

Abstract
Transient elevations in Ca2+ have previously been shown to promote focal adhesion disassembly and cell motility through an unknown mechanism. In this study, evidence is provided to show that CaMK-II, a Ca2+/calmodulin dependent protein kinase, influences fibroblast adhesion and motility. TIRF microscopy reveals a dynamic population of CaMK-II at the cell surface in migrating cells. Inhibition of CaMK-II with two mechanistically distinct, membrane permeant inhibitors (KN-93 and myr-AIP) freezes lamellipodial dynamics, accelerates spreading on fibronectin, enlarges paxillin-containing focal adhesions and blocks cell motility. In contrast, constitutively active CaMK-II is not found at the cell surface, reduces cell attachment, eliminates paxillin from focal adhesions and decreases the phospho-tyrosine levels of both FAK and paxillin; all of these events can be reversed with myr-AIP. Thus, both CaMK-II inhibition and constitutive activation block cell motility through over-stabilization or destabilization of focal adhesions, respectively. Coupled with the existence of transient Ca2+ elevations and a dynamic CaMK-II population, these findings provide the first direct evidence that CaMK-II enables cell motility by transiently and locally stimulating tyrosine dephosphorylation of focal adhesion proteins to promote focal adhesion turnover. Cell Motil.
Keywords: calmodulin, fibronectin, fibroblast, CaM kinase II, TIRF
Introduction
Cell migration occurs during embryonic development, wound healing, immune surveillance and cancer metastasis and involves dozens of molecular players [Lauffenburger and Horwitz, 1996; Ridley et al., 2003]. Essential to cell migration is the formation and turnover of structures known as focal adhesions. Focal adhesions are critical signaling and structural hubs found in migrating cells, which assemble in response to the interaction of extracellular matrix ligands with integrin receptors [Huttenlocher et al., 1995; Lauffenburger and Horwitz, 1996; Ridley et al., 2003].
Integrin engagement with the extracellular matrix stimulates the tyrosine phosphorylation of key cytoskeletal proteins, beginning with FAK autophosphorylation on Y397 [Mitra et al., 2005]. FAK autophosphorylation recruits src to focal adhesions, and the FAK/src complex phosphorylates two adapter proteins, paxillin and p130CAS [Bellis et al., 1995; Schaller and Parsons, 1995; Cary et al., 1998; Webb et al., 2004; Mitra et al., 2005]. Phosphorylated paxillin and p130CAS recruit proteins, such as Crk, to focal adhesions through SH2/ SH3 binding domains. Fibroblasts deficient in either FAK or paxillin exhibit defects in cell migration [Ilic et al., 1995; Hagel et al., 2002], suggesting that both proteins are essential for adhesion dynamics [Webb et al., 2004].
Transient elevations in Ca2+ have been shown to induce the disassembly of focal adhesions [Marks and Maxfield, 1990; Giannone et al., 2002, 2004; Conklin et al., 2005]. While calcineurin and calpain have been implicated as Ca2+ targets which influence focal adhesion dynamics [Franco et al., 2004; Conklin et al., 2005; Lee et al., 2005; Shao et al., 2006], the type II Ca2+/calmodulin-dependent protein kinase (CaMK-II) has also emerged as a potential target of fibronectin (FN)-induced Ca2+ transients [Blystone et al., 1999; Illario et al., 2005].
CaMK-II is encoded by four genes (α, β, γ and δ) to yield over three dozen splice variants [Tombes et al., 2003], some of which interact with the actin cytoskeleton [Shen et al., 1998; Faison et al., 2002; Fink et al., 2003; Lin et al., 2004; Easley et al., 2006]. CaMK-II has been shown to influence cell migration during embryogenesis and in cultured mammalian cells. Morphogens encoded by certain members of the wnt gene family activate CaMK-II and lead to convergent extension cell movements during and after gastrulation [Kuhl et al., 2000; Sheldahl et al., 2003; Kohn and Moon, 2005]. CaMK-II is also necessary for the attachment and motility of human mammary epithelial cells (HME), Chinese hamster ovary cells (CHO) and vascular smooth muscle cells (VSM) [Pauly et al., 1995; Bilato et al., 1997; Bouvard and Block, 1998; Bouvard et al., 1998; Lundberg et al., 1998; Suzuki and Takahashi, 2003; Pfleiderer et al., 2004], and has been implicated in integrin cross-talk [Blystone et al., 1999]. While these studies emphasize the importance of CaMK-II in cell motility, the mechanism by which CaMK-II influences motility and adhesion dynamics remains unknown.
To define the mechanism by which CaMK-II influences NIH/3T3 fibroblast cell motility, GFP-tagged wild type and constitutively active CaMK-IIs were used in conjunction with membrane permeant CaMK-II inhibitory drugs in localization, motility and focal adhesion assays. Even though a direct substrate has not yet been identified, the results of this study indicate that CaMK-II catalytic activity promotes focal adhesion disassembly and detachment from the extracellular matrix by inducing the tyrosine dephosphorylation of focal adhesion proteins, thus enabling cell motility.
Materials and Methods
NIH/3T3 Culture and Harvesting
NIH/3T3 cells were used in all studies and were maintained on tissue culture dishes (Nunc, Rochester, NY) at 37°C in DMEM with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA). Cells were sub-cultured every 3–4 days, never exceeding 95% confluency. When specified, dishes were pre-incubated with 1 μg/ml human fibronectin (Invitrogen) in PBS (Phosphate Buffered Saline) for 1 h at 37°C or overnight at 4°C, washed once with PBS and then placed into DMEM/10% FBS just prior to plating cells. Cells were harvested by trypsinization, washed in ice-cold PBS and then resuspended in homogenization buffer, which consisted of 30 mM Hepes pH 7.4, 20 mM MgCl2, 80 mM β-glycerol phosphate, 2.6 mM EGTA, 0.1 μM okadaic acid, 1 μg/ml each chymostatin, leupeptin, antipain, pepstatin and soybean trypsin inhibitor. Cells were lysed using two 4-s bursts from a probe sonicator (Misonix, Farmingdale, NY) and then centrifuged at 12,000g for 15 min at 4°C.
