) expression by Sp1 in the mammalian testes. Biol Reproduction. 2002;66:659–666. doi: 10.1095/biolreprod66.3.659. [DOI] [PubMed] [Google Scholar]
- 29.Cornil CA, Ball GF, Balthazart J. Functional significance of the rapid regulation of brain estrogen action: Where do the estrogens come from? Brain Research. 2006;1126:2–26. doi: 10.1016/j.brainres.2006.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zubenko GS, Hughes HB., III Effects of the G(-656)A variant on CREB1 promoter activity in a glial cell line: interactions with gonadal steroids and stress. Am J Med Genet (Neuropsychiatric Genet) 2008 doi: 10.1002/ajmg.b.30708. in press. [DOI] [PubMed] [Google Scholar]
- 31.Lau KF, Miller CC, Anderton BH, Shaw PC. Molecular cloning and characterization of the human glycogen synthase kinase-3 beta promoter. Genomics. 1999;60(2):121–128. doi: 10.1006/geno.1999.5875. [DOI] [PubMed] [Google Scholar]
- 32.Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: A drug target for CNS therapies. J Neurochem. 2004;89(6):313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- 33.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases, and therapeutics. Neurochem Res. 2007;32(4–5):577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schahab S, Heun R, Schmitz S, Maier W, Kölsch H. Association of polymorphism in the transcription factor LBP-1c/CP2/LSF gene with Alzheimer's disease and major depression. Dement Geriatr Cogn Disord. 2006;22:95–98. doi: 10.1159/000093460. [DOI] [PubMed] [Google Scholar]
- 35.Lambert J-C, Goumidi L, Wavrant-De Vrieze F, Frigard B, Harris JM, Cummings A, et al. The transcriptional factor LBP-1c/CP2/LSF gene on chromosome 12 is a genetic determinant of Alzheimer's disease. Hum Molec Genet. 2000;9:2275–2280. doi: 10.1093/oxfordjournals.hmg.a018918. [DOI] [PubMed] [Google Scholar]
- 36.McEwen BS, Alves SE. Estrogen actions in the central nervous system. Endocrine Rev. 1999;20(3):279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- 37.Perlis RH, Purcell S, Fagerness J, Cusin C, Yamaki L, Fava M, Smoller JW. Clinical and genetic dissection of anger expression and CREB1 polymorphisms in major depressive disorder. Biol Psychiatry. 2007;62:536–540. doi: 10.1016/j.biopsych.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 38.Perlis RH, Purcell S, Fava M, Fagerness J, Rush AJ, Trivedi MH, Smoller JW. Association between treatment-emergent suicidal ideation with citalipram and polymorphisms near cyclic adenosine monophosphate response element binding protein in the STAR*D Study. Arch Gen Psychiatry. 2007;64:689–697. doi: 10.1001/archpsyc.64.6.689. [DOI] [PubMed] [Google Scholar]