

Abstract
Dystrophic calcifications often occur after injury, infection, or onset of certain rheumatic diseases. Treatment has been limited to surgical removal following failure of medical therapy. In an attempt to establish a reproducible animal model for dystrophic calcification that permitted the screening of potential interventions, we evaluated cardiotoxin (injury)-induced calcifications in three murine strains at both the cellular and ultrastructural levels. All osteopontin null mice and tumor necrosis factor receptor null mice on a C57B6 background had calcifications at days 3 and 7 after injury compared to 75% of wild-type C57B6 mice. There was no difference in mineral content among calcifications from the three mouse strains. Osteogenesis was suggested by the expression of osteocalcin, osterix, and alkaline phosphatase in calcified murine muscle tissue. Osteoclast-like cells facilitated the removal of transient dystrophic deposits (<28 days) in all models. However, none of the models showed an association of mineral crystals with collagen, suggesting that the deposits were not bone-like. The dystrophic mechanism was validated as cell death, and mitochondrial calcifications occurred soon after skeletal muscle injury in the three murine strains.
Keywords: Osteogenesis, Mineral/matrix ratio, Dystrophic calcification, Mitochondria, Cell death
Dystrophic calcifications occur in soft tissue due to injury, infection, or rheumatic diseases [1]. Dystrophic calcifications differ from metabolic calcinosis, where abnormally elevated calcium and/or phosphate metabolism lead to systemic mineralization in that they are associated with cell death. They differ from ectopic bone formation, such as that seen in fibrous dysplasia ossificans [2], due to their association with cell death and the failure to form “bone.” Despite different underlying pathogeneses, there has not been an effective approach to removing the dystrophic calcifications other than surgical intervention [1]. An animal model of persistent and reproducible dystrophic calcification is needed to facilitate screening potential experimental therapeutics. The most common modes of injury related to models of calcifications in animals are cardiotoxin (CTX) and freeze–thaw damage. CTX induces injury in skeletal muscle through inhibiting Ca/Mg-ATPase in plasma membrane [3] and inducing calcium release by the sarcoplasmic reticulum [4]. Thus, necrosis of myofibrils and mitochondrial damage occur as early as 30 min after injection of CTX [5]. The murine models that have been reported to develop calcifications after injury by CTX include tumor necrosis factor (TNF)-α receptor double-knockout mice (p55−/−p75−/−) [6], fibroblast growth factor receptor 4 (FGFR4) null mice [7], and, as reported here, osteopontin (OPN) knockout mice. OPN is an inhibitor of soft tissue calcification [8]. Therefore, we hypothesized that the deposits would be more persistent in the OPN null animals. Other injuries, such as freeze–thaw injury directed to the myocardium, are reported to induce dystrophic cardiac calcification in a few rodent strains [9].
On the basis of prior reports, we selected two murine models and their background wild-type (WT) strain (C57B6) for further investigation of the properties of the dystrophic deposits—OPN null and TNFR null mice. We asked the following questions: (1) What is the composition of the deposit? (2) Does the calcification persist? (3) If the animal model calcifications do not persist, what factors were associated with the deposit and how was its removal facilitated? (4) In what cellular structure does the calcification start; i.e., is the calcium deposition associated with collagen, as it is in bone?
Materials and Methods
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee (2005-42) at Children's Memorial Hospital. TNF-α receptor p55−/−p75−/− mice (B6;129S-Tnfrsf1atm1Imx Tnfrsf1btm1Imx/J,TNFR double null mice), OPN null mice (B6.129S6[Cg]-Spp1tm1Blh/J), and WT control C57BL/6 J mice were purchased from Jackson Laboratory (Bar Harbor, ME) and used for breeding. Both OPN null mice and TNFR double null mice were back-crossed with B6 mice for over five generations to ensure a similar genetic background. Animals were housed in groups (two to six per cage), unless they were mating, with a 12-h dark/12-h light cycle. All animals were fed with standard rodent chow (Nestlé Purina PetCare, St. Louis, MO) after weaning.
Mice from each strain were injected with CTX (0.5 μM at 50 μl) into the tibialis anterior at 4 weeks of age to induce calcifications. Injected muscles were collected for micro-computed tomography (micro-CT), Fourier transform infrared imaging (FTIRI), and histology at 3, 7, 14, and 28 days postinjury (n = 4–5 for each time point per mouse strain). In a limited number of cases, the tissues were also analyzed at 56 days. The uninjected contralateral muscle was also collected to serve as control. A separate set of specimens were collected at baseline and at 1, 2, and 3 days postinjury for transmission electron microscopic (TEM) analysis (n = 2–3 for each time point per mouse strain). Muscles were fixed in 4% paraformaldehyde (pH 7.0) overnight and stored in saline before analysis.
Micro-CT
CTX-injected and contralateral control muscles were fixed in 70% ethanol and scanned by quantitative micro-CT (SkyScan 1174; SkyScan, Kontich, Belgium), which provided a three-dimensional representation of mineralized tissue. Specimens were scanned at 50 V and 800 μA. Isotropic voxel size was approximately 15 μm. Specimen-specific thresholds were determined by first selecting a volume of interest and identifying the threshold that differentiated the mineralized tissue from the background and soft tissue. Individual projections to CT volume data were reconstructed with Nrecon software (SkyScan). Reconstructed scans were analyzed using Ctan software (SkyScan). Volume was calculated as millimeters cubed per muscle.