Plasmid Constructs
EGFP-linked CaMK-II constructs used in this study were prepared as previously described [Lantsman and Tombes, 2005]. The δC CaMK-II variant used here represents the simplest splice variant and the most common form expressed in these cells [Tombes et al., 2003]. EGFP-paxillin, dsRed-paxillin and EGFP-FAK were prepared as described [Webb et al., 2004; Brown et al., 2006]. EGFP-talin and vinculin were generous gifts from Dr. Kenneth Yamada, National Institutes of Health, Bethesda, MD and Dr. Benjamin Geiger, Weizmann Institute of Science, Rehovot Israel, respectively.
Transfection and Microscopy
Freshly sub-cultured cells were transfected with Lipofectamine 2000 as specified (Invitrogen). Typically, transfections utilized 20 μg of total DNA for 100-mm plates and 4 μg for 6-well dishes. Co-transfections used equal amounts of each construct. Live or formaldehyde fixed cells were imaged in phase contrast, traditional fluorescence (Fm) or Total Internal Reflection Fluorescence microscopy (TIRFm) using an IX-70 inverted microscope equipped with a 12-bit black/white F-View CCD camera and processed using Microsuite-B3SV Version 3.2 software (Olympus, Melville, NY). TIRF Illumination utilized a 10 mW Argon-ion laser (Melles Griot, Carlsbad, CA) for 488 nm illumination via a 60×/1.45NA PlanApo objective (Olympus). Living cells were maintained at 37°C using a stage heater (20/20 Technology, Wilmington, NC)
Analysis of Cell Migration, Spreading and Focal Adhesion Size
NIH/3T3 cells were cultured just to confluency and then scratch-wounded using a single stroke from a fine pipette tip. Cells were immediately re-fed with fresh 10% FBS/DMEM containing either vehicle alone, 10 μM KN-93 or 20 μM myr-AIP. Cells were then imaged under phase contrast at 0, 3 and 6 h post wounding. The microscope stage was maintained at 37°C. Between imaging sessions, cells were returned to the incubator. Lamellipodial dynamics were captured over 10 min using phase contrast imaging and Olympus Microsuite-B3SV Version 3.2 software (Olympus) for cells plated on FN in the presence of vehicle control or 20 μM myr-AIP (supplemental videos 1 and 2). Cell migration rates were calculated from images taken 1 and 4 h after plating on FN with Microsuite-B3SV Version 3.2. Diameter measurements for cell spreading and focal adhesion size analysis were also computed with Microsuite-B3SV Version 3.2 software. Minimum and maximum threshold values for focal adhesion size were 1.7 and 35 μm2, respectively. T-tests were employed to determine statistically significant differences, as indicated by P-value thresholds.
Immunoblotting
Transfected NIH/3T3 cells were harvested as described above (see Cell Culture and Harvesting). Samples were subjected to SDS-PAGE on 4–15 or 10% polyacrylamide gels (Bio-Rad, Hercules, CA), transferred to 0.45-μm nitrocellulose sheets, and then blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.01% Tween-20 and 0.1% sodium azide (TBSTA) and 5% pre-immune goat serum. Blots were then incubated overnight with 1 μg/ml primary antibody in 5% BSA, TBSTA. After a 2-h incubation with 2 μg/ ml alkaline phosphatase conjugated goat anti-mouse or rabbit IgG, blots were developed with 0.25 mg/ml BCIP/ NBT (Roche, Indianapolis, IN) in phosphatase buffer, pH 9.4 [Lantsman and Tombes, 2005].
Reagents
Total FAK and paxillin antibodies were obtained from BD Biosciences (Rockville, MD), phospho-Y31 paxillin and phospho-S843 FAK antibodies from Biosource/Invitrogen, and phospho-Y925 FAK antibody from Cell Signaling Technology (Danvers, MA). All secondary antibodies were obtained from Jackson Immuno-Research Laboratories (West Grove, PA). KN-93 (CalBiochem, La Jolla, CA) is a calmodulin antagonist, specific for a subset of CaM kinases including CaMK-I, CaMK-II and CaMK-IV [Sumi et al., 1991; Hidaka and Ishikawa, 1992]. However, CaMK-I and CaMK-IV were undetectable in NIH/3T3 cells as determined using immunological methods (data not shown). KN-93 inhibits CaMK-II by 50% at ∼2 μM and by 90% at 10 μM in these cells [Tombes et al., 1995]. Myristoylated autoinhibitory peptide (KKALRRQEAVDAL) is based on the CaMK-II autoinhibitory domain [Laabich and Cooper, 2000]. Myr-AIP inhibits purified CaMK-II by 90% at 20 μM (data not shown) and was obtained from Biomol (Plymouth Meeting, PA). According to the manufacturer and consistent with our findings, this peptide has an effective treatment limit of 6-8 h.
Results
CaMK-II Inhibition Accelerates Cell Spreading on Fibronectin
Fibronectin (FN) binding to β1 integrin has been shown to induce localized transient Ca2+ elevations [Praetorius et al., 2004], which activate CaMK-II [Blystone et al., 1999; Illario et al., 2005]. To assess the role of CaMK-II in cell spreading and motility, two mechanistically distinct membrane-permeant CaMK-II inhibitors, myr-AIP (myristoylated autoinhibitory peptide) and KN-93 were used to disrupt endogenous CaMK-II activity. Myr-AIP directly blocks catalytic activity while KN-93 antagonizes CaM binding and thus prevents activation. Both compounds maximally reduce CaMK-II activity within 2 h of treatment [Easley et al., 2006]. NIH/3T3 cells were sub-cultured onto 1 μg/ml FN coated dishes in medium containing vehicle alone (Fig. 1A), 20 μM myr-AIP (Fig. 1B), or 10 μM KN-93 (Fig. 1C) and imaged at 5, 10, 15, 30, 45 and 60 min post plating. Cells treated with either KN-93 or myr-AIP spread more rapidly than untreated cells as shown at 30, 45 and 60 min. KN-92, an inactive analog of KN-93, had no effect on spreading (data not shown). The degree of spreading was quantified by averaging cell diameters (n > 100) for each condition, and both KN-93 and myr-AIP significantly accelerated spreading after 30 min (P < 0.01) (Fig. 1D). Once cells were fully spread (beyond 1 h), treatment with myr-AIP arrested lamellipodial dynamics as compared to untreated cells, as shown in supplemental movies 1 (control) and 2 (myr-AIP). The effects of CaMK-II inhibition on cell spreading were dependent on FN as cells cultured on laminin or on untreated dishes, attached slowly and did not exhibit enhanced spreading in response to KN-93 or myr-AIP (data not shown).
