

The gaseous radical nitric oxide (•NO, NO) is a signaling molecule that plays a vital role in biology. It facilitates vasodilation and inhibits platelet aggregation in the cardio-vascular system, initiates the pro-inflammatory immune response, and regulates neurotransmission.[1,2] Impaired NO bioavailability is associated with a wide variety of vascular pathologies, including endothelial cell dysfunction.[3] Conse-quently, there is considerable interest in NO donor molecules, such as 1-(N,N-dialkylamino)diazen-1-ium-1,2-diolates (R2N-NONOates; Figure 1), which spontaneously decompose by first-order acid-catalyzed processes to release up to two NO molecules and the corresponding amine.[4–6] R2N-NONOates are widely used as NO precursors in studies of NO-dependent biological processes[5,7] and as NO prodrugs with applications in NO-releasing biomaterials, wound healing, organ protection, and chemotherapy.[5,8] R2N-NONOates can also be combined with anti-inflammatory drugs[9] and incorporated into dendrimers, nanoparticles, or microspheres to optimize their delivery.[10–12] These species are typically synthesized by reacting amines with NO(g) at high pressures.[6,13,14] O2-alkylated R2N-NONOate conjugates with considerably enhanced stability have also been developed.[15–17]
Figure 1.

Structures of selected R2N-NONOates. (For definitions of the abbreviations, see the Supporting Information).
One important distinction between R2N-NONOates and the majority of other NO donor species is that they are generally regarded as being insensitive to the reaction medium and unreactive with most biomolecules, including thiols.[5,8,18] Reports on the reactions between R2N-NON-Oates and transition metal complexes are rare. X-ray structures of CuII/R2N-NONOate complexes show that NON-Oate can coordinate to metal centers through one or both oxygen atoms.[19–21] For a series of metal cations, it was also shown that the NO release rates from the NONOate adduct of the polyamine spermine is relatively insensitive to a range of metal cations (Fe3+, Al3+, La3+, Ca2+, Zn2+, Mg2+; rate of spontaneous NONOate decomposition up to 60 % slower, presumably via binding of the NONOates to the metal center[4]). However, by functionalizing one alkyl substituent of the R2N-NONOate dialkylamine moiety to provide additional donor atoms to the metal center, the rate of NO release from the metal-coordinated R2N-NONOate can be slowed by over an order of magnitude.[21]
Cobalamins (Cbls, vitamin B12 derivatives) are octahedrally coordinated cobalt(III) macrocycles, and can incorporate a range of ligands at the upper (β) axial site. These include aqua/hydroxy (H2OCbl+/HOCbl; pKa(H2OCbl+)=7.76±0.02 at 25.0°C, total ionic strength I=0.50 m (KNO3)[22]), cyanide (CNCbl), methyl (MeCbl), 5′-deoxyadenosyl (AdoCbl), nitro (NO2Cbl), and nitrosyl (NOCbl) ligands. In searching for an efficient method to synthesize nitroxylcobalamin (also referred to as nitrosylcobalamin, NOCbl) for X-ray diffraction studies, we found that it can be synthesized in high yield and purity by reacting HOCbl with diethylamine-NONOate (DEA-NONOate, Figure 1) under anaerobic conditions.[23] Like other R2N-NONOates, DEA-NONOate decomposes under anaerobic conditions by a clean, first-order, acid-catalyzed process to release NO and the corresponding amine, namely diethylamine (DEA).[4,24,25] Control experiments showed that DEA-NONOate decomposes cleanly into DEA alone (+ NO; pD 9.50 and 10.40; 1H NMR spectroscopy). It is, however, well established that neither H2OCbl+ nor HOCbl react with NO.[26] We therefore carried out kinetic and mechanistic studies on this intriguing reaction.