FTIRI
Aliquots of fixed tissue from the calcifications were embedded in Spurr (Electron Microscopy Sciences, Hatfield, PA; Low Viscosity Embedding kit, by Dr. Spurr). Sections of the embedded tissue were cut at 2–3 μm thickness and examined by infrared imaging spectroscopy using a Perkin-Elmer (Shelton, CT) Spotlight infrared imaging system. Areas with visible mineralized tissue were scanned with special resolution 6.25 μm and spectral resolution 8 cm−1. Interference from the embedding medium was subtracted from the spectra using ISYS software (Spectral Dimensions, Olney, MD). This same software was used to calculate spectral parameters, which included mineral/matrix ratio, collagen maturity, mineral crystallinity, and carbonate/phosphate ratio, as detailed elsewhere [10]. Images were displayed using the same color scale for each parameter, unless the values in the sample were more than five times those in the control.
TEM Analyses of Mineral
Samples collected in freshly prepared 4% formaldehyde were further dehydrated in increasing alcohol content and embedded in Spurr's resin, which is miscible with alcohol. Thin sections, 50–80 nm thick, were collected on pH 8–9 deionized water containing two drops of bromthymol blue indicator, using an ultramicrotome with a diamond knife (Ultracut; Reichert, Depew, NY). The use of alkaline water prevented hydroxyapatite dissolution. Sections were placed on copper (no supporting film) or stainless steel (formvar-coated) grids. For crystallite analysis, unstained sections were viewed at 50,000–100,000 magnification on a TEM (CM-12; Philips, Eindhoven, the Netherlands) at 80-100 kV, accelerating voltage. Mineral crystals were viewed while tilting the specimen stage through 20° [11] or by dark field imaging [12]. Using a combination of these methods often permitted us to determine whether we were observing single crystals or a cluster of crystallites. In the dark field mode, by using the 002 diffraction line from the selected area diffraction and reducing the aperture size, we could measure highly reflective individual crystallites. For cellular morphology, thin sections of formaldehyde-fixed tissues were collected on copper grids, stained with alcoholic uranyl acetate and aqueous lead citrate, and reviewed; and selected areas were photographed.
Histology
Samples embedded in paraffin, after fixation in formalin, were sectioned at 10 μm thickness. Alizarin red staining was used to confirm the presence of calcium. Tissue sections were deparaffinized before antigen retrieval (citrate buffer) for immunohistochemical staining with the following antibodies: bone, anti-mouse osteocalcin (AbD Serotec, Raleigh, NC), anti-mouse osterix (Abcam, Cambridge, MA), anti-mouse alkaline phosphatase (R&D Systems, Minneapolis, MN); mineral inhibitor anti-matrix gla protein (MGP) (Proteintech Group, Chicago, IL); and apoptotic marker anti-mouse poly (ADP-ribose) polymerase (PARP) (Promega, Madison, WI), with diaminobenzidine reagent kits (Vector Laboratories, Burlingame, CA) following protocols previously published [13]. Images of stained muscle sections were acquired using computer software (Openlab 4.04; Improvision, Lexington, MA) and a microscope (DMR-HC; Leica Microsystems, Wetzlar, Germany) coupled to a charge-coupled device camera (Photometric CoolSNAP; Roper Scientific, Tucson, AZ), then edited using digital imaging software (Photoshop CS2; Adobe Systems, San Jose, CA).
Tartrate-Resistant Acid Phosphatase Staining
Tissues for tartrate-resistant acid phosphatase (TRAP) staining were embedded in optimal cutting temperature (OCT) and snap-frozen in liquid nitrogen before storing in −80°C for sectioning. TRAP staining for identification of osteoclasts and macrophages was performed as previously described [14]. In brief, TRAP reaction solution was prepared by mixing 5 mg naphthol AS-MX phosphate dissolved in 0.5 ml N,N-dimethylformamide with 50 ml 0.1 M Na-acetate buffer containing 50 mM Na tartrate (pH 5.0) along with 30 mg fast violet LB salt. Tissue sections were incubated with TRAP reaction solution overnight before being counterstained with hematoxylin.
Data Analysis
Images of serial sections stained for each marker were examined by two independent observers blinded to the nature of the treatment and the mouse background. Positive staining was defined as areas of brown (or purple for TRAP) comparable to positive control slides. The presence or absence of each marker was recorded and graded as 0 (none), 1 (sporadically positive), 2 (moderately positive), or 3 (positive in most areas). The mean value of each marker among three mouse strains, the mineral volume among three mouse strains at each time point, and the FTIRI parameters including matrix/mineral ratios among the three mouse strains at 7 days post-CTX were compared using ANOVA followed by the least significant difference test, with P < 0.05 accepted as significant. Due to the small sample size (n = 4 in most cases), we used the most sensitive pair comparison test to identify the difference. SPSS 14.0 (SPSS, Inc., Chicago, IL) was used to perform all statistical analyses.