Fig. 1.
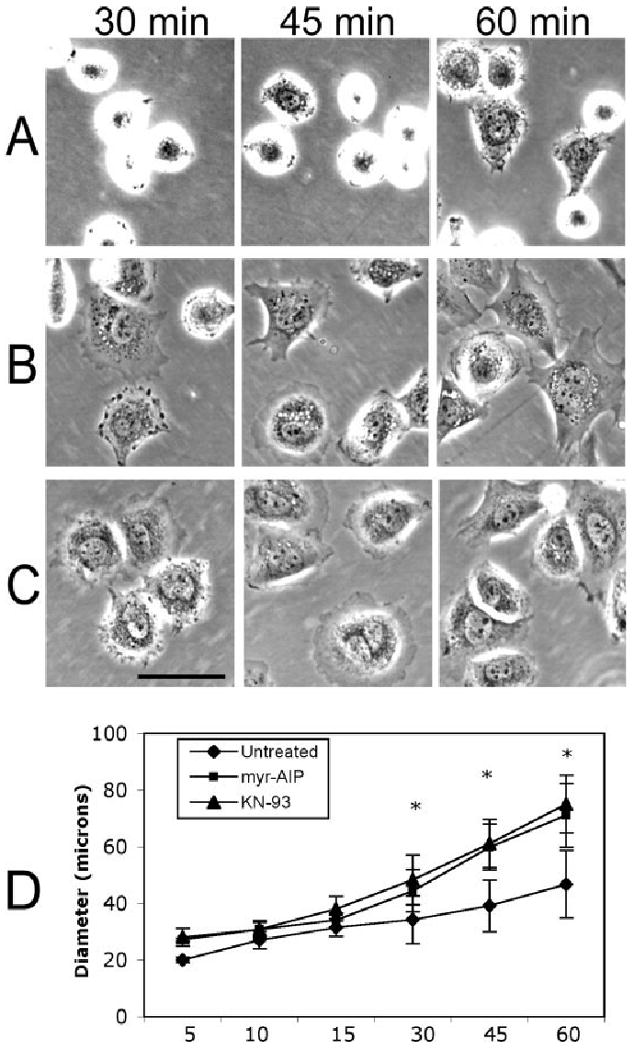
CaMK-II inhibition accelerates cell spreading. NIH/3T3 cells were plated on FN-coated dishes in the presence of vehicle alone (A), 20 μM myr-AIP (B) or 10 μM KN-93 (C). Cells were imaged live under phase contrast at the indicated times post plating. Scale bar, 50 μm. (D) Graphical representation of diameter measurements from cells imaged at each time point (in minutes) for each condition. n > 100 for each condition and each time point. * indicates P < 0.01.
CaMK-II Inhibition Reduces Cell Motility
Mechanical wounding of confluent cells has previously been shown to increase cytosolic Ca2+ levels in cells along the edge of the wound [Tran et al., 1999]. To evaluate whether CaMK-II inhibition impairs cell motility, separate dishes of NIH/3T3 cells were identically scratch-wounded, immediately treated with fresh medium containing vehicle alone, 20 μM myr-AIP or 10 μM KN-93 and imaged 0, 3 and 6 h post wounding (Fig. 2). Cells treated with myr-AIP or KN-93 retained a sizeable wound after 6 h while cells treated with vehicle alone exhibited >70% wound closure (Fig. 2). Both washout of KN-93 and the natural instability of myr-AIP beyond 6 h (see Materials and Methods) resulted in eventual wound closure (data not shown).
Fig. 2.
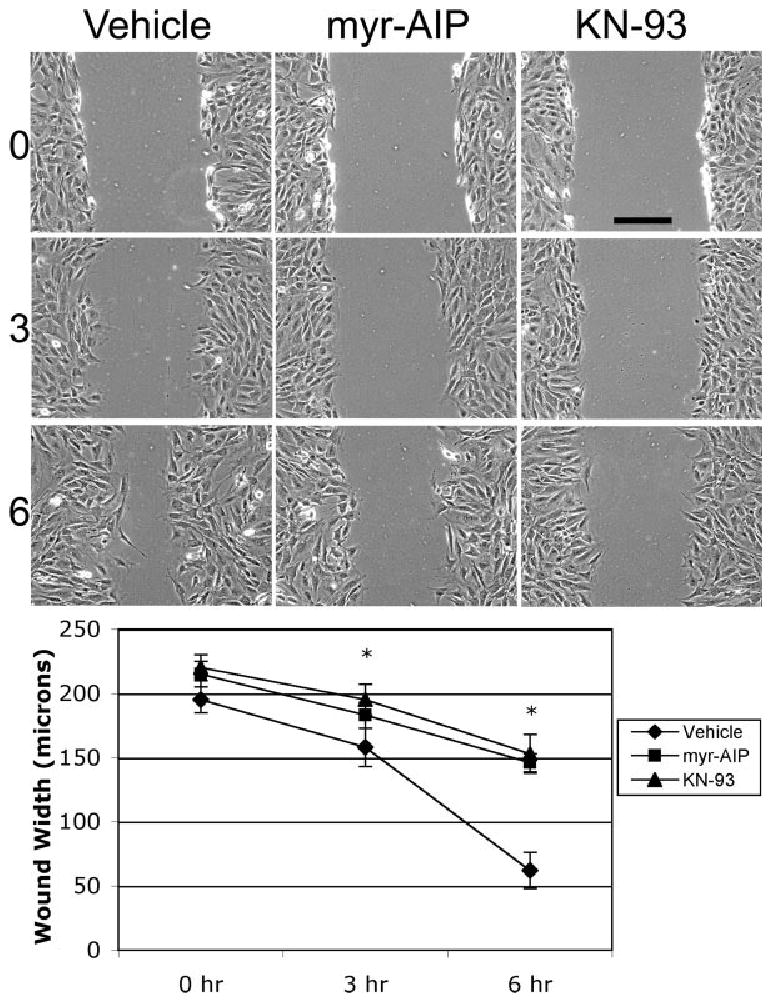
CaMK-II inhibition reduces the rate of wound closure. Confluent monolayers of cells were scratch-wounded and then imaged at 0, 3 and 6 h in the presence of vehicle alone, 20 μM myr-AIP or 10 μM KN-93. Scale bar, 100 μm. Wound size (in μm) was measured (n > 30) for each condition at each time point. * indicates P < 0.01.
