

Abstract
AMP-activated protein kinase (AMPK) plays a key role in the regulation of energy homeostasis and is activated in response to cellular stress, including hypoxia/ischemia and hyperglycemia. The stress events are accompanied by rapid release of extracellular nucleotides from damaged tissues or activated endothelial cells (EC) and platelets. We demonstrate that extracellular nucleotides (ATP, ADP and UTP, but not UDP) and adenosine independently induce phosphorylation and activation of AMPK in human umbilical vein EC (HUVEC) by the mechanism that is not linked to changes in AMP:ATP ratio. HUVEC express NTPDases, as well as 5′-nucleotidase, hence nucleotides can be metabolized to adenosine. However, inhibition of 5′- nucleotidase had no effect on ATP/ADP/UTP-induced phosphorylation of AMPK, indicating that AMPK activation occurred as a direct response to nucleotides. Nucleotide-evoked phosphorylation of AMPK in HUVEC was mediated by P2Y1, P2Y2 and/or P2Y4 receptors, while P2Y6, P2Y11 and P2X receptors were not involved. The nucleotide-induced phosphorylation of AMPK was affected by changes in the concentration of intracellular Ca2+ and by Ca2+/calmodulin-dependent kinase kinase (CaMKK), while most likely it was not dependent on LKB1 kinase. Adenosine-induced phosphorylation of AMPK was not mediated by P1 receptors but required adenosine uptake by equilibrative nucleoside transporters followed by its (intracellular) metabolism to AMP but not inosine. Moreover, adenosine effect was Ca2+- and CaMKK-independent while probably associated with upstream LKB1. We hypothesize that P2 receptors and adenosine transporters could be novel targets for the pharmacologic regulation of AMPK activity and its downstream effects on EC function.
Keywords: endothelial cells, AMP-activated protein kinase, P2 receptors, adenosine, CaMKK
Introduction
AMP-activated kinase (AMPK) is an evolutionarily conserved enzyme that acts as an ultrasensitive energy charge sensor and regulates cell energy metabolism 1. AMPK is a heterotrimeric Ser/Thr kinase consisting of a catalytic α subunit and regulatory β and γ subunits. AMPK is activated in response to ATP depletion associated with an increase in the intracellular AMP:ATP ratio. This ATP consumption can originate from pathologic cellular stress, such as heat shock, hypoxia or ischemia, and from physiologic exercise-induced skeletal muscle contraction 2. AMPK can also be phosphorylated and activated by the mechanism independent of changes in AMP:ATP levels 3. AMPK is activated allosterically by AMP (up to 5-fold) and by phosphorylation of Thr-172 on the á subunit (50 to 100-fold), which is catalyzed by upstream kinases, including LKB1 and CaMKK 4,5. Activated AMPK turns on catabolic pathways that generate ATP and turns off pathways that consume ATP by phosphorylation of multiple targets. These include glycogen synthase, acetyl-CoA carboxylase (ACC) and hydroxymethylglutaryl-CoA (HMG-CoA) reductase. Since AMPK is central in controlling the metabolism of glucose and fatty acids, its role in obesity and type 2 diabetes is of major importance. AMPK is expressed in skeletal muscle, brain, liver and pancreas. AMPK has been also localized in EC 6, however pathways of its activation, as well as the functions of this kinase in the endothelium are still not well understood and remains to be determined.
Extracellular tri- and diphosphate nucleosides are released into tissue fluids and plasma from aggregated platelets and activated leukocytes and endothelial cells (EC), in responses to various pro-inflammatory stimuli, tissue damage and cell death 7. They generate a variety of responses within the vasculature, including platelet activation, changes in smooth muscle contractility and vasodilation.
Early cellular responses to extracellular nucleotides include elevation of the intracellular free calcium ion concentration ([Ca2+]i) due to calcium influx or release from intracellular stores. Extracellular nucleotides exert their effects through purinergic P2 receptors, which are classified into two main groups: P2X, that are ligand-gated ion channels, and P2Y, G-protein-coupled receptors 8,9. Analyses of P2 receptors at the RNA and protein levels, as well as evaluation of their pharmacological characteristics, indicate that P2X4, P2Y11, P2Y1 and P2Y2 are the most abounded in HUVEC 10. However, other receptors, including P2Y4, P2Y6, P2X7 and some other P2X receptors have also been identified in EC 11. ADP is a specific agonist of P2Y1 receptors, UTP is known to stimulate P2Y2, and P2Y4 receptors, while UDP is an agonist of P2Y6 receptors 12-14. ATP can stimulate P2Y2 receptors, while ATP and its analog BzATP are ligands for P2Y11 and most of P2X receptors 12-14.
EC express CD39, a NTPDase that hydrolyzes tri- and di-phosphate nucleosides to mono-nucleosides, as well as 5′-nucleotidase (CD73), which can further hydrolyze AMP to adenosine 15,16. This adenosine can exert cellular responses employing two mechanisms, acting externally through P1 receptors or after being internalized through nucleoside transporters.
The aim of the present study was to determine whether extracellular nucleotides and their metabolites that can be released in response to cellular stress, are involved in activation of AMPK. We demonstrate that phosphorylation and activation of AMPK is induced by two independent pathways: one mediated by extracellular nucleotides, associated with P2Y1, P2Y2 and/or P2Y4 receptors, and the second being initiated by adenosine, with the involvement of adenosine transporters and its metabolism by adenosine kinase. Our results suggest that extracellular nucleotides, P2 receptors and adenosine transporters could be novel factors modulating EC energy metabolism through AMPK activation.
Materials and Methods
Reagents
Antibodies to human AMPK-α and phospho-AMPK-α (Thr-172) (P-AMPK) were purchased from Cell Signaling Technology (Beverly, MA). Rabbit anti-human phospho-ACC(Ser79) (P-ACC) was from Upstate Laboratories (San Diego, CA). Streptavidin-horseradish peroxidase (HRP)-conjugated antibody was from Amersham Biosciences Co. (Piscataway, NJ). Secondary donkey anti-rabbit-HRP-conjugated antibody was purchased from Pierce Biotechnology (Rockford, IL). Bisindolylmaleimide I (BIM) was from Calbiochem (La Jolla, CA). ATP, UTP, ADP, adenosine, inosine, hypoxanthine, 8-(sulfophenyl) theophylline (8-SPT), nitrobenzylthioinosine (NBTI), 2′,3′-O-(4-benzoylbenzoyl)-ATP (BzATP), adenosine-5′α,β-methylene diphosphate (AOPCP), 5′-amino-5′deoxyadenosine p-toluensulfonate (AMDA), erythro-9-(2-hydroxy-3-nonyl) adenosine hydrochloride (EHNA), 1,8-Naphthoylene benzimidazole-3-carboxylic acid (STO-609), 1-[N,O-bis(5-isoquinolinesulphonyl)-N -methyl-L-tyrosyl]-4-phenylpiperazine (KN62), 1,2-bis-(o-Aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetraacetoxy-Methyl ester (BAPTA-AM), 2-(4-Morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one Hydrochloride (LY294002), and wortmannin were from Sigma-Aldrich Co. (St. Louis, MO). Enhanced Chemiluminescence (ECL) kit was obtained from Perkin Elmer Life Science (Boston, MA).
Cell Culture
HUVEC (Cambrex, Walkersville, MD) were cultured in EGM-2 Bullet Kit medium containing human epidermal growth factor, hydrocortisone, human fibroblast growth factor, vascular endothelial growth factor, ascorbic acid, gentamicin, amphotericin-B, human insulin-like growth factor, heparin and 2% (v/v) fetal bovine serum, at 37°C in a 5% CO2 humidified air incubator. Confluent cells at passage 4 were used for all experiments. HeLa cells were grown in DMEM containing 10% fetal bovine serum.
Cell Treatment and Cell Lysates Preparation
Cells were incubated with ATP, ADP, UTP or adenosine (1 to 250 μmol/L) for various time periods (1 to 30 minutes). In some experiments cells were pre-incubated with inhibitors for 15 to 30 minutes. After incubation, cells were placed on ice, washed with ice-cold Tris-buffered saline (TBS) (20 mmol/L Tris, pH 7.5, 150 mmol/L NaCl), suspended in cell lysis buffer (20 mmol/L Tris, pH 7.5, 100 mmol/L NaCl, 1% (v/v) Nonidet P40, 1 mmol/L sodium orthovanadate, 100 mmol/L sodium fluoride, 2 μg/ml aprotinin, 1 μg/ml leupeptin and 1 mmol/L phenylmethylsulfonyl fluoride), scraped, incubated on ice for 20 minutes and centrifuged at ∼14,000xg for 5 minutes at 4°C. Supernatants were kept at -80°C until used for Western blot. Protein concentration was measured by the modified method of Lowry 17, using a detergent compatible protein assay kit (Bio-Rad, Hercules, CA).