Results
All groups of mice showed evidence of calcification in the injected site at early time points. There was no detectable calcification in the noninjected muscles at any time point in any mouse strain.
The Composition of the Deposits: Mineral
FTIRI analysis showed that deposits had spectra characteristic of poorly crystalline hydroxyapatite (Fig. 1), as is seen in mouse bone, only the dystrophic deposits were more punctate, had areas of mineral/matrix ratio appreciably higher than bone, and had crystallinity and carbonate/phosphate values comparable to normal rodent bone [15–17].
Fig. 1.

FTIR images of CTX-injured mouse muscles. In each image white indicates the highest mineral/matrix ratio and black, the lowest mineral/matrix ratio. Gray scales are different among images
Tissues from the CTX-injected side and contralateral side (control) were analyzed by FTIRI for mineral/matrix ratio. In some C57B6 mice, injured muscles (day 3 or 7) had a similarly high ratio (∼59), while the maximal ratio was lower in OPN null mice (∼31) and in TNFR null mice (∼6) (Fig. 1). At day 7, CTX-induced calcifications in all three mouse strains had similar mineral/matrix ratio, carbonate/phosphate ratio, and collagen maturity except that calcifications in OPN null mice had lower crystallinity than those in the other two strains (Table 1). Furthermore, with time, the deposits seemed to coalesce, with the highest mineral/matrix ratio in the center of small punctate deposits (Fig. 1, OPN null mice). To determine whether repeated injury would increase the volume and mineral/matrix ratio of the deposits, TNFR null mice received weekly CTX injection into the tibialis anterior two or three times before being killed. At days 7 and 14, the volume of the calcifications was not increased, nor was the mineral/matrix ratio of these deposits higher.
Table 1.
Comparison of mineral/matrix ratio (M/M), carbonate/phosphate ratio, collagen maturity, and crystallinity in CTX-induced calcifications among C57B6 (WT) mice, TNFR double null mice, and OPN null mice at day 7 postinjection
| Model | C57B6 mice (n = 4) | TNFR p55−/−p75−/− mice (n = 5) | OPN null mice (n = 4) |
|---|---|---|---|
| Average M/M | 5.17 ± 0.14 | 4.82 ± 0.50 | 3.46 ± 2.00 |
| Max. M/M | 13.4 ± 0.4 | 11.9 ± 1.1 | 11.9 ± 6.7 |
| Carbonate/phosphate | 0.0057 ± 0.0015 | 0.0040 ± 0.0005 | 0.0051 ± 0.0015 |
| Collagen maturity | 4.27 ± 0.44 | 4.65 ± 0.26 | 4.64 ± 0.57 |
| Crystallinity | 1.07 ± 0.02 | 1.08 ± 0.01 | 1.04 ± 0.01* |
Values are means ± SD. There was no difference among the three strains except that the crystallinity was significantly higher in C57B6 mice and TNFR null mice than in OPN null mice
P < 0.05
The Composition of the Deposits: The Matrix
All injured muscle showed the presence of collagen, which is a muscle component. In injured mouse muscles, osteocalcin, osterix, and alkaline phosphatase were expressed in mineralized deposits, regardless of strain, at day 7 (Fig. 2). The expression of these markers was primarily located within mineralized areas, while some vascular endothelial cells and muscle fibers were positive for osterix. Staining with the appropriate antibody isotype control showed absence of nonspecific staining for tested tissues. The mineralization inhibitor MGP and the apoptotic marker PARP were present in all deposits obtained from the three mouse strains (Fig. 3), and there was no observable difference among them (Table 2).
Fig. 2.
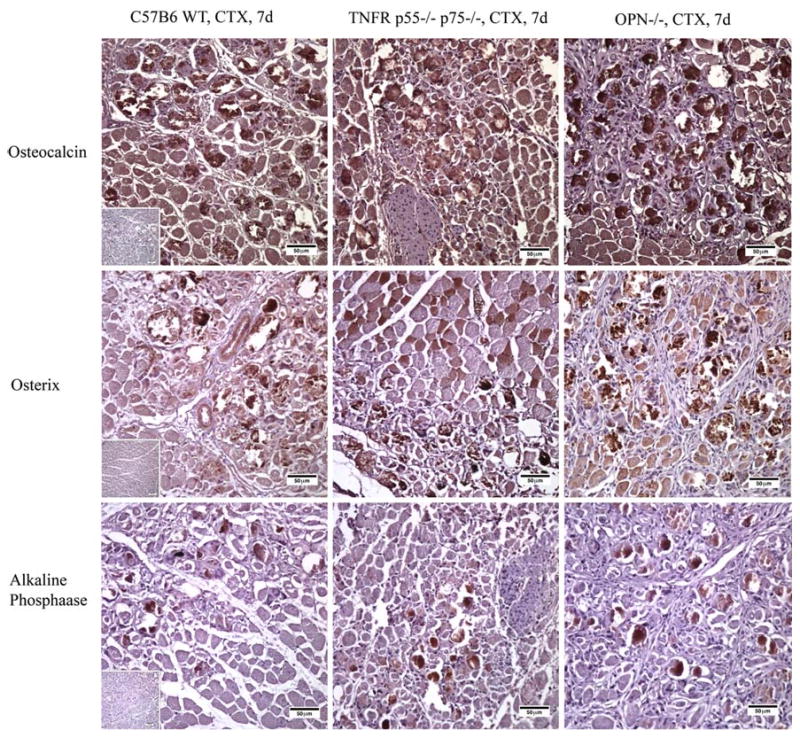
Immunohistochemistry of osteocalcin, osterix, and alkaline phosphatase in CTX-injured mouse muscles at day 7. Brown is positive staining. Insets are images from negative isotype controls
Fig. 3.