CaMK-II Localizes to the Surface of Motile NIH/3T3 Cells
Because CaMK-II inhibition profoundly affected cell spreading and motility, Total Internal Reflection Fluorescence microscopy (TIRFm) was used to examine the localization of CaMK-II at the cell surface. NIH/3T3 cells were transfected with GFP-tagged δC CaMK-II and then sub-cultured onto FN-coated coverslips. Conventional fluorescence (Fm) confirmed the perinuclear distribution of δC CaMK-II, as previously reported in these cells [Tombes et al., 1999; Caran et al., 2001]. TIRFm of the same cell indicated that a substantial portion of δC CaMK-II localizes to the cell surface (Fig. 3A). Time-lapse TIRFm demonstrated that CaMK-II is dynamically enriched at the leading edge of migrating cells (Fig. 3A) and in discrete patterns throughout the cell (supplemental movie 3). This novel finding supports a role for CaMK-II in adhesive and migratory events.
Fig. 3.
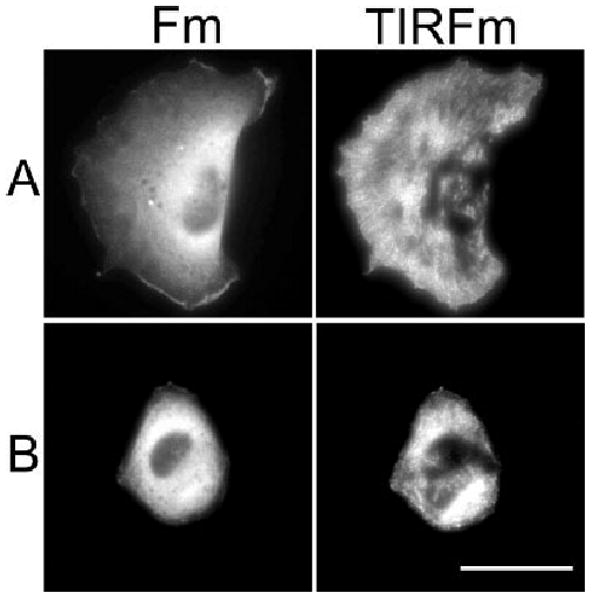
CaMK-II is dynamically localized at the surface of motile fibroblasts. Cells were transfected with either GFP-labeled wild-type δC CaMK-II (A) or constitutively active (CON) δC CaMK-II (B) and sub-cultured on FN for 3 h. Cells were imaged using either traditional fluorescence microscopy (Fm) or total internal reflection fluorescence microscopy (TIRFm). Scale bar, 50 μm. Supplemental videos 3 and 4 of live TIRF imaged cells correspond to these conditions.
Constitutively Active CaMK-II Causes Cell Rounding and Inhibits Cell Motility
In a complementary approach to CaMK-II inhibition, the effect of constitutively active CaMK-II on cell motility was investigated. CaMK-II can be made constitutively active (CON) through a phosphomimetic point mutation (T287D) in the autoinhibitory domain [Johnson et al., 2000]. Cells which were transfected with GFP-tagged CON δC CaMK-II, sub-cultured on FN-coated coverslips and imaged using Fm and TIRFm exhibited a rounded morphology (Fig. 3B) and overall were less adherent than cells transfected with WT CaMK-II. These cells also failed to migrate on FN (supplemental movie 4).
Both Constitutive Activation and Inhibition of CaMK-II Disrupt Cell Motility
To further assess the effect of CaMK-II activity on cell motility, cells were sub-cultured on FN in the presence of CaMK-II inhibitors as indicated above or were transfected with WT or CON δC CaMK-II 24 h before sub-culture. After incubation on FN for 1 h to allow for attachment, cell motility was tracked over a 3-h time-course (n > 15 cells for each condition). Over this time course, motility rates were constant and in the same direction. Both conditions (CaMK-II inhibition and constitutive activation) significantly reduced motility rates (Fig. 4) as compared to either untreated cells or cells expressing WT CaMK-II, respectively (P < 0.01).
Fig. 4.
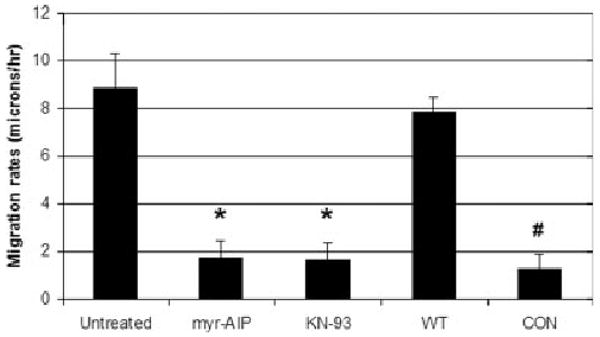
Both CaMK-II inhibition and expression of CON CaMK-II block cell motility. NIH/3T3 cells were transfected with either GFP-labeled wild-type or CON δC CaMK-II and sub-cultured on FN for 1 h. Untransfected cells were sub-cultured on FN in the presence or absence of the indicated inhibitor. After incubation on FN for 1 h, cells were imaged under phase contrast over 3 h. Migration rates (in microns/hour) were determined using Olympus Microsuite software. *P < 0.01 compared to Untreated. # P < 0.01 compared to WT.