Western Blot
Cell lysates (30 μg protein/well) were analyzed under reducing conditions by SDS-polyacrylamide gel electrophoresis performed according to Laemmli 18. Proteins were separated on 4-15% polyacrylamide gel and transferred to PVDF membrane by semi-dry electroblotting. Membranes were blocked with 5% (w/v) nonfat dry milk and probed with specific antibodies overnight at 4°C. Membranes were incubated with HRP-conjugated goat anti-rabbit IgG (diluted 1:5000) for 1 hour at room temperature. Total ACC was visualized with streptavidin-HRP (diluted 1:1000 in TBS buffer 0.4% Tween 20), which detects the biotin part in the ACC molecule 19. Proteins were detected with ECL followed by exposure to the autoradiography film. Immunoblot images were scanned and bands were densitometrically analyzed using ImageJ 1.33u software (NIH). The P-AMPK/total AMPK ratio was determined and results were expressed as percentage of the control (untreated cells) AMPK relative phosphorylation.
Measurement of intracellular nucleotides
Cells were harvested and levels of intracellular nucleotides were measured by high performance liquid chromatography (HPLC), using a C18 reverse-phase column, as described earlier20.
Statistical Analysis
Results are presented as means ± standard error. Data were analyzed by one-way analysis of variance (ANOVA) followed by the post-hoc Duncan multiple range test when F was significant. Concentration dependent effects were tested by regression analysis. Differences between groups were rated significant at a probability error (P) of less than 0.05.
Results
Extracellular Nucleotides and Adenosine Induce Phosphorylation and Activation of AMPK in HUVEC
We examined the effect of adenosine and extracellular nucleotides on phosphorylation of AMPK in HUVEC. Cells were incubated for 0 to 10 minutes in presence of ATP, ADP, UTP (100 μmol/L) or adenosine (25 μmol/L) and cell lysates were analyzed by Western blotting with antibodies to human P-AMPK-α (Thr-172) and total AMPK-α. We observed a transient, statistically significant increase of phosphorylation of AMPK induced by extracellular ATP, ADP, UTP and adenosine (Figure 1). The increase of AMPK phosphorylation was very rapid, reaching maximum stimulation after 1 minute for ATP (up to 3.5-fold), UTP (up to 3.0-fold) and ADP (up to 2.6-fold) and the P-AMPK levels were still significantly higher than control after 10 minutes incubation with all nucleotides. Adenosine-induced AMPK phosphorylation was slower than that observed with nucleotides, and was significantly increased after 5 minutes (2.1-fold).
Figure 1.
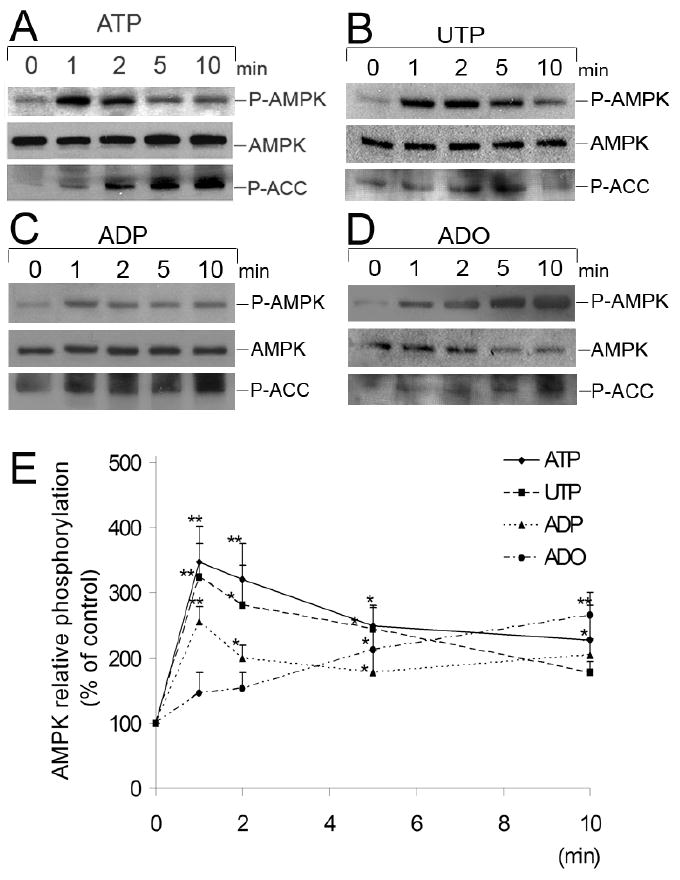
Time-course studies of phosphorylation of AMPK induced by nucleotides and adenosine in HUVEC. Representative Western blots of HUVEC stimulated by (A) ATP 100 μmol/L, (B) UTP 100 μmol/L, (C) ADP 100 μmol/L or (D) adenosine (ADO) (25 μmol/L) from 0 to 10 minutes. Cell lysates were immunoblotted with anti-phospho-AMPK (Thr-172) (P-AMPK), anti-AMPK-α and anti-P-ACC antibodies. (E) Results are expressed as percentage of the control (0 minutes) AMPK relative phosphorylation. Values are presented as means ± S.E.M of 3-5 independent experiments. *P<0.05, ** P< 0.01 compared to the control.
In addition, incubation of HUVEC for 5 minutes with varying concentrations of nucleotides (1 to 250 μmol/L) showed that ATP, ADP and UTP at 50 μmol/L and higher, induced phosphorylation of AMPK in a concentration-dependent manner [β = 0.849, P< 0.001; β= 0.751, P< 0.001; β= 0.714, P< 0.001, respectively] (Figure 2). Adenosine-dependent phosphorylation of AMPK was maximal at 25 μmol/L of adenosine (Figure 2).
Figure 2.
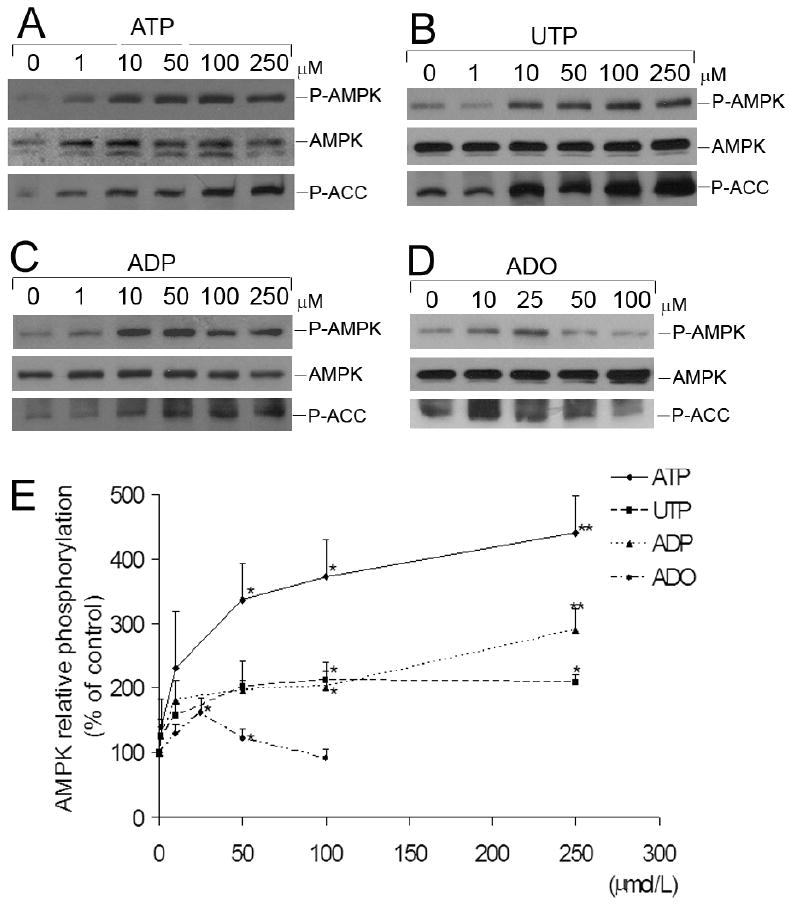
Concentration-dependence of nucleotide- and adenosine-induced phosphorylation of AMPK in HUVEC. Representative Western blots of HUVEC stimulated for 5 minutes with the indicated concentrations of (A) ATP, (B) UTP, (C) ADP or (D) adenosine (ADO), ranging from 0 to 250 μmol/L. Cell lysates were immunoblotted with anti-P-AMPK (Thr-172), anti-AMPK-α and anti-P-ACC antibodies. (E) Results are expressed as percentage of the control (0 μmol/L) AMPK relative phosphorylation. Values are means ± S.E.M of 3-4 independent experiments. *P<0.05, **P< 0.01 compared to control.
It is well established that phosphorylation of AMPK on Thr-172 is associated with AMPK activation 4. Active AMPK phosphorylates its various downstream targets, including ACC 6. Western blot analysis of lysates from HUVEC treated with ATP, ADP, UTP or adenosine demonstrated phosphorylation of ACC (Figure 3) and hence additionally confirmed activation of AMPK by nucleotides and adenosine.