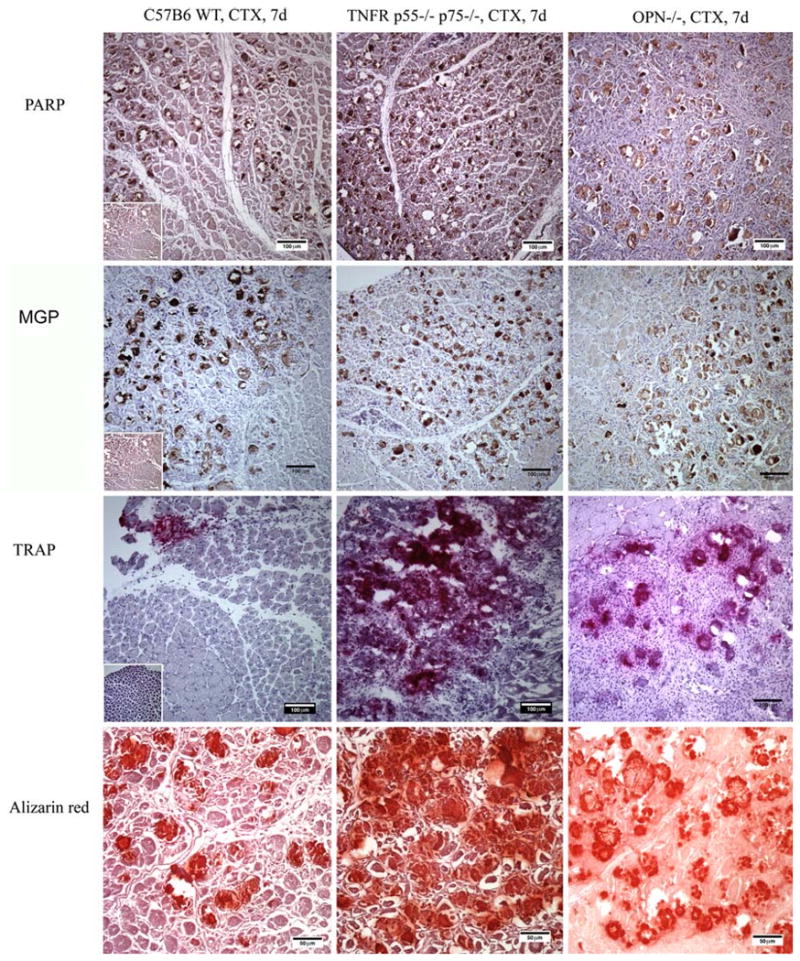
Immunohistochemistry of cell death marker PARP (brown), mineral deposition inhibitor MGP, osteoclast marker TRAP (purple), and alizarin red identification of mineral (red) in CTX-injured mouse muscles at day 7. Insets are images from negative isotype controls
Table 2.
Comparison of presence of bone markers alkaline phosphatase (ALP), osteocalcin, and osterix; mineral deposition inhibitor MGP; cell death marker PARP; and osteoclast marker TRAP in CTX-induced calcifications among C57B6 (WT) mice, TNFR double null mice, and OPN null mice at day 7 postinjection
| Models | C57B6 mice (n = 3) | TNFR p55−/−p75−/− mice (n = 4) | OPN null mice (n = 3) |
|---|---|---|---|
| ALP | 1.33 ± 0.29 | 1.75 ± 0.58 | 2.2 ± 0.63 |
| Osteocalcin | 2.87 ± 0.23 | 3 ± 0 | 3 ± 0 |
| Osterix | 2.61 ± 0.54 | 2.76 ± 0.17 | 3 ± 0 |
| MGP | 2.95 ± 0.09 | 2.9 ± 0.2 | 3 ± 0 |
| PARP | 2.84 ± 0.24 | 2.9 ± 0.2 | 2.84 ± 0.24 |
| TRAP | 0.58 ± 0.51 | 2.5 ± 1.0 | 0.9 ± 1.43 |
There was no significant difference in the expression of any of the markers among the three mouse strains (P < 0.05)
The Transient Nature of the Deposits
Based on micro-CT, calcifications were identified in 75% of C57B6 mice and 100% of TNFR and OPN null mice at day 3. As shown in Fig. 4, the mineral volume was significantly higher in TNFR null mice than C57B6 mice at day 3. Within each strain, the mineral volume peaked at day 7 and then decreased at days 14 and 28. At day 14, there was significantly higher mineral volume in OPN null mice than in the other two strains. At day 28, there was slightly more calcification in muscles from OPN null mice than in those from the two other strains. At an extended time point, 56 days, there was little calcification detectable in the muscle of the injected OPN null mice (data not shown), indicating that in all three murine strains CTX-induced calcification is transient in nature.