Constitutively Active CaMK-II Disrupts Paxillin Incorporation into Focal Adhesions
Because CaMK-II localizes to the cell surface and influences cell spreading and motility, the effect of CaMK-II activity on focal adhesion proteins was next evaluated. GFP-linked focal adhesion proteins were used for localization and time-lapse studies since CON CaMK-II cells adhere poorly to FN and wash away when endogenous proteins are immunolocalized. Cells were co-transfected with either non-fluorescent WT or CON δC CaMK-II and one GFP-tagged focal adhesion protein (paxillin, FAK, vinculin or talin) as indicated (Fig. 5). After 24 h, cells were sub-cultured on FN-coated coverslips for 3 h and imaged using conventional fluorescent microscopy. WT CaMK-II expressing cells exhibited robust adhesions as visualized with each of the four focal adhesion proteins (Fig. 5). In contrast, cells expressing CON CaMK-II exhibited a near-complete loss of paxillin-containing focal adhesions, while FAK, vinculin and talin remained in focal adhesions. All transfected cells exhibited the typical rounded morphology characteristic of CON CaMK-II expressing cells. Paxillin was re-incorporated into focal adhesions when cells expressing CON CaMK-II were sub-cultured on FN in the presence of 20 μM myr-AIP (CON/myr-AIP). WT CaMK-II cells also exhibited enhanced focal adhesions when treated with myr-AIP, although it was a less significant increase (data not shown), since focal adhesions were already abundant.
Fig. 5.
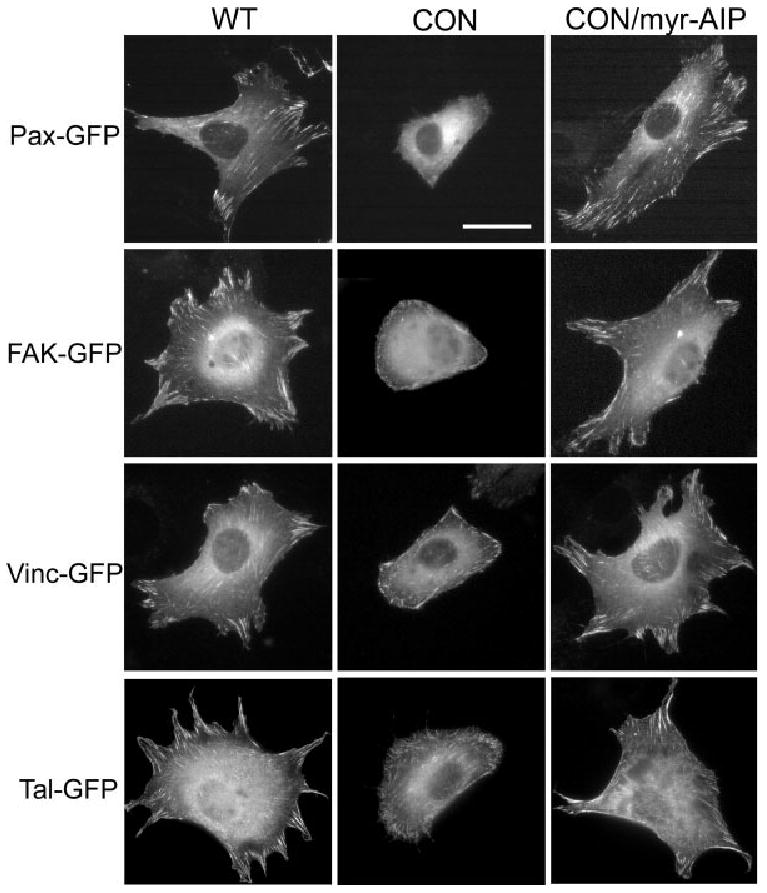
Constitutively Active CaMK-II impairs focal adhesion formation. Cells were co-transfected with either unlabeled wild-type or CON δC CaMK-II and the indicated GFP-tagged protein. Cells were sub-cultured on FN for 3 h in the presence or absence of 20 μM myr-AIP and then imaged under traditional fluorescence microscopy. Scale bar, 50 μm.
Because paxillin was preferentially, but reversibly eliminated from focal adhesions in cells expressing CON CaMK-II, the rate of paxillin recovery to focal adhesions after myr-AIP treatment was assessed in single cells using GFP-paxillin time lapse microscopy. Adjacent non-transfected and transfected (CON CaMK-II and GFP-paxillin) cells were imaged under phase contrast and conventional fluorescent microscopy after sub-culture on FN and treatment with myr-AIP (Fig. 6A). At the earliest time point, the transfected cell was rounded and lacked detectable paxillin-containing adhesions but exhibited GFP-paxillin fluorescence within the cytosol. Within 1 h of myr-AIP treatment, paxillin re-incorporation into focal adhesions was evident (Fig. 6A). This increased over 3 h as both cells spread. To further examine the specific effect of CON CaMK-II on focal adhesions, cells were co-transfected with non-fluorescent CON δC CaMK-II, GFP-vinculin and dsRed-paxillin, sub-cultured on FN and then imaged before and 1 h after treatment with myr-AIP. Within 1 h of treatment, paxillin containing focal adhesions preferentially re-appeared (arrowheads, Fig. 6B), many of which co-localized with vinculin-containing adhesions. These results suggest that CaMK-II activity preferentially affects paxillin dynamics in focal adhesions.
Fig. 6.
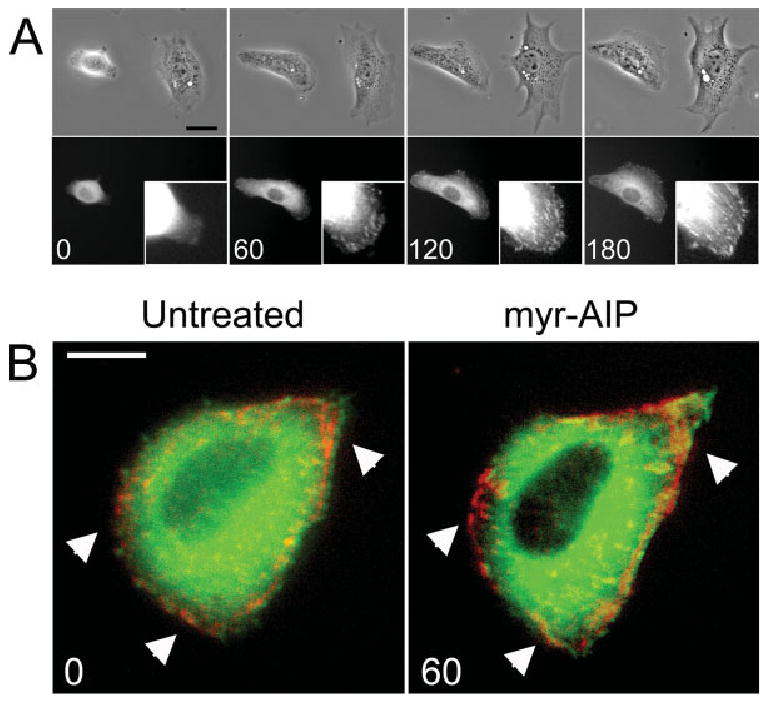
Inhibition of CON δC CaMK-II restores paxillin containing adhesions within 1 h. (A) Cells were co-transfected with unlabeled CON δC CaMK-II and GFP-paxillin. Cells were plated on FN for 3 h, re-fed with medium containing 20 μM myr-AIP and then imaged under phase contrast and traditional fluorescence microscopy 0, 60, 120 and 180 min later. Insets represent the lower right-hand corner of the transfected cell. Scale bar, 50 μm. (B) Cells were transfected with CON δC CaMK-II, GFP-vinculin and dsRed-paxillin and then sub-cultured on FN. Cells were then imaged under traditional fluorescent microscopy directly before and 1 h after treatment with 20 μM myr-AIP. Arrowheads indicate regions where myr-AIP treatment induces increased paxillin localization in focal adhesions. Scale bar, 10 μm.