Figure 3.
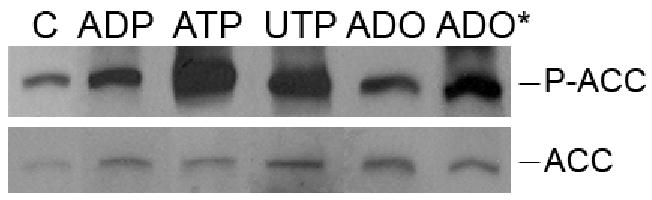
Extracellular nucleotides and adenosine induce phosphorylation of acetyl-CoA carboxylase (ACC). Western blot analysis of HUVEC stimulated by ADP (100 μmol/L), ATP (100 μmol/L), UTP (100 μmol/L) or adenosine (ADO) (25 μmol/L) for 5 minutes or ADO* for 30 minutes. Cell lysates were immunoblotted with anti-phospho-ACC (Ser-79) (P-ACC) or Streptavidin-HRP antibody (total ACC). Results are representative of 4-5 independent experiments.
HUVEC express various NTPDases, including NTPDase1 (described as CD39) that can hydrolyze ATP and ADP to AMP and UTP to UDP and UMP 21. However, extracellular AMP (97.5±15.5% of control; n=2, P= 0.718) or UDP (106±12.08% of control; n=5 P=0.549) (100 μmol/L, 5 minutes incubation) had no effect on phosphorylation of AMPK.
HUVEC also express 5′-nucleotidase, which can hydrolyze AMP to adenosine 22. To exclude the possible adenosine effect in nucleotide-induced AMPK activation, we inhibited 5′- nucleotidase with its specific inhibitor, adenosine-5′α,β-methylene diphosphate (AOPCP) 23. Pre-incubation of HUVEC with AOPCP (100 μmol/L, 15 minutes) did not reduce the nucleotide-induced phosphorylation of AMPK and ACC (Figure 4), indicating that this effect is in fact nucleotide-dependent.
Figure 4.
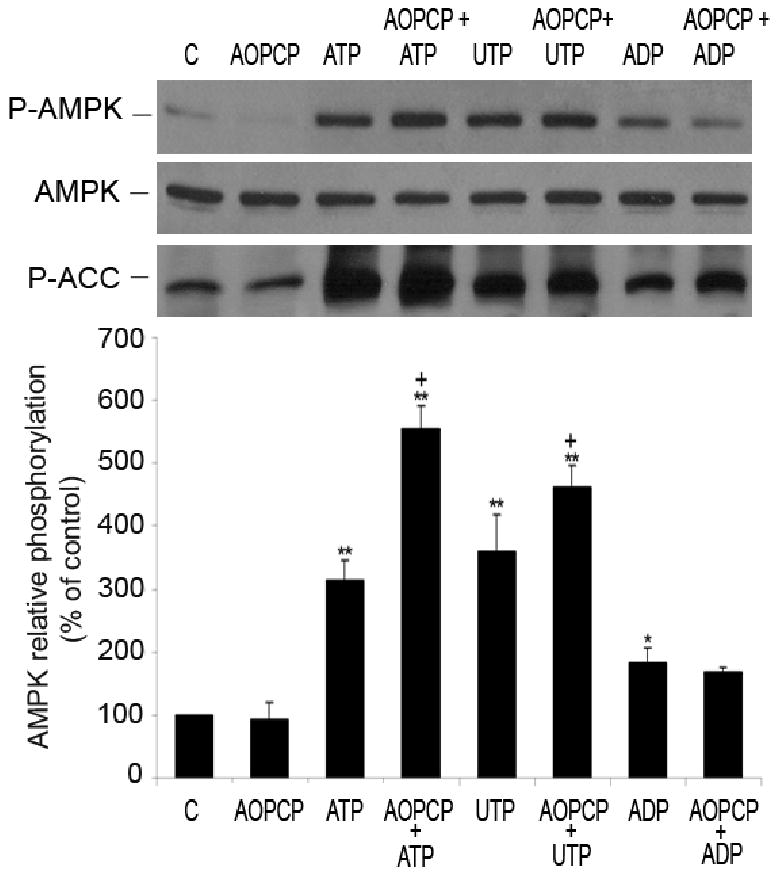
Nucleotide-induced phosphorylation of AMPK is not adenosine-dependent. Representative Western blot of HUVEC pre-treated for 15 minutes with inhibitor of 5′nucleotidase, AOPCP (100 μmol/L) and incubated with or without nucleotides (100 μmol/L, 5 minutes). Cell lysates were immunoblotted with anti-P-AMPK (Thr-172), anti-AMPK-α and anti-P-ACC antibodies. After quantification, values were statistically analyzed and are expressed as means ± S.E.M of 2-4 independent experiments. *P<0.05, **P< 0.01 compared to control, +P< 0.05 compared to non pre-incubated cells, treated with respective nucleotide.
Mechanism of AMPK activation can be linked to the increase in the intracellular AMP:ATP ratio. We measured levels of intracellular AMP and ATP by HPLC in EC stimulated with ADP (100 μmol/L) and adenosine (25 μmol/L) for 5 and 10 minutes. No statistically significant changes in the AMP:ATP ratio (∼1.3-fold increase) were observed (Figure 5), even though some tendency to increase in AMP level (50%) was visible for ADP (after 5 minutes) or adenosine (after 10 minutes).
Figure 5.
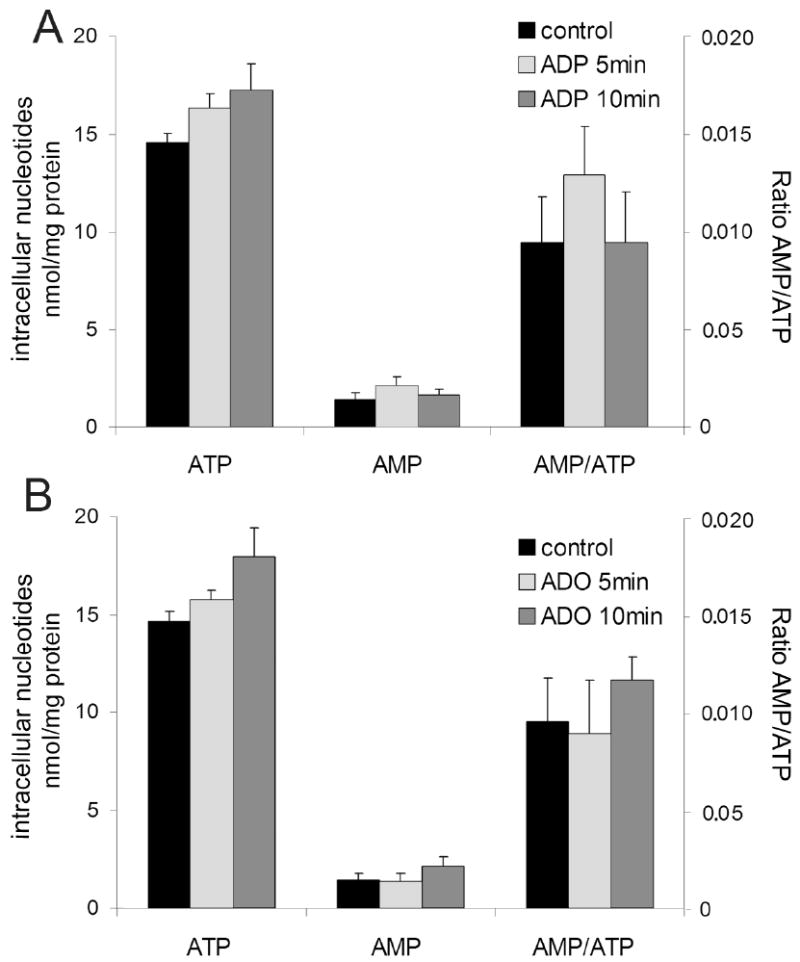
Changes in AMP:ATP ratio do not affect AMPK activation by extracellular nucleotides and adenosine. HUVEC were stimulated with (A) 100 μmol/L ADP or (B) 25 μmol/L adenosine for 5 and 10 minutes and concentrations of intracellular AMP and ATP were measured by HPLC. Data are expressed as means ± S.E.M of 3 independent experiments. P>0.05 compared to control.
Identification of P2 Receptors Involved in Nucleotide-Induced Phosphorylation of AMPK
It is well known that nucleotides exert their effects on cells through purinergic P2X and P2Y receptors and that EC express many subtypes of these receptors. To identify the receptors responsible for nucleotide-induced activation of AMPK, pharmacological analysis was done. As shown in Figures 1-3, ADP and UTP induced phosphorylation of AMPK and its downstream target, ACC. These data indicate that P2Y1, P2Y2 and/or P2Y4 receptors are involved in the activation of the AMPK signaling pathway. UDP had no effect on AMPK phosphorylation hence the involvement of the P2Y6 receptors can be excluded.