Figure 2 gives typical UV/Vis spectra for the reaction between HOCbl (0.050 mm) and excess DEA-NONOate (10.0 mm) as a function of time under anaerobic conditions at pH 10.80 (0.30 m CAPS buffer, 25.0°C, I=1.0 m (NaCF3SO3)). High buffer concentrations (0.30 m) were necessary to ensure a stable pH was maintained during the reaction. Figure 2, Inset a, shows a comparison between the initial and final spectrum of the reaction in which HOCbl is cleanly converted into NOCbl (λmax=256, 278 (shoulder), 289, 315, and 478 nm) with sharp isosbestic points observed at 341, 370, and 498 nm, which is in agreement with literature values.[27,28] In Figure 2, Inset b, the best fit of the absorbance data at 356 nm versus time to a first-order rate equation is superimposed upon the experimental data, giving kobs=(1.91±0.01) × 10−3 min−1. The rate of spontaneous acidcatalyzed decomposition of DEA-NONOate to NO and DEA was found to be more than one order of magnitude slower than the reaction of DEA-NONOate with HOCbl (half-life for the spontaneous decomposition t1/2≥ 1 week, pH 10.80; Table 1). This result suggests that HOCbl reacts directly with the DEA-NONOate complex to give NOCbl, and that decomposition of DEA-NONOate to form NO is not a prerequisite for the reaction to occur. The direct transfer of a nitroxyl group (NO−) from R2N-NONOate to a transition metal center to yield the corresponding nitroxyl complex is, to our knowledge, unprecedented.
Figure 2.

UV/Vis spectra for the reaction between HOCbl (0.050 mm) and DEA-NONOate (10.0 mm) at pH 10.80. (Spectra taken every 10.0 min, 0.30 m CAPS, I=1.0m (NaCF3SO3), 25.0°C.) Isosbestic points occur at 341, 370, and 498 nm. Inset a) First and last spectra. The final product is NOCbl. b) Fit of the absorbance data at 356 nm (A356) versus time to a first-order rate equation, giving kobs=(1.91±0.01)×10−3 min−1.
Table 1.
Apparent rate constants kapp for the reaction between HOCbl and DEA-NONOate, and observed rate constants for the spontaneous decomposition of HOCbl (kHOCbl) and DEA-NONOate (kL)
| pH | kapp [L mol−1 min−1] | 103kHOCbl [min−1] | 103kL [min−1] |
|---|---|---|---|
| 9.50 | 0.68±0.02 | 0.33±0.01 | 1.24±0.01 |
| 9.80 | 0.29±0.03 | 0.97±0.02 | 0.555±0.008 |
| 10.40 | 0.14±0.01 | 1.43±0.05 | 0.197±0.001 |
| 10.80 | 0.056±0.002 | 1.32±0.03 | t½≥1 week |
[a]I=1.0m (NaCF3SO3), 0.30m buffer, 25°C.
To further probe for formation of intermediate(s), the reaction was followed using 1H NMR spectroscopy. Cbl complexes have five corrin and nucleotide protons that resonate in the aromatic region with chemical shifts dependent on the β-axial ligand. Observation of the reaction of HOCbl (2.96 mm) with DEA-NONOate (4.44 mm) at pD 11.30 by 1H NMR spectroscopy showed that HOCbl (δ=7.17, 6.70, 6.49, 6.23, and 6.04 ppm) was cleanly converted into NOCbl (δ=7.44, 7.27, 6.80, 6.35, and 6.25, in agreement with literature values[27,29]), without any detectable Cbl intermediate (Supporting Information, Figure S1). Alkaline conditions were used to ensure that the spontaneous decomposition of DEA-NONOate is negligible. The reaction stoichiometry was determined by recording 1H NMR spectra of the products of the reaction between HOCbl and 0.55, 1.1, 1.2, 1.5, or 2.2 mol equivalents of DEA-NONOate at pD 10.42. With 0.55 equivalents of DEA-NONOate, a mixture of NOCbl (ca. 55 %) and unreacted HOCbl (approx. 45 %) was observed, whereas with 1.2 equivalents of DEA-NONOate, HOCbl was essentially completely converted into NOCbl (Figure 3 a). Because approximately 1.2 equivalents of DEA-NONOate is required for the reaction to proceed to completion, this observation suggests that only one of the two nitric oxide moieties in the parent NONOate reacts with the cobalamin to form NOCbl.
Figure 3.