Inhibition of Endogenous CaMK-II Increases the Size of Paxillin-GFP Labeled Focal Adhesions
The influence of endogenous CaMK-II on focal adhesions was next evaluated. Cells were transfected with GFP-paxillin alone and sub-cultured on FN for 3 h in the presence or absence of 20 μM myr-AIP. Compared to untreated cells (Fig. 7A), treatment with myr-AIP increased the size of GFP-paxillin adhesions (Fig. 7B). Focal adhesion size was quantified for each condition (n > 600 focal adhesions) and confirmed this distinct shift to larger focal adhesions in cells treated with myr-AIP (Fig. 7C). Furthermore, the effects of myr-AIP treatment were specific to paxillin as FAK, vinculin and talin showed no significant change in focal adhesion size in the presence of myr-AIP (Fig. 7D). Thus, the inhibition of CaMK-II disrupts cell motility by overstabilizing paxillin in focal adhesions whereas constitutively active CaMK-II blocks cell motility and attachment by eliminating paxillin from focal adhesions.
Fig. 7.
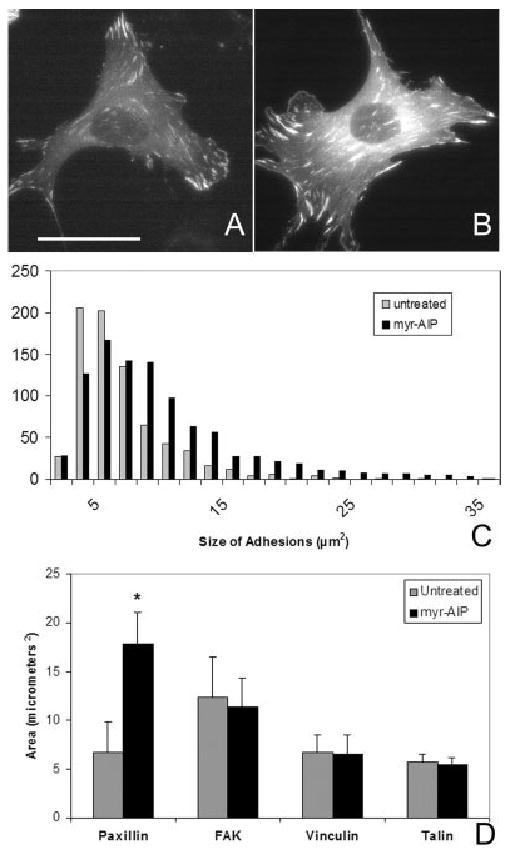
Inhibition of endogenous CaMK-II enlarges paxillin containing adhesions. GFP-paxillin expressing cells were sub-cultured on FN for 3 h in the presence of vehicle alone (A) or 20 μM myr-AIP (B) and imaged using traditional fluorescence microscopy. Scale bar, 50 μm. (C) The area of each GFP-paxillin adhesion (from >10 cells and >600 adhesions for each condition) was calculated. The threshold area for focal adhesions was 1.7 μm2. (D) The average focal adhesion size was determined in cells transfected with either GFP-labeled paxillin, FAK, vinculin or talin after sub-culturing on FN for 3 h in the presence or absence of 20 μM myr-AIP.
Constitutively Active CaMK-II Decreases Phospho-Tyrosine Levels on FAK and Paxillin
β1 integrin-mediated cell attachment stimulates a series of tyrosine phosphorylations on adhesion proteins, which form phosphorylated SH2 and SH3 domains necessary for focal adhesion assembly [Bellis et al., 1995; Schaller and Parsons, 1995; Cary et al., 1998; Mitra et al., 2005]. In particular, tyrosine phosphorylation of FAK on Y925 and paxillin on Y31 promotes focal adhesion assembly [Bellis et al., 1995; Schlaepfer et al., 2004; Sieg et al., 2000]. Because CaMK-II activity disrupted paxillin incorporation into focal adhesions, we evaluated whether CaMK-II influences tyrosine phosphorylation of FAK and paxillin by co-transfecting GFP-FAK or GFP-paxillin with either WT or CON CaMK-II. After sub-culturing transfected cells on FN for 3 h either with or without 20 μM myr-AIP, as described above, GFP-FAK and paxillin lysates were immunoblotted with residue-specific phospho-tyrosine antibodies and integrated densitometrically (Figs. 8A and 8B). CON CaMK-II expression significantly reduced FAK phospho-Y925 levels as compared to cells transfected with WT CaMK-II, but did not impair FAK autophosphorylation (Y397). Similarly, CON CaMK-II expression significantly decreased paxillin Y31 phosphorylation. Both FAK phospho-Y925 and paxillin phospho-Y31 levels were partially restored when CON CaMK-II expressing cells were treated with 20 μM myr-AIP (CON/myr-AIP).
Fig. 8.