ATP significantly induced AMPK phosphorylation (Figures 1 and 2). However, ATP can activate a broad spectrum of P2 receptors expressed on HUVEC, including P2Y2, P2Y11 and all P2X receptors. Among P2X receptors, P2X7 is rather unique since it responds to high concentrations of ATP (mmolar range) 24,25. Therefore, to study the involvement of P2X7 receptors in AMPK activation, we used 1 mmol/L ATP and additionally, treated EC with KN62, the antagonist of P2X7 receptors 26. Even though high concentrations of ATP induced phosphorylation of AMPK, pre-incubation with KN62 (1 μmol/L, 20 minutes) did not affect the increase of P-AMPK and P-ACC levels (Figure 6), suggesting that P2X7 receptors are not involved in AMPK phosphorylation. Furthermore, we treated cells with BzATP, a potent agonist of P2X7 receptors, and a ligand for some other P2X and P2Y11 receptors 27-29. The treatment with BzATP did not trigger phosphorylation of AMPK and ACC (Figure 6) confirming that these subtypes of P2 receptors do not participate in ATP-induced activation of AMPK in HUVEC.
Figure 6.
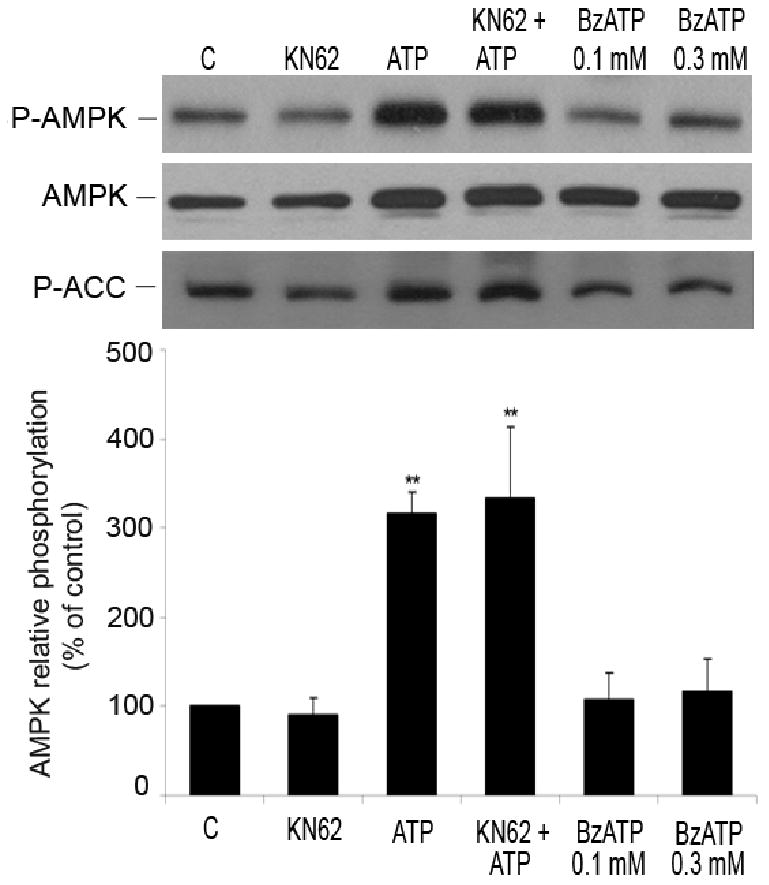
Role of P2X and P2Y11 receptors in AMPK phosphorylation. HUVEC were stimulated with ATP (1 mmol/L, 5 minutes) with and without pre-incubation with KN62 (1 μmol/L, 20 minutes), the antagonist of P2X7 receptor. Cells were also treated with BzATP (100 and 300 μmol/L, 5 minutes), an agonist for P2X and P2Y11 receptors. Cell lysates were analyzed by Western blotting using antibodies to P-AMPK (Thr-172), AMPK-α and P-ACC. Results obtained from 3 independent experiments are presented as means ± S.E.M. **P< 0.01 compared to control.
The data suggest that P2Y1, P2Y2 and/or P2Y4 receptors are involved in phosphorylation of AMPK in HUVEC, while P2Y6, P2Y11 and P2X receptors probably do not play a role in this activation.
Mechanism of Nucleotide-Induced Phosphorylation of AMPK
One of the first indicators of P2 receptor stimulation is an increase in [Ca2+]i Therefore, we investigated the role of Ca2+ in nucleotide-induced AMPK phosphorylation. HUVEC were pre-incubated with a chelator of intracellular Ca2+, BAPTA-AM (20 minutes, 10 μmol/L), followed by stimulation with 100 μmol/L of ATP or UTP for 1 minute. BAPTA-AM significantly attenuated nucleotide-induced phosphorylation of AMPK and ACC (Figure 7), suggesting that Ca2+ plays an important role in this activation.
Figure 7.
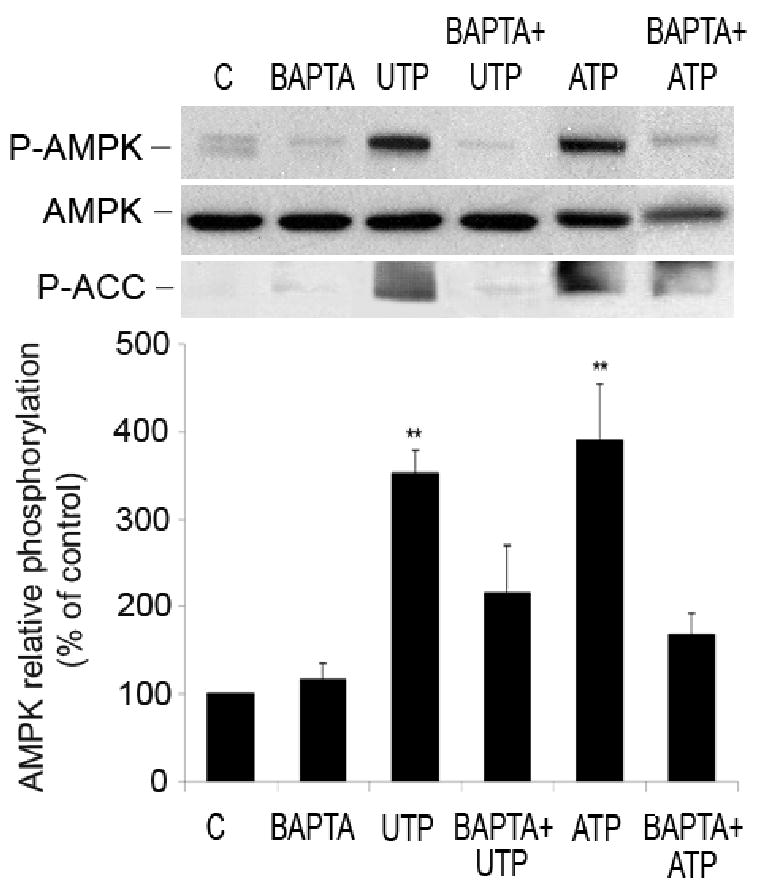
Phosphorylation of AMPK induced by nucleotides depends on Ca2+ originating from intracellular stores. Representative Western blots showing the effect of BAPTA-AM on nucleotide-induced AMPK and ACC phosphorylation. HUVEC were pre-treated with BAPTA-AM (20 minutes, 10 μmol/L) and stimulated with ATP and UTP (1 minute, 100 μmol/L). Cell lysates were immunoblotted with antibodies to P-AMPK, AMPK-α and P-ACC. After quantification, values were statistically analyzed and are expressed as means ± S.E.M of 3-6 independent experiments. *P < 0.05 and **P < 0.01 compared to untreated cells.
PI3K has been demonstrated as an up-stream activator of AMPK 30. Moreover, we have shown earlier that PI3K pathway was involved in extracellular nucleotide-induced EC migration 31. Thus, to establish the role of PI3K in nucleotide-induced activation of AMPK, two inhibitors of PI3K, LY294002 and wortmannin, were applied. Cell pre-incubation with LY294002 (LY, 10 μmol/L, 20 minutes) or wortmannin (W, 100 nmol/L, 20 minutes) prior to stimulation with ATP, UTP or ADP, had no significant effects on P-AMPK levels (ATP: 329±25, LY+ATP: 278±31, W+ATP: 340±10, UTP: 298±14, LY+UTP: 270±21, W+UTP: 298±17, ADP: 205±22, LY+ADP: 252±24, W+ADP: 221±38, results expressed as % of control ± SEM, n=3-6 P=0.179).
Stimulation of G-protein coupled P2Y receptors is linked to PKC activation. Furthermore, it has been shown that ischemic preconditioning induced AMPK activation in the PKC-dependent manner 4. To evaluate a role of PKC in the nucleotide-induced activation of AMPK we used an inhibitor of PKC, bisindolylmaleimide I (BIM). Preincubation of EC with BIM (20 minutes, 100 μmol/L) did not change nucleotide-induced phosphorylation of AMPK (data not shown), indicating that PKC is not involved in the purinergic activation of AMPK.