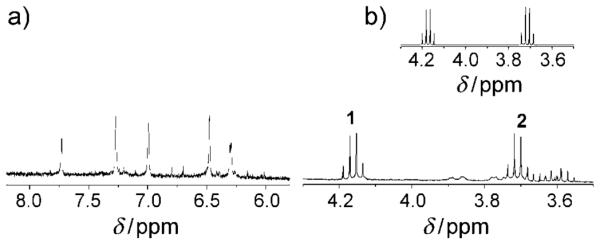
1H NMR spectrum of the products of the reaction between HOCbl and 1.2 equiv DEA-NONOate after 5 days (pD 10.42, 0.50 m CAPS). a) Aromatic region showing the 5 characteristic signals of NOCbl at δ=7.73, 7.27, 7.00, 6.48, and 6.31 ppm. The small peaks are impurities arising from Cbl decomposition at the high pD conditions. b) 4.3–3.5 ppm region, showing signals attributable to DEA-NO (δ=4.20, 4.18, 4.16, 4.15, 3.74, 3.72, 3.71, and 3.69 ppm) overlapping with NOCbl signals. These signals were not observed in the 1H NMR spectrum of the reactant HOCbl. Inset: 1H NMR spectrum of authentic DEA-NO (0.50 m CAPS, pD=10.42).
Experiments were carried out to identify the non-cobalamin reaction product(s). It was determined that nitrite was not a reaction product (Griess assay; see Supporting Information). Two other potential non-Cbl reaction products, diethylamine (DEA) and N-nitrosodiethylamine (DEA-NO), are individually distinguishable by 1H NMR spectroscopy. The 1H NMR spectrum of the products of the reaction of HOCbl and 1.2 equivalents of DEA-NONOate in alkaline solution (pD 10.42) in the 3.5–4.3 ppm region revealed DEA-NO to be the non-Cbl reaction product (Figure 3 b). Therefore, DEA-NONOate reacts directly with HOCbl to produce NOCbl and DEA-NO. Formation of the corresponding nitrosamine is undesirable from a biological and pharmaceutical view point, given that many of these species, including DEA-NO, are carcinogenic.[30] Nitrosamines are also products of R2N-NONOate photolysis and R2N-NONOate decomposition under aerobic conditions.[30,32] Although it has been previously suggested that DEA-NO rapidly decompose to DEA and NO,[4,25] in this study it was a stable species.
To confirm that DEA-NONOate, rather than its decomposition products, react with H2OCbl+/HOCbl, H2OCbl+/HOCbl was added to a solution of DEA-NONOate that had fully decomposed to DEA and NO(g) at pH 7.40, 8.50, or 9.80, and the reaction was followed by UV/Vis spectroscopy. No reactions were observed after 16 h under any of the three pH conditions.
If indeed HOCbl reacts directly with DEA-NONOate, the observed rate constant should depend on the DEA-NON-Oate concentration. The dependence of kobs on DEA-NON-Oate concentration (2.5–25.0 mm) at pH 10.80 was therefore determined (Figure 4). Measurements at higher DEA-NON-Oate concentrations were not possible owing to the limited solubility of DEA-NONOate. Fitting the data to a straight line gives a second-order rate constant, kapp=(0.056±0.002) L mol−1 min−1 (=kK’; see below) at pH 10.80 with an intercept of (1.31±0.02) × 10−3 min−1. A control experiment showed that HOCbl itself slowly decomposes at pH 10.80 (kHOCbl=(1.32±0.03) × 10−3 min−1; Table 1, which, within experimental error, is the same as the intercept. Note that HOCbl decomposition is not strictly first-order and does not proceed to completion at lower pH values. The self-reduction of HOCbl in alkaline solution has been reported previously.[32,33]
Figure 4.
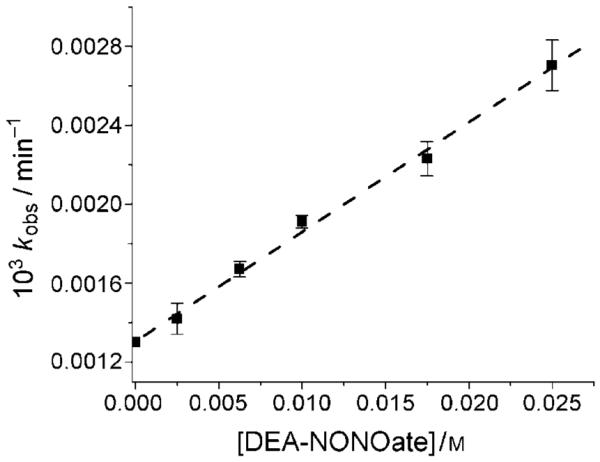
Plot of observed rate constant kobs versus NONOate concentration for the reaction between HOCbl and DEA-NONOate at pH 10.80. The best fit of the data gives kapp =(0.056±0.002) Lmol−1 min−1 (slope) and kHOCbl =(1.32±0.03)×10−3 min−1 (intercept, 0.30m CAPS, I=1.0m (NaCF3SO3), 25.0°C).