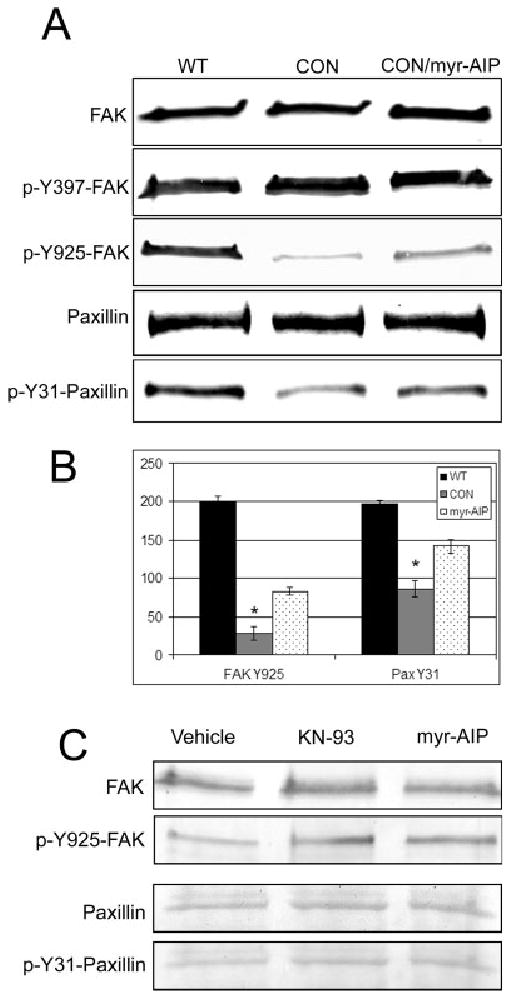
Constitutively active CaMK-II decreases phospho-tyrosine levels on FAK and paxillin. (A) Cells were co-transfected with GFP-FAK or GFP-paxillin and either wild-type (WT) δC CaMK-II or constitutively active (CON) δC CaMK-II and then sub-cultured on FN for 3 h. Cells expressing CON CaMK-II were treated with vehicle alone or with 20 μM myr-AIP during sub-culture (as indicated). GFP-FAK and GFP-paxillin lysates were then probed with the indicated antibody directed against the total protein or phosphorylated residue. GFP-FAK and GFP-paxillin possess Mr of 132 and 95 kDa, respectively. (B) Densitometric analysis was averaged from three different immunoblots for FAK phospho-Y925 and paxillin phospho-Y31. *P < 0.01 compared to WT. (C) Untransfected cells were sub-cultured on FN for 3 h in the presence of either vehicle control, 10 μM KN-93 or 20 μM myr-AIP. Cells were harvested and probed with the indicated antibodies.
The effects of CaMK-II inhibition on endogenous FAK and paxillin phospho-tyrosine levels were also examined. Cells were sub-cultured on FN for 3 h in the presence of either vehicle control, 10 μM KN-93 or 20 μM myr-AIP (Fig. 8C). Although FAK and paxillin phospho-tyrosine levels are known to already be elevated in adherent cells [Bellis et al., 1995; Schaller and Parsons, 1995; Cary et al., 1998; Vadlamudi et al., 2002; Webb et al., 2004; Mitra et al., 2005], inhibition of CaMK-II induced a slight increase in phospho-Y925 on FAK and phospho-Y31 on paxillin, as compared to vehicle control cells, without affecting total expression levels of FAK and paxillin (Fig. 8C). Together, these results indicate that CaMK-II promotes focal adhesion disassembly by stimulating tyrosine dephosphorylation of FAK and paxillin.
Discussion
This study provides the first direct evidence of a link between CaMK-II activity and focal adhesion turnover as a means of influencing cell motility. Previous studies have shown that CaMK-II influences cell adhesion by disrupting β1 integrin clustering through either direct phosphorylation of β1 integrin [Wennerberg et al., 1998; Takahashi, 2001] or phosphorylation of integrin cytoplasmic associated protein-1 alpha, ICAP-1α [Blystone et al., 1999]. However, FAK autophosphorylation, which reflects β1 integrin engagement [Mitra et al., 2005], was not affected by CON CaMK-II or CaMK-II inhibition. Furthermore, NIH/3T3 embryonic fibroblasts, lack ICAP-1α [Bouvard et al., 2003]. Our results are consistent with previous findings implicating Ca2+ transients in focal adhesion disassembly [Conklin et al., 2005] and suggest a novel means by which CaMK-II influences focal adhesion turnover by transiently promoting the tyrosine dephosphorylation of both FAK and paxillin.
While the results presented here support a role for CaMK-II in focal adhesion turnover, the relevant substrate has not been determined. The loss of lamellipodial dynamics by CaMK-II inhibition is consistent with the ability of CaMK-II to phosphorylate and activate Tiam1, a Rac GEF [Fleming et al., 1999; Hamelers et al., 2005]. However, constitutively active Rac increases cell spreading [Kerr et al., 2008], while constitutively active CaMK-II causes cell rounding. Because of the profound effect of CaMK-II activity on paxillin localization to focal adhesions, we suggest that CaMK-II mediates both focal adhesion turnover and Rac activation to drive cell motility (Fig. 9).
Fig. 9.
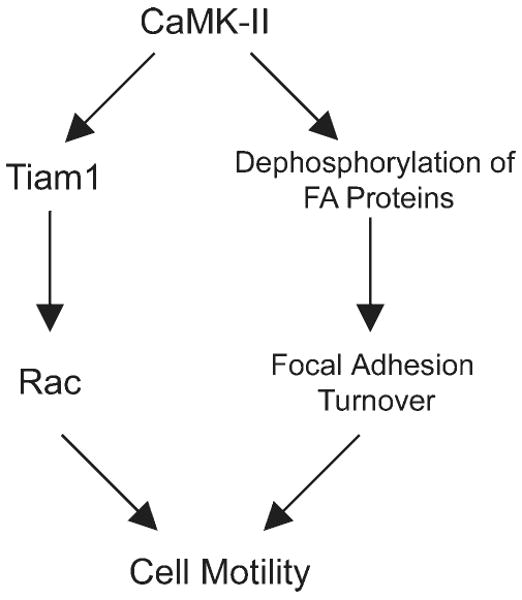
Dual role for CaMK-II in Cell Motility. Schematic demonstrating a possible mechanism by which CaMK-II enables cell motility by activating Rac and inducing focal adhesion turnover.