Fig. 4.
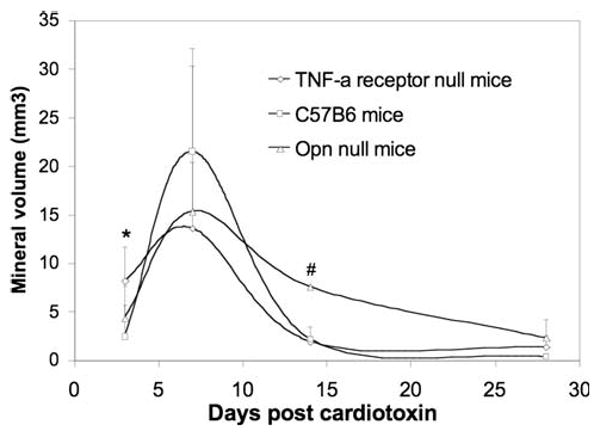
Temporal change of mineral volume as determined by micro-CT analyses in each strain of mice. * Significant difference between TNFR null mice and C57B6 mice. # OPN null mice had significantly higher mineral volume than TNFR null mice and C57B6 mice
How Are the Deposits Removed?
TRAP was present near the site of mineralization in all three strains (Fig. 3), and its expression gradually increased from day 3 to day 7 and stabilized at day 14 (data not shown). Among the three mouse strains, C57B6 mice and OPN null mice muscle appeared to show lower expression of TRAP than TNFR null mice on day 7.
In What Cellular Structure Does the Calcification Start?
Injured muscles from the three mouse strains collected at baseline and at days 1, 2, and 3 were analyzed by TEM. Calcific nodules (dark deposits) were observed in mitochondria from all strains as early as day 1 in TNFR null mice and day 2 in OPN null mice and in C57B6 mice (Fig. 5). The skeletal muscle structure was obliterated after CTX injection. Swollen mitochondria were aggregated, exhibiting a range of sizes, while nonstructured calcific deposits were present. Starting from day 3, remnants of mineral deposits in macrophages migrating into the necrotic area suggested that macrophages participated in the phagocytosis of the mineral. Unlike bone, where mineral is directly deposited oriented upon a type I collagen, mouse muscle calcified deposits did not display an association of mineral with fibrillar collagen.
Fig. 5.
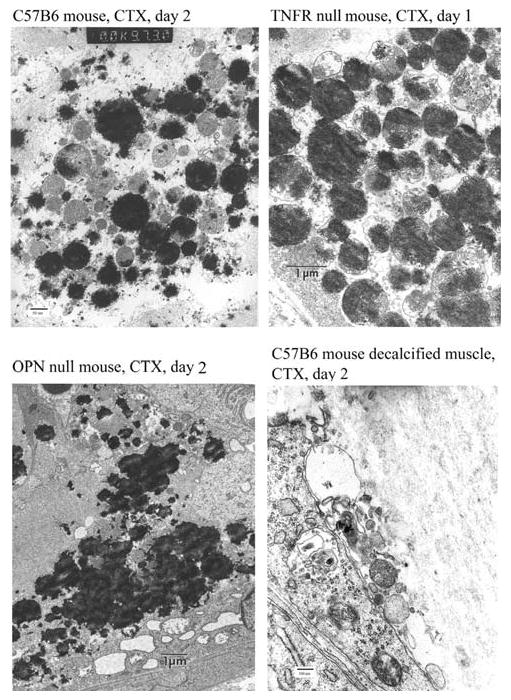
TEM images of CTX-injected mouse muscles. There was electron-dense mineral scattered throughout the mitochondria a in C57B6 mice on day 2, b in TNFR null mice on day 1, and c in OPN null mice on day 2. d A decalcified sample of CTX-injected C57B6 mice on day 2 showed the absence of collagen
Discussion
This is the first report comparing injury-based murine models of CTX-induced calcifications from several points of view: the properties of the mineral, the ultrastructural morphology, and the presence of markers of bone formation and resorption. These models provide new insights into possible calcification mechanisms and potential therapeutic approaches. While it has long been known that during dystrophic calcification the death of the cell leads to a release of catabolic enzymes and calcium, detailed knowledge of the mechanism of the formation and development of dystrophic calcifications is still limited. An animal model that even approximated the dystrophic calcifications seen in human diseases, such as old injection sites [18] and juvenile dermatomyositis [19, 20], would be useful to effectively screen potential medical interventions.
Compared with WT C57B6 mice, the TNFR null mice and OPN null mice were more likely to develop calcification after an injury to skeletal muscle. However, although the mineral deposits were present at early stages (days 3 and 7), they were significantly smaller by day 14 and entirely disappeared at day 28 (C57B6 mice and TNFR null mice) or day 56 (OPN null mice). This transient nature of CTX-induced calcification has not been previously reported, partly because most studies [6, 7] did not involve the longer period of observation used in the present study. More importantly, these data, when compared to those seen in WT mice, provide insight into the resorption of the deposits in the mutants.