The dependence of kobs on DEA-NONOate concentration (2.50–25.0 mm) was studied at three other pH conditions (pH 9.50, 9.80 and 10.40). The data is plotted in Figure S2 in the Supporting Information and the rate constants are summarized in Table 1. It was necessary to take into account spontaneous DEA-NONOate decomposition in the treatment of the kinetic data at pH 9.50 and 9.80 at the lower DEA-NONOate concentrations by fitting to Equation (1):
| (1) |
where Aobs, A0, and A∞ are the observed, initial, and final absorbances respectively, kapp is the (pH-dependent) rate constant, kL is the observed rate constant for spontaneous NONOate decomposition, and [L]0 is the initial NONOate concentration. The derivation of this equation is given in the Supporting Information.
Above pH 10.80, the rate of reaction between HOCbl and DEA-NONOate is extremely slow, whereas at pH values below 9.50, the spontaneous decomposition of DEA-NON-Oate was found to be within one order of magnitude or even faster than the reaction of interest. For example, at pH 9.30, the observed rate constant for the reaction between DEA-NONOate (2.5 mm) and HOCbl (0.050 mm) is 3.0 × 10−3 min−1, whereas that for spontaneous decomposition of DEA-NONOate is 1.6 × 10−3 min−1. At higher NONOate concentrations, although the rate of the Cbl/NONOate reaction is faster, considerable interference occurs from gas evolution despite gentle stirring with stir bars at the bottom of the cuvettes. The gas arises from acid-catalyzed spontaneous DEA-NONOate decomposition to NO(g) and DEA, leading to unreliable data. Furthermore, a second reaction was observed below pH 10, which was subsequently shown to arise from excess NO(g) from decomposed DEA-NONOate reacting with NOCbl to form nitrocobalamin (NO2Cbl). This reaction becomes increasingly important at lower pH values and higher DEA-NONOate conditions. Further details are given in the Supporting Information.
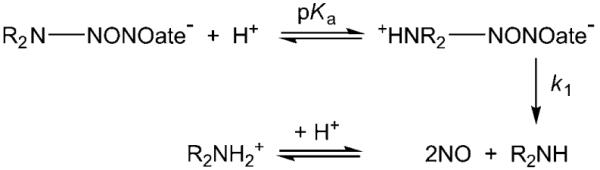
From Table 1, it can be seen that the second-order rate constant kapp increases with decreasing pH. The pKa of DEA-NONOate is 5.0,[4] and a 1H NMR titration experiment showed no further deprotonation for DEA-NONOate in the range pH 8.5–12.5. Control experiments showed that DEA-NONOate does not react with methylcobalamin (MeCbl), cyanocobalamin (CNCbl) or adenosylcobalamin (AdoCbl). This result suggests that a labile β-axial ligand, such as the aqua ligand of H2OCbl+, is required for the reaction between Cbls and NONOates to proceed. It is well established that HOCbl is inert to substitution.[22] Using the value of kapp at pH 9.50 (0.68 Lmol−1 min−1) and pKa(H2OCbl+)=7.76,[22] a second-order rate constant for the reaction between H2OCbl+ and DEA-NONOate of approximately 38 Lmol −1 min−1 (ca. 0.63 Lmol −1 s−1) was estimated. However, rate constants for ligand substitution of the β-axial ligand of H2OCbl+ are typically two to four orders of magnitude larger than this.[34] The simplest mechanism consistent with the experimental data is given in Scheme 1, in which a rapid pre-equilibrium to form the NONOate-Cbl complex precedes rate-determining nitrogen-nitrogen bond cleavage to give NOCbl and R2N-NO. The corresponding rate equation is given in Equation (2) where K’=K[H+]/([H+]+Ka(H2OCbl+):
| (2) |
Equation (2) reduces to kobs=kK’[NONOate]+kHOCbl when K’ is small, which is consistent with the Cbl reactant being predominantly HOCbl, not H2OCbl+, and a reaction intermediate not being observable. Although it was not possible to obtain rate data for the reaction between H2OCbl+/HOCbl at pH values close to the pKa value of H2OCbl+, and therefore show that H2OCbl+ (pKa 7.76[22]), not HOCbl, reacts with DEA-NONOate, the increase in kapp as the pH is decreased (Table 1) is consistent with this interpretation.