Thus, we conclude that nucleotide-induced activation of AMPK in HUVEC is PI3K-and PKC-independent, however increase in [Ca2+]i is crucial for this pathway. Ca2+ interacts with calmodulin and activates Ca2+/calmodulin-dependent kinase kinase (CaMKK) and Ca2+/calmodulin-dependent kinase II (CaMK II), potential upstream regulators of AMPK, both expressed in HUVEC 32 33,34. KN62 (10 μmol/L), an inhibitor of CaMK II had no effect on nucleotide-induced phosphorylation of AMPK (data not shown), while STO-609, a specific inhibitor of CAMKK (10 μmol/L), attenuated this response (and ACC phosphorylation) (Figure 8), indicating that CaMKK is the upstream kinase involved in extracellular nucleotides-mediated activation of AMPK.
Figure 8.
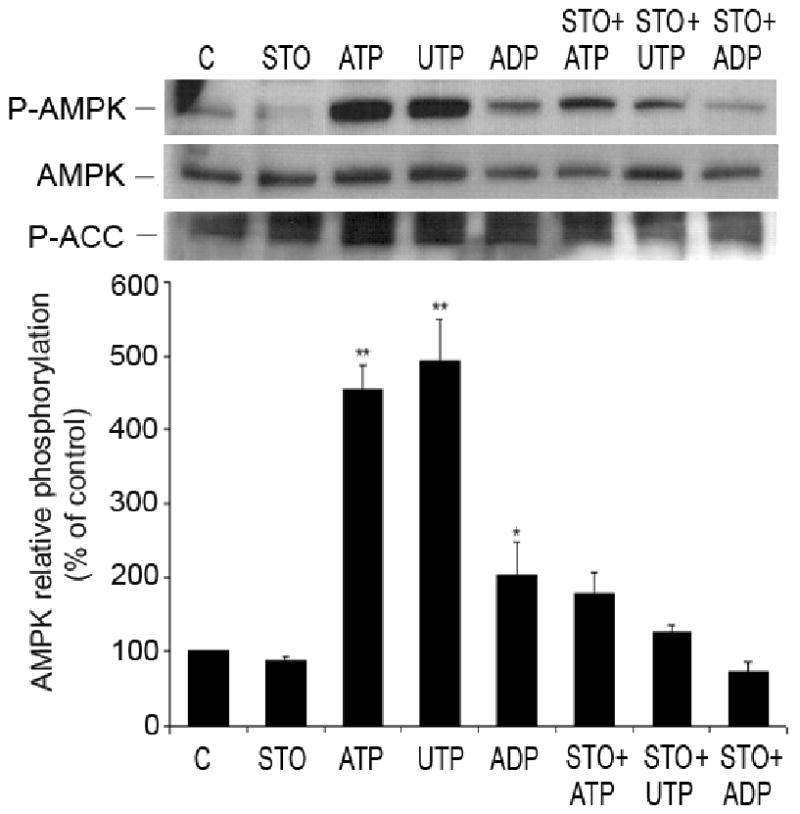
Nucleotide-induced phosphorylation of AMPK is mediated by CAMKK in HUVEC. Representative Western blot of HUVEC pre-treated for 30 minutes with STO-609 (1μg/μL), inhibitor of CAMKK, and incubated with or without nucleotides (100 μmol/L, 1 minute). Cell lysates were immunoblotted with anti-P-AMPK (Thr-172), anti-AMPK-α and anti-P-ACC antibodies. After quantification, values were statistically analyzed and are expressed as means ± S.E.M of 3 independent experiments. *P<0.05, **P< 0.01 compared to control.
Finally, to investigate a role of LKB1 in nucleotide-induced phosphorylation of AMPK, we used HeLa cells that are deficient in LKB1 35,36. All studied nucleotides (ATP, UTP and ADP) were still able to stimulate the AMPK phosphorylation even in the absence of LKB1. The nucleotide effects on AMPK were also inhibited by pre-incubation of HeLa cells with STO-609 (Figure 9). These results probably exclude LKB1 as an upstream kinase and confirm the involvement of CaMKK in nucleotide-induced AMPK phosphorylation.
Figure 9.
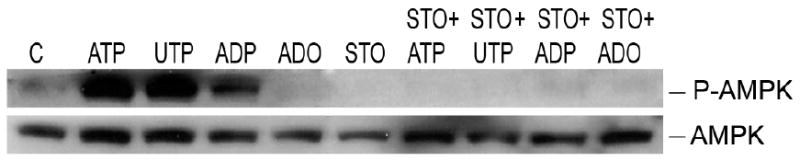
Role of LKB1 in nucleotide and adenosine-mediated activation of AMPK. HeLa cells were incubated with extracellular nucleotides, ATP, UTP and ADP (100 ìmol/L; 2 minutes) or adenosine (25 ìmol/L; 5 minutes). Representative Western blots show cell lysates immunoblotted with antibodies to P-AMPK (Thr-172) and AMPK-α. Representative Western blots of 3 independent experiments.
Mechanism of Adenosine-Induced Phosphorylation of AMPK
As we demonstrated in Figures 1 and 2, adenosine induced phosphorylation of AMPK. EC are known to have a very active adenosine metabolism, characterized by a large capacity for uptake and release of the nucleoside. Extracellular adenosine can be metabolized to inosine and hypoxanthine. To evaluate the possible effect of adenosine metabolites, HUVEC were incubated for 5 minutes with 25 μmol/L inosine or hypoxanthine. Neither of them had an effect on AMPK phosphorylation (inosine: 118.3±16.84%, n=3, P=0.344; hypoxanthine: 100.3±14.4% of control, n=3, P= 0.705). These data suggest that the observed adenosine-induced AMPK phosphorylation is indeed adenosine specific.
Extracellular adenosine can exert its effect on cells either by P1 receptors or after entering the cells, which is mediated by adenosine transporters 37. To identify the mechanism of adenosine-induced AMPK phosphorylation, HUVEC were pre-treated for 20 minutes with a non-selective antagonist of P1 receptors, 8-SPT (30 μmol/L) 38 and with an analogue of adenosine, NBTI (10 μmol/L), which is a competitive inhibitor of the equilibrative adenosine transporter system 39. 8-SPT did not affect adenosine-induced phosphorylation of AMPK and ACC while NBTI (Figure 10) and dipyridamole (data not shown) significantly prevented it. These results indicate that adenosine-induced AMPK phosphorylation depends on adenosine uptake via nucleoside transporters.
Figure 10.
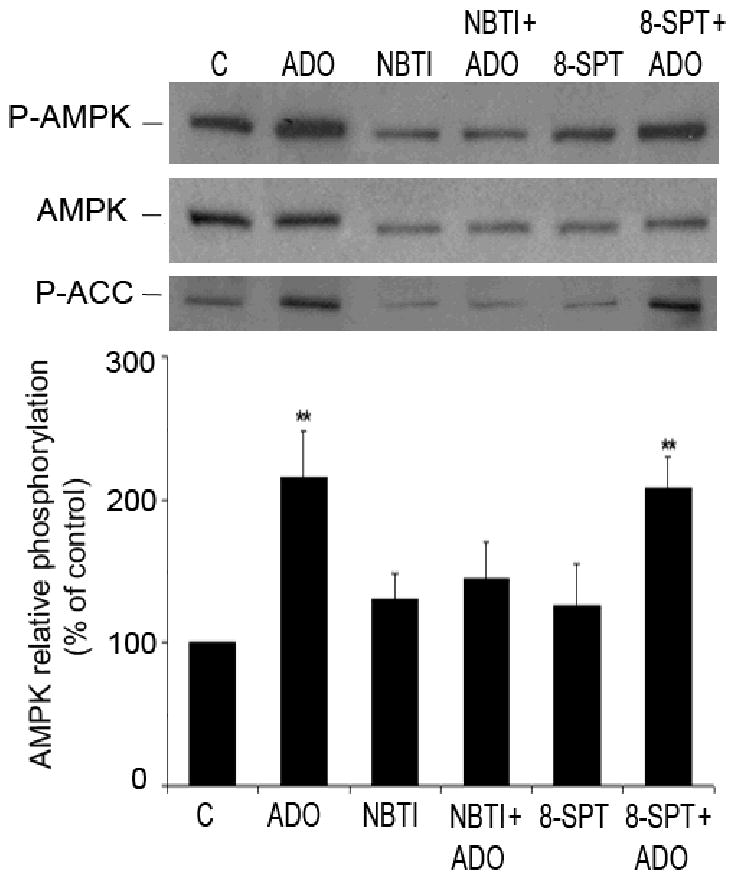
Adenosine transporters and not P1 receptors are involved in adenosine-induced phosphorylation of AMPK. Representative Western blots of HUVEC pre-treated for 20 minutes with either an antagonist of P1 receptors, 8-SPT (30 μmol/L), or inhibitor of adenosine transporter NBTI (10 μmol/L), and incubated with adenosine (ADO) (25 μmol/L, 5 minutes). Cell lysates were immunoblotted with antibodies to P-AMPK (Thr-172), AMPK-α and P-ACC. After quantification, values were statistically analyzed and are expressed as means ± S.E.M of 5-7 independent experiments. **P< 0.01 compared to the control.