In mice, osteoclast-like cells were involved in resorbing the muscle calcific deposits, as evidenced by TRAP staining. There were no osteoclasts present in control mouse tissue or noncalcified tissue (i.e., 28 days post-CTX injection in WT mice). This observation raised the consideration that osteoclasts differentiated or were attracted to the deposited mineral once it appeared. The osteoclast resorptive activity, although present as early as day 3, did not appear to match the extent of mineral deposition. The increased osteoclast activity after day 7 may have accounted for the resolution of minerals in WT mice, TNFR null mice, and OPN null mice; but there also may have been a corresponding reduction in the stimulus for mineral deposition as the inflammatory injury diminished.
We further speculate that in normal WT mice and TNFR null mice the deposits disappeared more rapidly because chemotactic OPN was present. OPN null mice had the slowest rate of resolving calcifications, possibly because, in the absence of OPN, macrophage adhesion and activation were impaired [21]. Chemotaxis of OPN-null macrophages to monocyte chemoattractant protein-1 (MCP-1) can be abrogated and rescued by an OPN substratum or by addition of high levels of OPN in solution [22]. Osteoclasts bind to a variety of extracellular matrix proteins, including vitronectin, OPN, and bone sialoprotein through the αvβ3 integrin [23].
OPN is known to be an inhibitor of calcification [24]. Therefore, the absence of OPN in OPN null mice may have partially facilitated the growth of calcification and delayed subsequent resolution of calcification. Although the calcific deposits in OPN null mice were transient, the deposit-loading period was long enough (>14 days) to allow testing potential therapeutic interventions.
A key difference between the dystrophic calcifications and bone is the high mineral/matrix ratio observed by FTIRI (∼60 compared with ∼6 for bone). Although TNFR null mice developed volumetrically larger calcifications than WT mice and OPN null mice, their mineral/matrix ratio was similar to that of the other two strains. The mineral deposits in C57B6 mice and OPN null mice tended to be more punctate. Calcifications from OPN null mice had the smallest crystallinity. There was no apparent association between mineral volume and mineral/matrix ratio. Other modifiers, such as repeated injections of CTX and injection into fat tissue (foot pads), did not alter either the mineral/matrix ratio or the mineral volume in TNFR null mice. Zhao et al. [7] reported that FGFR4 null mice also developed calcifications with modestly elevated mineral/matrix ratio after CTX injection.
Other ectopic calcifications (i.e., end-stage atherosclerotic plaques) are also associated with markers of bone formation [25]. However, in contrast to atherosclerosis, our results revealed that in CTX-induced mouse calcifications there was little bone-like formation, as evidenced by the lack of association of mineral with type I collagen in mineral deposits and the absence of bone-like trabeculae. This lack of association between mineral and collagen was also found in calcifications from children with juvenile dermatomyositis [26]. The presence of “osteoblast markers” osteocalcin, osterix, and alkaline phosphatase may be due to the known affinity of these proteins for the mineral in the deposits; or alternatively, osteoblast-like cells might be involved in the process. However, the absence of trabecular bone structures in injury-induced calcifications in our animal models indicated a misregulated mineralization. Cell death, as shown by PARP staining and TEM, was present in CTX-injected mouse muscles. From the time course study with TEM, disintegration of muscle fibers occurred on day 1 in all three strains. However, there were no mineral deposits in muscles of C57B6 mice or OPN null mice until day 2. In contrast, TNFR null mice developed calcification as early as day 1. These results suggested that cell death may have preceded mineralization. During the mineral resolution phase (days 7–28) associated with lower mineral volume, cell death in CTX-injected mouse muscle was much less pronounced than at the earlier stage.
Cell death mediated through mitochondrial injury may be a common mechanism in the initiation of calcification in our mouse models. Using TEM analysis, early mineral deposits were observed in the mitochondria of TNFR null mice, OPN null mice, and WT C57B6 mice at days 1 and 2, respectively. It has been reported that necrosis of myofibrils and mitochondrial damage occur as early as 30 minutes after injection of CTX [5]. The mechanism was thought to be related to the contracture injury [27] induced by the accumulation of calcium in sarcolemmal membrane vesicles with the action of CTX on Ca2+/Mg2+-ATPase [28]. Thus, accumulated calcium may precipitate with phosphate in compromised mitochondria, which is also observed in untreated muscle from children with active juvenile dermatomyositis (Pachman, personal communication). It is not clear, however, whether the mitochondrial deposits precede the altered function or cause it.
The limitations of the current study include the small sample size in each experiment, our inability to track the longitudinal change of calcifications in the same animal without access to in vivo micro-CT, semiquantitative results based on immunostaining, and the fact that we tested only young female mice.
Conclusion
CTX-induced calcifications in OPN null mice and C57B6 mice may provide a useful model for the study of the evolution of dystrophic calcifications. Cell death and mitochondrial calcifications accounted for, at least partially, mineralization in injured skeletal muscles from all three strains of mice.