Scheme 1.

Proposed mechanism for the reaction of H2OCbl+/HOCbl with R2N-NONOate.
The reactions between H2OCbl+/HOCbl and three other R2N-NONOates were also investigated. DETA-NONOate (Figure 1) was chosen for studies at lower pH conditions to further probe whether the increase in the apparent rate constant kapp with decreasing pH arises from H2OCbl+/HOCbl speciation, because DETA-NONOate is remarkably stable to acid-catalyzed decomposition when compared with other R2N-NONOates (Table 2). However, at pH 7.4, the reaction between H2OCbl+/HOCbl and DETA-NONOate was extremely slow and incomplete, with a rate constant of the same order as that for the spontaneous rate of decom-position of the NONOate (kobs≈8×10−4 min−1, kL=1.6 × 10−4 min−1; [DETA-NONOate]=7.5 mm, ca. 70% of HOCbl/H2OCbl+ converted into NOCbl over 2.5 days). Although the reaction was faster at pH 5.88, once again H2OCbl+ was only partially converted into NOCbl and kL and kobs were within a factor of two of each other. Similar results were obtained for the reaction of H2OCbl+/HOCbl with DPTA-NONOate (pH 9.30, 8.00, 7.40, and 6.80), with the extent of H2OCbl+/HOCbl conversion into NOCbl decreasing with decreasing pH. At pH 6.80, practically no reaction occurred between H2OCbl+ and DPTA-NONOate, with the latter species apparently decomposing before it could react with H2OCbl+. Therefore, it appears that having two sterically bulky substituents on the secondary amine of R2N-NON-Oates leads to both thermodynamically and kinetically unfavorable reactions of these species with H2OCbl+/HOCbl, although further studies are required to confirm this. Sterically demanding substituents also stabilize R2N-NONOates with respect to acid-catalyzed decomposition (Table 2).[6,35]
Table 2.
pKa values and rate constants for spontaneous, acid-catalyzed decomposition of R2N-NONOates (37°C)
Kinetic studies on the reaction between H2OCbl+/HOCbl and MAHMA-NON-Oate were also carried out. As with DEA-NONOate above, HOCbl reacts with MAMHA-NONOate to produce NOCbl (Supporting Information, Figure S3). Interestingly, although the plot of kobs versus NONOate concentration was linear at pH 10.80, curvature was observed at lower pH values (Supporting Information, Figure S4). Additional studies revealed that the most likely explanation for the curvature is a nitrite impurity (approx. 10 %) in commercial MAHMA-NONOate, which is not easily removed. Nitrite reacts rapidly with H2OCbl+ to form nitrocobalamin (NO2Cbl),[36,37] and the formation of NO2Cbl slows down the apparent rate of the reaction between HOCbl and MAHMA-NONOate. Further details are given in the Supporting Information.
Finally, if a labile axial ligand is required for the transfer of NO− from R2N-NONOates to cobalt(III) corrinoids to produce the corresponding nitroxyl complex, then R2N-NONOates should also react with the closely related CoIII cobinamide derivatives (Cbi) in which the nucleotide is cleaved at the phosphodiester.[38] In support of this, aquahy-droxycobinamide was found to react rapidly with DEA-NONOate to produce the corresponding NOCbi complex (kobs=0.5 min−1, [DEA-NONOate]=1.0 mm, pH 9.30, 25.0°C). Detailed kinetic studies on this system are currently underway.
To summarize, UV/Vis and 1H NMR spectroscopy studies on the reaction between R2N-NONOates and HOCbl/H2OCbl+ suggest that H2OCbl+ reacts directly and essentially stoichiometrically with DEA-NONOate to give NOCbl and the corresponding toxic nitrosoamine DEA-NO. Decomposition of the NONOate to produce NO is not a prerequisite for the reaction to occur. To our knowledge, a direct reaction between R2N-NONOate and a transition metal complex to produce a nitroxyl complex is unprecedented. Given the widespread use of R2N-NONOates as NO donors and the interest in these compounds as pharmaceuticals, further studies on the potential biological relevance of this type of reaction would be of great interest.