Intracellular adenosine can be metabolized by either adenosine deaminase that converts adenosine to inosine or adenosine kinase that converts adenosine to AMP. Inhibition of adenosine kinase with AMDA (10 ìmol/L, 30 minutes) completely blocked AMPK activation and ACC phosphorylation, while inhibition of adenosine deaminase with EHNA (10 ìmol/L, 30 minutes) did not affect adenosine-induced AMPK activation (Figure 11). Therefore, in adenosine-initiated pathway of AMPK phosphorylation, the metabolism of intracellular adenosine to AMP seems to play an important role.
Figure 11.
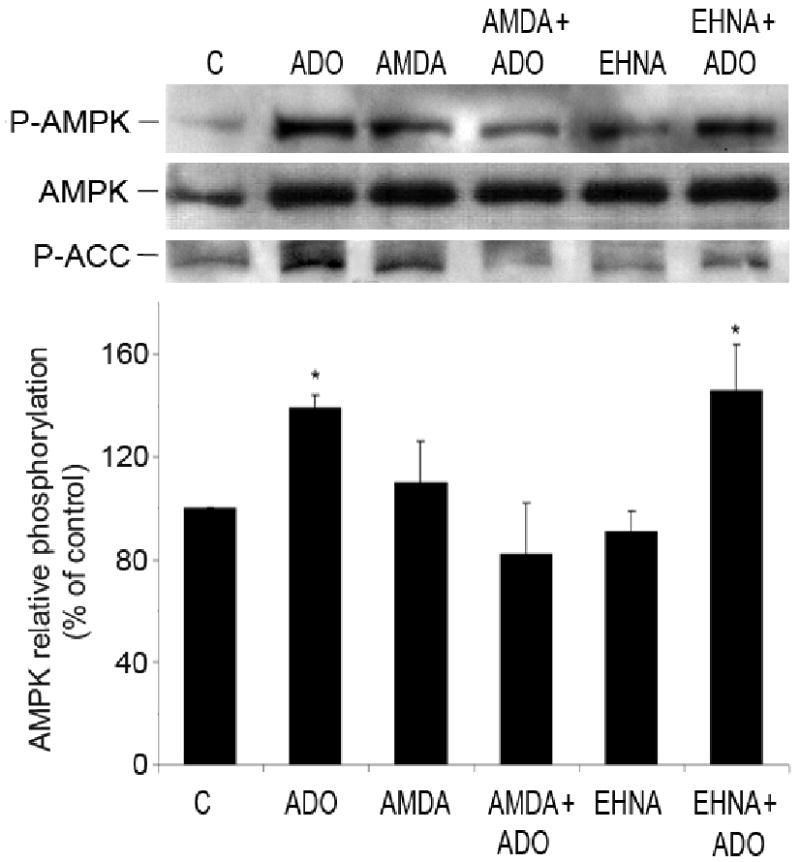
Intracellular metabolism of adenosine to AMP is essential for phosphorylation of AMPK. Representative Western blots of HUVEC pre-treated for 30 minutes with either an inhibitor of adenosine kinase, AMDA (10 μmol/L) or inhibitor of adenosine deaminase, EHNA (10 μmol/L), and stimulated with adenosine (ADO) (25 μmol/L, 5 minutes). Cell lysates were immunoblotted with anti-P-AMPK (Thr-172), anti-AMPK-α and anti-P-ACC antibodies. Values are means ± S.E.M of 3-4 independent experiments. *P<0.05, compared to control.
In addition, we examined a role of possible upstream activators of AMPK, in adenosine-induced AMPK phosphorylation. Wortmannin and LY294002 (inhibitors of PI3K), KN62 (10 μM; CaMK II inhibitor) or STO-609 (1 μg/mL; CaMKK inhibitor) did not change the effect of adenosine (25 μmol/L, 5 minutes) on AMPK phosphorylation in HUVEC (Figure 12). Therefore we conclude that PI3K, CaMK II and CaMKK were not mediators of adenosine-initiated pathway of AMPK activation.
Figure 12.
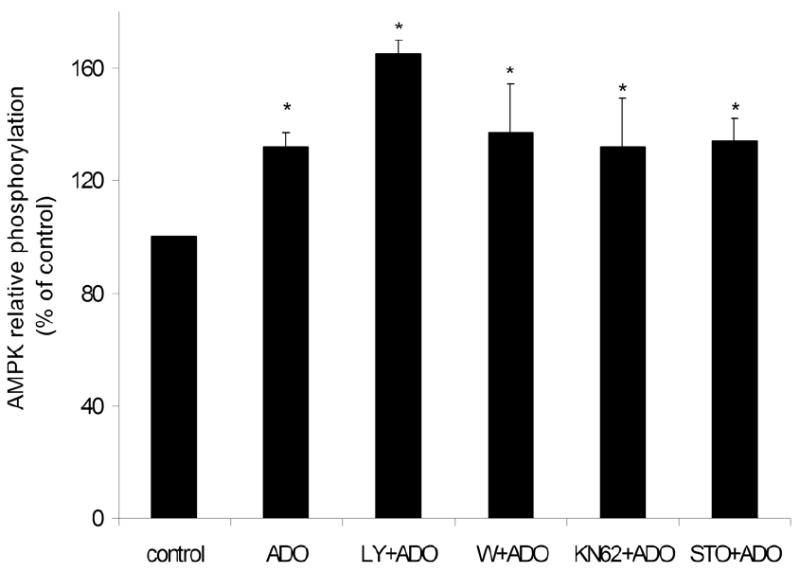
Adenosine-induced AMPK phosphorylation is independent on CaMK II, CaMKK and PI3K. HUVEC were pre-treated for 20 minutes with inhibitors of PI3K (LY294002, 10 μmol/L or wortmannin (W); 100 nmol/L), CaMK II (KN62, 10 μmol/L) and CaMKK (STO-609, 1 μg/μL) and stimulated with adenosine (ADO) (25 μmol/L, 5 minutes). Cell lysates were immunoblotted with anti-P-AMPK (Thr-172) and anti-AMPK-α antibodies and the bands were densitometrically quantified. Results are expressed as means ± S.E.M of 3-4 independent experiments. *P<0.05, compared to control.
In many cell systems LKB1 has been described as the AMPK upstream kinase 35. Since nucleotide-induced phosphorylation of AMPK is calcium dependent while adenosine effect on AMPK is not, as observed by experiments with BAPTA-AM (data not shown), we hypothesized that while CaMKK mediates nucleotide activation of AMPK, LKB1 might be involved in adenosine-induced pathway of AMPK phosphorylation. To test this hypothesis HeLa cells (deficient in LKB1) were examined. Adenosine (25 μmol/L) did not induce AMPK phosphorylation in these cells (Figure 9) suggesting that LKB1 could be involved in adenosine-initiated pathway of AMPK activation.
Discussion
In the present study, we show for the first time that in HUVEC, extracellular nucleotides and adenosine independently induce phosphorylation of AMPK via P2Y receptors and nucleoside transporters, respectively.
AMPK can be activated by the allosteric action of AMP and by phosphorylation of the Thr-172 on the catalytic α subunit by up-stream kinases 5. Extracellular nucleotides and adenosine induced phosphorylation of AMPK on Thr-172 in a time- and concentration-dependent manner (Figures 1 and 2) and thus activated it. This functional AMPK activation has been additionally validated by experiments showing phosphorylation of the AMPK downstream target, ACC (Figures 1-10). While nucleotides significantly phosphorylated AMPK at concentrations of 50 μmol/L and higher, the effect of adenosine was maximal at 25 μmol/L and decreased at 50 and 100 μmol/L. Adenosine that is transported into cells can be metabolized by different enzymes, including adenosine kinase and adenosine deaminase. At lower concentrations, adenosine is converted by adenosine kinase to AMP. However, adenosine kinase is substrate-inhibited, thus adenosine at concentrations exceeding several μmolar is metabolized via adenosine deaminase to inosine 40, which has no effect on AMPK activation. Hence, our data are consistent with the kinetics of adenosine metabolism.
Extracellular nucleotides are sequentially hydrolyzed by NTPDases and 5′-nucleotidase to adenosine, which can also activate AMPK. However, phosphorylation of AMPK induced by ATP, UTP and ADP was not diminished the inhibitor of 5′-nucleotidase, AOPCP (Figure 4), therefore we conclude that extracellular nucleotides and not adenosine, originating from nucleotide hydrolysis, activate AMPK.