Acknowledgments
We are grateful for technical support from Lyudmila Lukashova and Nicholas Geraci. We also thank Dr. Paula Stern for the MC3T3 cells and advice on TRAP staining. This work was supported by the National Institutes of Health (R0-1AR48289) and the Cure JM Program of Excellence in Juvenile Myositis Research (both to L. M. Pachman) and by the Hospital for Special Surgery, Musculoskeletal Repair and Regeneration Core Center Grant (AR046121, to A. L. Boskey).
Footnotes
This work was presented partially at the American Society of Bone and Mineral Research Annual Conference 2007 and American College of Rheumatology Annual Conference 2007.
Contributor Information
Yongdong Zhao, Email: yongdong.zhao@cchmc.org, Center of Excellence in Clinical Immunology, Children's Memorial Research Center, 2300 Children's Plaza, Box 212, Chicago, IL 60614, USA, Department of Pediatrics, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
Annette L. Urganus, Email: aurganus@childrensmemorial.org, Center of Excellence in Clinical Immunology, Children's Memorial Research Center, 2300 Children's Plaza, Box 212, Chicago, IL 60614, USA.
Lyudmila Spevak, Email: spevakm@hss.edu, Hospital for Special Surgery, Weill Cornell Medical College, New York, NY, USA.
Sheela Shrestha, Email: shshrestha@childrensmemorial.org, Center of Excellence in Clinical Immunology, Children's Memorial Research Center, 2300 Children's Plaza, Box 212, Chicago, IL 60614, USA.
Stephen B. Doty, Email: dotys@hss.edu, Hospital for Special Surgery, Weill Cornell Medical College, New York, NY, USA.
Adele L. Boskey, Email: boskey@hss.edu, Hospital for Special Surgery, Weill Cornell Medical College, New York, NY, USA.
Lauren M. Pachman, Email: pachman@northwestern.edu, Center of Excellence in Clinical Immunology, Children's Memorial Research Center, 2300 Children's Plaza, Box 212, Chicago, IL 60614, USA, Department of Pediatrics, Rheumatology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA.
References
- 1.Hussmann J, Russell RC, Kucan JO, Khardori R, Steinau HU. Soft-tissue calcifications: differential diagnosis and therapeutic approaches. Ann Plast Surg. 1995;34:138–147. [PubMed] [Google Scholar]
- 2.de la Pena LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- 3.Fourie AM, Meltzer S, Berman MC, Louw AI. The effect of cardiotoxin on (Ca2+ + Mg2+)-ATPase of the erythrocyte and sarcoplasmic reticulum. Biochem Int. 1983;6:581–591. [PubMed] [Google Scholar]
- 4.Fletcher JE, Jiang MS, Gong QH, Yudkowsky ML, Wieland SJ. Effects of a cardiotoxin from Naja naja kaouthia venom on skeletal muscle: involvement of calcium-induced calcium release, sodium ion currents and phospholipases A2 and C. Toxicon. 1991;29:1489–1500. doi: 10.1016/0041-0101(91)90005-c. [DOI] [PubMed] [Google Scholar]
- 5.Ownby CL, Fletcher JE, Colberg TR. Cardiotoxin 1 from cobra (Naja naja atra) venom causes necrosis of skeletal muscle in vivo. Toxicon. 1993;31:697–709. doi: 10.1016/0041-0101(93)90376-t. [DOI] [PubMed] [Google Scholar]
- 6.Chen SE, Gerken E, Zhang Y, Zhan M, Mohan RK, Li AS, Reid MB, Li YP. Role of TNF-α signaling in regeneration of cardiotoxin-injured muscle. Am J Physiol Cell Physiol. 2005;289:C1179–C1187. doi: 10.1152/ajpcell.00062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, Pachman LM, Sartorelli V, Hoffman EP. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD–Tead2–Fgfr4 transcriptional pathway. J Biol Chem. 2006;281:429–438. doi: 10.1074/jbc.M507440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giachelli CM, Speer MY, Li X, Rajachar RM, Yang H. Regulation of vascular calcification: roles of phosphate and osteopontin. Circ Res. 2005;96:717–722. doi: 10.1161/01.RES.0000161997.24797.c0. [DOI] [PubMed] [Google Scholar]
- 9.Brunnert SR. Morphologic response of myocardium to freeze–thaw injury in mouse strains with dystrophic cardiac calcification. Lab Anim Sci. 1997;47:11–18. [PubMed] [Google Scholar]
- 10.Boskey AL, Mendelsohn R. Infrared spectroscopic characterization of mineralized tissues. Vib Spectrosc. 2005;38:107–114. doi: 10.1016/j.vibspec.2005.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bocciarelli DS. Morphology of crystallites in bone. Calcif Tissue Res. 1970;5:261–269. doi: 10.1007/BF02017554. [DOI] [PubMed] [Google Scholar]