Supplementary Material
Footnotes
We thank Prof. Rudi van Eldik, University of Erlangen-Nürnberg (Germany) and Prof. István Fábián, University of Debrecen, Hungary for useful discussions. This research was funded by the Egyptian Ministry of Higher Education (PhD scholarship to H.A.H.), and the NSF (CHE-0848397, N.E.B.). R2N-NONOates = 1-(N,N-dialkylamino)diazen-1-ium-1,2-diolates, nitroxyl=NO−.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200904360.
Contributor Information
Hanaa A. Hassanin, Department of Chemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44242 (USA); Department of Chemistry, Faculty of Science, Ain Shams University, Abbassia, Cairo (Egypt)
Luciana Hannibal, School of Biomedical Sciences, Kent State University, Kent, OH 44242 (USA) and Department of Cell Biology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195 (USA).
Dr. Donald W. Jacobsen, School of Biomedical Sciences, Kent State University, Kent, OH 44242 (USA) and Department of Cell Biology, Lerner Research Institute, Cleveland Clinic, Cleveland, OH 44195 (USA)
Dr. Mohamed F. El-Shahat, Department of Chemistry, Faculty of Science, Ain Shams University, Abbassia, Cairo (Egypt)
Dr. Mohamed S. A. Hamza, Department of Chemistry, Faculty of Science, Ain Shams University, Abbassia, Cairo (Egypt)
Dr. Nicola E. Brasch, Department of Chemistry and School of Biomedical Sciences, Kent State University, Kent, OH 44242 (USA).
References
- [1].Ignarro LJ, Cirino G, Casini A, Napoli C. J. Cardiovasc. Pharmacol. 1999;34:879–886. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- [2].Bian K, Murad F. Front. Biosci. 2003;8:d264–278. doi: 10.2741/997. [DOI] [PubMed] [Google Scholar]
- [3].Yetik-Anacak G, Catravas JD. Vasc. Pharmacol. 2006;45:268–276. doi: 10.1016/j.vph.2006.08.002. [DOI] [PubMed] [Google Scholar]
- [4].Davies KM, Wink DA, Saavedra JE, Keefer LK. J. Am. Chem. Soc. 2001;123:5473–5481. doi: 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]
- [5].Keefer LK. Curr. Top. Med. Chem. 2005;5:625–636. doi: 10.2174/1568026054679380. [DOI] [PubMed] [Google Scholar]
- [6].Konter J, Abuo-Rahma GE-DA, El-Emam A, Lehmann J. Eur. J. Org. Chem. 2007:616–624. [Google Scholar]
- [7].Song T, Hatano N, Kambe T, Miyamoto Y, Ihara H, Yamamoto H, Sugimoto K, Kume K, Yamaguchi F, Tokuda M, Watanabe Y. Biochem. J. 2008;412:223–231. doi: 10.1042/BJ20071195. [DOI] [PubMed] [Google Scholar]
- [8].Miller MR, Megson IL. Br. J. Pharmacol. 2007;151:305–321. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Velázquez CA, Rao PNP, Citro ML, Keefer LK, Knaus EE. Bioorg. Med. Chem. 2007;15:4767–4774. doi: 10.1016/j.bmc.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Polizzi MA, Stasko NA, Schoenfisch MH. Langmuir. 2007;23:4938–4943. doi: 10.1021/la0633841. [DOI] [PubMed] [Google Scholar]
- [11].Shin JH, Schoenfisch MH. Chem. Mater. 2008;20:239–249. doi: 10.1021/cm702526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stasko NA, Schoenfisch MH. J. Am. Chem. Soc. 2006;128:8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- [13].Hrabie JA, Klose JR, Wink DA, Keefer LK. J. Org. Chem. 1993;58:1472–1476. [Google Scholar]