Extracellular nucleotides do not cross the cell membrane, but exert their biological action as extracellular signaling molecules through ubiquitously expressed purinergic P2X and P2Y receptors 9. Our data showed a role for P2Y1 and P2Y2 and/or P2Y4 receptors in the activation of AMPK since ADP and UTP, the primary agonists of these receptors, induced phosphorylation of AMPK. Lack of response to UDP suggested that P2Y6 receptors were not involved in AMPK activation. Experiments with BzATP and KN62, an agonist and a noncompetitive antagonist of P2X7 receptors, respectively, excluded the role of P2X7 receptor in phosphorylation of AMPK (Figure 6). BzATP has been recognized as a selective agonist of P2X7 receptors, however it can also activate P2Y11 receptors, as well as other P2X receptors 27-29. Lack of AMPK phosphorylation by BzATP excluded P2X and P2Y11 receptors from the list of receptors, potentially involved in the AMPK activation.
We have previously shown that in HUVEC, extracellular nucleotide-induced increases in [Ca2+]i were linked to P2Y receptors 31. Here, we observed significant attenuation of ATP and UTP-induced phosphorylation of AMPK by BAPTA-AM (Figure 7), which indicates that Ca2+ released from the endoplasmic reticulum is essential for the activation of AMPK. These results confirm that P2Y (and not P2X) receptors are indeed responsible for nucleotide-induced activation of AMPK in HUVEC.
The tumor-suppressor kinase, LKB1, as well as CaMKK have been identified in several studies as the most important upstream activators of AMPK 33,41. Other kinases, including PI3K 30, PKC 42 and CaMK II 34 have been identified as potential upstream activators of AMPK. However by using their pharmacological inhibitors we excluded these three kinases as upstream activators of AMPK in purinergic signaling. We identified CaMKK as the kinase involved in nucleotide-mediated AMPK activation in EC (Figure 8), while LKB1 seems not to participate in AMPK activation with extracellular nucleotides. As shown in Figure 9, extracellular nucleotides induced AMPK phosphorylation in HeLa cells, which are deficient in LKB1.
The pathway of adenosine-induced activation of AMPK differs from the one described for extracellular nucleotides. We established that extracellular adenosine, and not its metabolites, inosine or hypoxanthine, can stimulate activation of AMPK and phosphorylation of ACC (Figure 1-3). Furthermore, we showed that, in HUVEC, the effect of adenosine was not mediated by P1 receptors, but by adenosine uptake facilitated by nucleoside equilibrative transporters (Figure 10). In addition, our data strongly suggest that a conversion of adenosine to AMP is important for AMPK activation (Figure 11). We hypothesize that AMP generated from adenosine by adenosine kinase, allosterically activates and modifies AMPK so it is more prone to further activation by the upstream kinase.
Our additional experiments showed that adenosine-induced phosphorylation of AMPK, was independent of CaMKK, as well as PI3K (Figure 12). Therefore, we examined the role of LKB1, another AMPK upstream kinase, which appears to regulate AMPK activity under energy stress conditions 41. In HeLa cells, that do not express LKB1, adenosine did not phosphorylate AMPK (Figure 10), suggesting that LKB1 might be an important component of adenosine signaling pathway leading to phosphorylation of AMPK. This indirect evidence should be confirmed by further studies.
Initially, the mechanism of AMPK activation was associated with an increase in the AMP:ATP ratio that originated from ATP depletion 43. Further investigations revealed that certain stimuli, including osmotic shock, pH changes and an anti-diabetic drug, metformin, might induce AMPK activation that is not dependent on changes in the AMP and ATP levels 44. We expected that the mechanism of AMPK activation induced by adenosine, which is converted to AMP within the cell, would be dependent on the changes in the AMP:ATP ratio. However, this was not confirmed by our HPLC data (Figure 5). We do observe some tendency of AMP levels to increase in response to adenosine, however this is assisted by an increase in the ATP level. Such increase in the concentration of intracellular ATP was observed earlier in cells treated with extracellular nucleotides or adenosine 45,46. Therefore, the ratio of AMP to ATP does not change significantly, comparing to other studies showing 6 or 25-fold increase in the AMP:ATP ratio 47,48. We propose that this increasing AMP level may affect AMPK activation, and that the actual level of AMP is more important for activation of AMPK than the AMP:ATP ratio 49. In our experiments, AMP levels were measured in total cell lysates. However, the concentration of newly generated AMP might increase in some cellular compartments where AMPK activation takes place, and this could be a mechanism of adenosine-mediated AMPK activation. We hypothesize that the mechanism of adenosine-induced activation of AMPK is linked to LKB1, with possible effect of an increase in the intracellular AMP level.
There is also some (statistically insignificant) increase in the AMP level and in the AMP:ATP ratio in EC activated with ADP (Figure 5). We assume that the changes in intracellular nucleotide levels observed after incubation with ADP might be, in fact, related to adenosine, originating from hydrolysis of ADP. This hypothesis is supported by published data 45,46, showing that inhibitor of adenosine transporter, dipyridamole, entirely blocks changes in the intracellular nucleotide levels in response to extracellular nucleotides.
AMPK can play an important role in the endothelium associated with regulation of EC metabolism and angiogenesis and can protect the endothelium from various stresses, including high levels of glucose and fatty acids. It regulates fatty acid metabolism in EC and therefore is involved in the cellular energy homeostasis 50. AMPK also induces NO generation, enhances the response to insulin and protects EC from hyperglicemia-induced apoptosis 51,52.
AMPK has been shown to be activated by hypoxia and AMPK signaling was identified as a new regulator of angiogenesis that is specifically required for endothelial cell migration and differentiation under conditions of hypoxia 53. Other study indicates that AMPK activated under hypoxia can be involved in co-regulation of hypoxia induced factor-1 (HIF-1), a transcription factor that is critical for hypoxic induction of physiologically important genes, as well as secretion of vascular endothelial growth factor (VEGF) and glucose uptake 54. Further studies on the AMPK role in physiology and patho-physiology of the endothelium seem to be of great importance.
In summary, our findings demonstrate two distinct, but converging pathways of AMPK activation in HUVEC: one induced by extracellular nucleotides, linked to P2Y1, P2Y2 and/or P2Y4 receptors, dependent on Ca2+ and CaMKK, and the second pathway, induced by adenosine uptake with the involvement of adenosine transporters, followed by the generation of (intracellular) AMP, and activation of AMPK with LKB1 as a putative upstream kinase. In an in vivo situation, nucleotides are finally hydrolyzed to adenosine. Therefore, the two mechanisms of AMPK activation, nucleotide and adenosine-dependent, can operate at the same time. Extracellular nucleotides and adenosine whose local concentrations under pathophysiological conditions can be substantially increased, could play an important role in restoring the energy balance by AMPK activation that modulates glucose uptake and fatty acid metabolism. We propose that P2Y receptors and nucleoside transporters could be novel targets for a pharmacological intervention to regulate AMPK and hence EC metabolism and function.
Acknowledgments
This work was funded by a grant # HL66167 from the NIH.
Contributor Information
Cleide Gonçalves da Silva, Email: cdasilva@bidmc.harvard.edu, Beth Israel Deaconess Medical Center, Harvard Medical School, 99 Brookline Avenue, Boston, MA 02215 Tel: 617 632 0883; Fax: 617 632 0880.
Robert Jarzyna, Email: rjarzyna@biol.uw.edu.pl, Beth Israel Deaconess Medical Center, Harvard Medical School, 99 Brookline Avenue, Boston, MA 02215 Tel: 617 632 0883; Fax: 617 632 0880 Warsaw University, Miecznikowa 1, Warsaw, Poland Tel: 22 554 3204; Fax: 22 554 3221.
Anke Specht, Email: anke.specht@web.de, Beth Israel Deaconess Medical Center, Harvard Medical School, 99 Brookline Avenue, Boston, MA 02215; Tel: 617 632 0883; Fax: 617 632 0880.
Elzbieta Kaczmarek, Email: ekaczmar@bidmc.harvard.edu, Beth Israel Deaconess Medical Center, Department of Medicine, Harvard Medical School, 99 Brookline Avenue, Boston, MA 02215 Tel: 617 632 0827; Fax: 617 632 0880.