- 12.Jackson SA, Cartwright AG, Lewis D. The morphology of bone mineral crystals. Calcif Tissue Res. 1978;25:217–222. doi: 10.1007/BF02010772. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y, Fedczyna TO, McVicker V, Caliendo J, Li H, Pachman LM. Apoptosis in the skeletal muscle of untreated children with juvenile dermatomyositis: impact of duration of untreated disease. Clin Immunol. 2007;125:165–172. doi: 10.1016/j.clim.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woo JT, Nakagawa H, Krecic AM, Nagai K, Hamilton AD, Sebti SM, Stern PH. Inhibitory effects of mevastatin and a geranylgeranyl transferase I inhibitor (GGTI-2166) on mononuclear osteoclast formation induced by receptor activator of NF kappa B ligand (RANKL) or tumor necrosis factor-alpha (TNF-alpha) Biochem Pharmacol. 2005;69:87–95. doi: 10.1016/j.bcp.2004.08.036. [DOI] [PubMed] [Google Scholar]
- 15.Verdelis K, Ling Y, Sreenath T, Haruyama N, MacDougall M, van der Meulen MC, Lukashova L, Spevak L, Kulkarni AB, Boskey AL. DSPP effects on in vivo bone mineralization. Bone. 2008;43:983–990. doi: 10.1016/j.bone.2008.08.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ling Y, Rios HF, Myers ER, Lu Y, Feng JQ, Boskey AL. DMP1 depletion decreases bone mineralization in vivo: an FTIR imaging analysis. J Bone Miner Res. 2005;20:2169–2177. doi: 10.1359/JBMR.050815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boskey AL, Moore DJ, Amling M, Canalis E, Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wildtype controls. J Bone Miner Res. 2003;18:1005–1011. doi: 10.1359/jbmr.2003.18.6.1005. [DOI] [PubMed] [Google Scholar]
- 18.Wei BP, Somers GR, Castles L. Dystrophic calcification and amyloidosis in old subcutaneous injection sites. Aust N Z J Surg. 2003;73:556–558. doi: 10.1046/j.1445-1433.2003.02665.x. [DOI] [PubMed] [Google Scholar]
- 19.Stock S, Ignatiev K, Lee P, Abbott K, Pachman L. Pathological calcification in juvenile dermatomyositis (JDM): microCT and synchrotron X-ray diffraction reveal hydroxyapatite with varied microstructures. Connect Tissue Res. 2004;45:248–256. doi: 10.1080/03008200490903066. [DOI] [PubMed] [Google Scholar]
- 20.Pachman LM, Veis A, Stock S, Abbott K, Vicari F, Patel P, Giczewski D, Webb C, Spevak L, Boskey AL. Composition of calcifications in children with juvenile dermatomyositis: association with chronic cutaneous inflammation. Arthritis Rheum. 2006;54:3345–3350. doi: 10.1002/art.22158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faccio R, Grano M, Colucci S, Zallone AZ, Quaranta V, Pelletier AJ. Activation of alphavbeta3 integrin on human osteoclast-like cells stimulates adhesion and migration in response to osteopontin. Biochem Biophys Res Commun. 1998;249:522–525. doi: 10.1006/bbrc.1998.9180. [DOI] [PubMed] [Google Scholar]
- 22.Zhu B, Suzuki K, Goldberg HA, Rittling SR, Denhardt DT, McCulloch CA, Sodek J. Osteopontin modulates CD44-dependent chemotaxis of peritoneal macrophages through G-protein-coupled receptors: evidence of a role for an intracellular form of osteopontin. J Cell Physiol. 2004;198:155–167. doi: 10.1002/jcp.10394. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura I, Rodan GA, le Duong T. Regulatory mechanism of osteoclast activation. J Electron Microsc (Tokyo) 2003;52:527–533. doi: 10.1093/jmicro/52.6.527. [DOI] [PubMed] [Google Scholar]
- 24.Pampena DA, Robertson KA, Litvinova O, Lajoie G, Goldberg HA, Hunter GK. Inhibition of hydroxyapatite formation by osteopontin phosphopeptides. Biochem J. 2004;378:1083–1087. doi: 10.1042/BJ20031150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doherty TM, Asotra K, Fitzpatrick LA, Qiao JH, Wilkin DJ, Detrano RC, Dunstan CR, Shah PK, Rajavashisth TB. Calcification in atherosclerosis: bone biology and chronic inflammation at the arterial crossroads. Proc Natl Acad Sci USA. 2003;100:11201–11206. doi: 10.1073/pnas.1932554100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urganus AL, Zhao YD, Pachman LM. Juvenile dermatomyositis calcifications selectively displayed markers of bone formation. Arthritis Rheum. 2009;61:501–508. doi: 10.1002/art.24391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fletcher JE, Lizzo FH. Contracture induction by snake venom cardiotoxin in skeletal muscle from humans and rats. Toxicon. 1987;25:1003–1010. doi: 10.1016/0041-0101(87)90164-4. [DOI] [PubMed] [Google Scholar]
- 28.Huang JL, Trumble WR. Cardiotoxin from cobra venom affects the Ca–Mg-ATPase of cardiac sarcolemmal membrane vesicles. Toxicon. 1991;29:31–41. doi: 10.1016/0041-0101(91)90037-r. [DOI] [PubMed] [Google Scholar]