- [14].Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, Cologna SM, Dutton AS, Champion HC, Mancardi D, Tocchetti CG, Saavedra JE, Keefer LK, Houk KN, Fukuto JM, Kass DA, Paolocci N, Wink DA. J. Med. Chem. 2005;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- [15].Valdez CA, Saavedra JE, Showalter BM, Davies KM, Wilde TC, Citro ML, Barchi JJ, Jr., Deschamps JR, Parrish D, El-Gayar S, Schleicher U, Bogdan C, Keefer LK. J. Med. Chem. 2008;51:3961–3970. doi: 10.1021/jm8000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chakrapani H, Wilde TC, Citro ML, Goodblatt MM, Keefer LK, Saavedra JE. Bioorg. Med. Chem. 2008;16:2657–2664. doi: 10.1016/j.bmc.2007.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chakrapani H, Goodblatt MM, Udupi V, Malaviya S, Shami PJ, Keefer LK, Saavedra JE. Bioorg. Med. Chem. Lett. 2008;18:950–953. doi: 10.1016/j.bmcl.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mason RP, Cockcroft JR. J. Clin. Hypertens. 2006;8:40–52. doi: 10.1111/j.1524-6175.2006.06041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schneider JL, Halfen JA, Young VG, Jr., Tolman WB. New J. Chem. 1998;22:459–466. [Google Scholar]
- [20].Schneider JL, Young VG, Jr., Tolman WB. Inorg. Chem. 1996;35:5410–5411. doi: 10.1021/ic960414m. [DOI] [PubMed] [Google Scholar]
- [21].Ziche M, Donnini S, Morbidelli L, Monzani E, Roncone R, Gabbini R, Casella L. ChemMedChem. 2008;3:1039–1047. doi: 10.1002/cmdc.200700354. [DOI] [PubMed] [Google Scholar]
- [22].Xia L, Cregan AG, Berben LA, Brasch NE. Inorg. Chem. 2004;43:6848–6857. doi: 10.1021/ic040022c. [DOI] [PubMed] [Google Scholar]
- [23].Hannibal L, Smith CA, Jacobsen DW, Brasch NE. Angew. Chem. 2007;119:5232–5235. doi: 10.1002/anie.200701131. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:5140–5143. doi: 10.1002/anie.200701131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. J. Med. Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- [25].Ramamurthi A, Lewis RS. Chem. Res. Toxicol. 1997;10:408–413. doi: 10.1021/tx960183w. [DOI] [PubMed] [Google Scholar]
- [26].Wolak M, Stochel G, Hamza M, van Eldik R. Inorg. Chem. 2000;39:2018–2019. doi: 10.1021/ic991266d. [DOI] [PubMed] [Google Scholar]
- [27].Wolak M, Zahl A, Schneppensieper T, Stochel G, van Eldik R. J. Am. Chem. Soc. 2001;123:9780–9791. doi: 10.1021/ja010530a. [DOI] [PubMed] [Google Scholar]
- [28].Zheng D, Birke RL. J. Am. Chem. Soc. 2001;123:4637–4638. doi: 10.1021/ja015682k. [DOI] [PubMed] [Google Scholar]
- [29].Brasch NE, Finke RG. J. Inorg. Biochem. 1999;73:215–219. doi: 10.1016/s0162-0134(99)00018-5. [DOI] [PubMed] [Google Scholar]
- [30].Chakrapani H, Maciag AE, Citro ML, Keefer LK, Saavedra JE. Org. Lett. 2008;10:5155–5158. doi: 10.1021/ol8020989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Srinivasan A, Kebede N, Saavedra JE, Nikolaitchik AV, Brady DA, Yourd E, Davies KM, Keefer LK, Toscano JP. J. Am. Chem. Soc. 2001;123:5465–5472. doi: 10.1021/ja002898y. [DOI] [PubMed] [Google Scholar]
- [32].Yamada RH, Kato T, Shimizu S, Fuki S. Biochim. Biophys. Acta Gen. Subj. 1966;117:113. doi: 10.1016/0304-4165(66)90147-4. [DOI] [PubMed] [Google Scholar]
- [33].Lee LP, Schrauzer GN. J. Am. Chem. Soc. 1968;90:5274–5276. doi: 10.1021/ja01021a043. [DOI] [PubMed] [Google Scholar]
- [34].Marques HM, Knapton L. J. Chem. Soc. Dalton Trans. 1997:3827–3832. [Google Scholar]
- [35].Horstmann A, Menzel L, Gaebler R, Jentsch A, Urban W, Lehmann J. Nitric Oxide. 2002;6:135–141. doi: 10.1006/niox.2001.0398. [DOI] [PubMed] [Google Scholar]
- [36].Marques HM, Knapton L. J. Chem. Soc. Dalton Trans. 1997:3827–3833. [Google Scholar]
- [37].Knapton L, Marques HM. Dalton Trans. 2005:889–895. doi: 10.1039/b416083e. [DOI] [PubMed] [Google Scholar]
- [38].Friedrich W, Bernhauer K. Chem. Ber. 1956;89:2507–2512. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.