References
- 1.Hardie DG, Salt IP, Hawley SA, Davies SP. AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J. 1999;338(Pt 3):717–22. [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie DG, Salt IP, Davies SP. Analysis of the role of the AMP-activated protein kinase in the response to cellular stress. Methods Mol Biol. 2000;99:63–74. doi: 10.1385/1-59259-054-3:63. [DOI] [PubMed] [Google Scholar]
- 3.Daniel T, Carling D. Expression and regulation of the AMP-activated protein kinase-SNF1 (sucrose non-fermenting 1) kinase complexes in yeast and mammalian cells: studies using chimaeric catalytic subunits. Biochem J. 2002;365:629–38. doi: 10.1042/BJ20020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–87. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 5.Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345(Pt 3):437–43. [PMC free article] [PubMed] [Google Scholar]
- 6.Dagher Z, Ruderman N, Tornheim K, Ido Y. The effect of AMP-activated protein kinase and its activator AICAR on the metabolism of human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1999;265:112–5. doi: 10.1006/bbrc.1999.1635. [DOI] [PubMed] [Google Scholar]
- 7.Gordon EL, Pearson JD, Slakey LL. The hydrolysis of extracellular adenine nucleotides by cultured endothelial cells from pig aorta. Feed-forward inhibition of adenosine production at the cell surface. J Biol Chem. 1986;261:15496–507. [PubMed] [Google Scholar]
- 8.Burnstock G, Kennedy C. Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol. 1985;16:433–40. doi: 10.1016/0306-3623(85)90001-1. [DOI] [PubMed] [Google Scholar]
- 9.Dubyak GR, el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Karlsson L, Moses S, Hultgardh-Nilsson A, Andersson M, Borna C, Gudbjartsson T, Jern S, Erlinge D. P2 receptor expression profiles in human vascular smooth muscle and endothelial cells. J Cardiovasc Pharmacol. 2002;40:841–53. doi: 10.1097/00005344-200212000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Burnstock G. Introduction: P2 receptors. Curr Top Med Chem. 2004;4:793–803. doi: 10.2174/1568026043451014. [DOI] [PubMed] [Google Scholar]
- 12.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–92. [PubMed] [Google Scholar]
- 13.von Kugelgen I, Wetter A. Molecular pharmacology of P2Y-receptors. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:310–23. doi: 10.1007/s002100000310. [DOI] [PubMed] [Google Scholar]
- 14.White PJ, Webb TE, Boarder MR. Characterization of a Ca2+ response to both UTP and ATP at human P2Y11 receptors: evidence for agonist-specific signaling. Mol Pharmacol. 2003;63:1356–63. doi: 10.1124/mol.63.6.1356. [DOI] [PubMed] [Google Scholar]
- 15.Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–22. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- 16.Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
- 17.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Lopaschuk GD, Witters LA, Itoi T, Barr R, Barr A. Acetyl-CoA carboxylase involvement in the rapid maturation of fatty acid oxidation in the newborn rabbit heart. J Biol Chem. 1994;269:25871–8. [PubMed] [Google Scholar]
- 20.Picher M, Sevigny J, D'Orleans-Juste P, Beaudoin AR. Hydrolysis of P2-purinoceptor agonists by a purified ectonucleotidase from the bovine aorta, the ATP-diphosphohydrolase. Biochem Pharmacol. 1996;51:1453–60. doi: 10.1016/0006-2952(96)00086-x. [DOI] [PubMed] [Google Scholar]
- 21.Koziak K, Sevigny J, Robson SC, Siegel JB, Kaczmarek E. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb Haemost. 1999;82:1538–44. [PubMed] [Google Scholar]
- 22.Niemela J, Henttinen T, Yegutkin GG, Airas L, Kujari AM, Rajala P, Jalkanen S. IFN-alpha induced adenosine production on the endothelium: a mechanism mediated by CD73 (ecto-5′-nucleotidase) up-regulation. J Immunol. 2004;172:1646–53. doi: 10.4049/jimmunol.172.3.1646. [DOI] [PubMed] [Google Scholar]
- 23.Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Moe SJ, Carnes DK, Mrsny RJ, Madara JL. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J Clin Invest. 1997;99:2588–601. doi: 10.1172/JCI119447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–6. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- 25.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–8. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 26.Burnstock G, Williams M. P2 purinergic receptors: modulation of cell function and therapeutic potential. J Pharmacol Exp Ther. 2000;295:862–9. [PubMed] [Google Scholar]
- 27.Liu C, Mather S, Huang Y, Garland CJ, Yao X. Extracellular ATP facilitates flow-induced vasodilatation in rat small mesenteric arteries. Am J Physiol Heart Circ Physiol. 2004;286:H1688–95. doi: 10.1152/ajpheart.00576.2003. [DOI] [PubMed] [Google Scholar]
- 28.Boeynaems JM, Communi D, Gonzalez NS, Robaye B. Overview of the P2 receptors. Semin Thromb Hemost. 2005;31:139–49. doi: 10.1055/s-2005-869519. [DOI] [PubMed] [Google Scholar]
- 29.Inoue K, Denda M, Tozaki H, Fujishita K, Koizumi S. Characterization of multiple P2X receptors in cultured normal human epidermal keratinocytes. J Invest Dermatol. 2005;124:756–63. doi: 10.1111/j.0022-202X.2005.23683.x. [DOI] [PubMed] [Google Scholar]
- 30.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem. 2003;278:34003–10. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 31.Kaczmarek E, Erb L, Koziak K, Jarzyna R, Wink MR, Guckelberger O, Blusztajn JK, Trinkaus-Randall V, Weisman GA, Robson SC. Modulation of endothelial cell migration by extracellular nucleotides: involvement of focal adhesion kinase and phosphatidylinositol 3-kinase-mediated pathways. Thromb Haemost. 2005;93:735–42. doi: 10.1267/THRO05040735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stagliano NE, Healy A, Acton SL, Galvin KM, Donoghue MA, Rodrigue-Way A, Tomlinson JE. Methods and compositions for treating cardiovascular disease using 1722, 10280, 59917, 85553, 10653, 9235, 21668, 17794, 2210, 6169, 10102, 21061, 17662, 1468, 12282, 6350, 9035, 1820, 23652, 7301, 8925, 8701, 3533, 9462, 9123, 12788, 17729, 65552, 1261. 2005 [Google Scholar]
- 33.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–6. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 34.Wright DC, Hucker KA, Holloszy JO, Han DH. Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes. 2004;53:330–5. doi: 10.2337/diabetes.53.2.330. [DOI] [PubMed] [Google Scholar]
- 35.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–8. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 36.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stiles GL. Adenosine receptors. J Biol Chem. 1992;267:6451–4. [PubMed] [Google Scholar]
- 38.Coney AM, Marshall JM. Role of adenosine and its receptors in the vasodilatation induced in the cerebral cortex of the rat by systemic hypoxia. J Physiol. 1998;509(Pt 2):507–18. doi: 10.1111/j.1469-7793.1998.507bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thorn JA, Jarvis SM. Adenosine transporters. Gen Pharmacol. 1996;27:613–20. doi: 10.1016/0306-3623(95)02053-5. [DOI] [PubMed] [Google Scholar]
- 40.Archer S, Juranka PF, Ho JH, Chan VL. An analysis of multiple mechanisms of adenosine toxicity in baby hamster kidney cells. J Cell Physiol. 1985;124:226–32. doi: 10.1002/jcp.1041240209. [DOI] [PubMed] [Google Scholar]
- 41.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishino Y, Miura T, Miki T, Sakamoto J, Nakamura Y, Ikeda Y, Kobayashi H, Shimamoto K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res. 2004;61:610–9. doi: 10.1016/j.cardiores.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 43.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–20. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 44.Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–32. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 45.Andreoli SP, Liechty EA, Mallett C. Exogenous adenine nucleotides replete endothelial cell adenosine triphosphate after oxidant injury by adenosine uptake. J Lab Clin Med. 1990;115:304–13. [PubMed] [Google Scholar]
- 46.Lasso de la Vega MC, Terradez P, Obrador E, Navarro J, Pellicer JA, Estrela JM. Inhibition of cancer growth and selective glutathione depletion in Ehrlich tumour cells in vivo by extracellular ATP. Biochem J. 1994;298(Pt 1):99–105. doi: 10.1042/bj2980099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol. 1994;4:315–24. doi: 10.1016/s0960-9822(00)00070-1. [DOI] [PubMed] [Google Scholar]
- 48.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–55. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 49.Witters LA, Kemp BE, Means AR. Chutes and Ladders: the search for protein kinases that act on AMPK. Trends Biochem Sci. 2006;31:13–6. doi: 10.1016/j.tibs.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 50.Dagher Z, Ruderman N, Tornheim K, Ido Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ Res. 2001;88:1276–82. doi: 10.1161/hh1201.092998. [DOI] [PubMed] [Google Scholar]
- 51.Ruderman NB, Cacicedo JM, Itani S, Yagihashi N, Saha AK, Ye JM, Chen K, Zou M, Carling D, Boden G, Cohen RA, Keaney J, Kraegen EW, Ido Y. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans. 2003;31:202–6. doi: 10.1042/bst0310202. [DOI] [PubMed] [Google Scholar]
- 52.Ido Y, Carling D, Ruderman N. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes. 2002;51:159–67. doi: 10.2337/diabetes.51.1.159. [DOI] [PubMed] [Google Scholar]
- 53.Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–6. doi: 10.1074/jbc.M300643200. [DOI] [PubMed] [Google Scholar]
- 54.Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278:39653–61. doi: 10.1074/jbc.M306104200. [DOI] [PubMed] [Google Scholar]