Another potential CaMK-II target, FAK, has been shown to be phosphorylated by CaMK-II on S843 within minutes of G-protein receptor activation [Fan et al., 2005]. However, the phosphorylation status of FAK S843 was not altered upon CaMK-II overexpression or inhibition (data not shown), and therefore it is unlikely that CaMK-II directly phosphorylates FAK under the conditions described here. Rather, the reduction in FAK and paxillin phospho-tyrosine levels caused by constitutively active CaMK-II is most consistent with the activation of a tyrosine phosphatase.
The most likely candidate tyrosine phosphatase downstream of CaMK-II activity is the SH2 domain-containing protein tyrosine phosphatase 2 (SHP-2), also known as PTPN11 or PTP1D. CaMK-II has been implicated upstream from SHP-2 in T-cell signaling [McGargill et al., 2005], but this link has not been investigated with respect to cell motility and/or focal adhesion turnover. SHP-2, a non-receptor tyrosine phosphatase, translocates from the ER to focal adhesions in response to src signaling [Wang et al., 2006] to dephosphorylate FAK on Y925 and paxillin on Y31, thus promoting detachment from the extracellular matrix [Vadlamudi et al., 2002]. Like CaMK-II inhibition, the knockout or inhibition of SHP-2 increases focal adhesion size and blocks cell motility in both cells in culture and in mouse embryos [Yu et al., 1998; Manes et al., 1999; Inagaki et al., 2000; Saxton et al., 2000; MacGillivray et al., 2003; Wang et al., 2005]. While SHP-2 activity is known to be regulated by tyrosine phosphorylation, this enzyme can also be phosphorylated on serine/threonine residues [Poole and Jones, 2005]. However, SHP-2 does not appear to be a direct substrate of CaMK-II [McGargill et al., 2005], therefore CaMK-II may phosphorylate undetermined SHP-2 regulators to influence its activation or localization.
As with CaMK-II and SHP-2 perturbations, FAK knockout fibroblasts exhibit decreased focal adhesion turnover [Ilic et al., 1995], while hyperactive FAK accelerates focal adhesion turnover [von Wichert et al., 2003]. Results such as these, which suggest that there are optimal activity levels for CaMK-II, SHP-2 and FAK, support common roles for each enzyme in promoting the cyclical formation and breakdown of focal adhesions as cells migrate.
In summary, the attachment of cells to FN triggers focal adhesion assembly, which eventually yields a localized, transient Ca2+ release to activate CaMK-II. Activated CaMK-II then directly or indirectly activates a protein tyrosine phosphatase, most likely SHP-2, to dephosphorylate a limited number of tyrosine residues on FAK and paxillin, thus promoting focal adhesion turnover. CaMK-II simultaneously drives cell motility by activating Rac through Tiam1 to induce lamellipodial extension (Fig. 9). Such a molecular mechanism may explain the role of CaMK-II in convergent extension cell movements during embryogenesis [Kuhl et al., 2000], in which FAK, paxillin, SHP-2 and Rac have all been implicated [Saxton et al., 2000; Henry et al., 2001; Crawford et al., 2003; Tahinci and Symes, 2003].
Supplementary Material
Acknowledgments
The authors thank Colleen Brosnahan and Jamie McLeod for their assistance with sample preparations.
Contract grant sponsor: National Science Foundation; Contract grant number: IOS-0238821.
Footnotes
This article contains supplementary material available via the Internet at http://www.interscience.wiley.com/jpages/0886-1544/suppmat.
References
- Bellis SL, Miller JT, Turner CE. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270(29):17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- Bilato C, Curto KA, Monticone RE, Pauly RR, White AJ, Crow MT. The inhibition of vascular smooth muscle cell migration by peptide and antibody antagonists of the alphavbeta3 integrin complex is reversed by activated calcium/calmodulin- dependent protein kinase II. J Clin Invest. 1997;100(3):693–704. doi: 10.1172/JCI119582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blystone SD, Slater SE, Williams MP, Crow MT, Brown EJ. A molecular mechanism of integrin crosstalk: Alphavbeta3 suppression of calcium/calmodulin-dependent protein kinase II regulates alpha5beta1 function. J Cell Biol. 1999;145(4):889–897. doi: 10.1083/jcb.145.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard D, Block MR. Calcium/calmodulin-dependent protein kinase II controls integrin alpha5beta1-mediated cell adhesion through the integrin cytoplasmic domain associated protein-1alpha. Biochem Biophys Res Commun. 1998;252(1):46–50. doi: 10.1006/bbrc.1998.9592. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Molla A, Block MR. Calcium/calmodulin-dependent protein kinase II controls alpha5beta1 integrin-mediated inside-out signaling. J Cell Sci. 1998;111(Partt 5):657–665. doi: 10.1242/jcs.111.5.657. [DOI] [PubMed] [Google Scholar]
- Bouvard D, Vignoud L, Dupe-Manet S, Abed N, Fournier HN, Vincent-Monegat C, Retta SF, Fassler R, Block MR. Disruption of focal adhesions by integrin cytoplasmic domain-associated protein-1 alpha. J Biol Chem. 2003;278(8):6567–6574. doi: 10.1074/jbc.M211258200. [DOI] [PubMed] [Google Scholar]
- Brown CM, Hebert B, Kolin DL, Zareno J, Whitmore L, Horwitz AR, Wiseman PW. Probing the integrin-actin linkage using high-resolution protein velocity mapping. J Cell Sci. 2006;119(Part 24):5204–5214. doi: 10.1242/jcs.03321. [DOI] [PubMed] [Google Scholar]
- Caran N, Johnson LD, Jenkins KJ, Tombes RM. Cytosolic targeting domains of gamma and delta calmodulin-dependent protein kinase II. J Biol Chem. 2001;276(45):42514–42519. doi: 10.1074/jbc.M103013200. [DOI] [PubMed] [Google Scholar]
- Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140(1):211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin MW, Lin MS, Spitzer NC. Local calcium transients contribute to disappearance of pFAK, focal complex removal and deadhesion of neuronal growth cones and fibroblasts. Dev Biol. 2005;287(1):201–212. doi: 10.1016/j.ydbio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Crawford BD, Henry CA, Clason TA, Becker AL, Hille MB. Activity and distribution of paxillin, focal adhesion kinase, and cadherin indicate cooperative roles during zebrafish morphogenesis. Mol Biol Cell. 2003;14(8):3065–3081. doi: 10.1091/mbc.E02-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley CA, Faison MO, Kirsch TL, Lee JA, Seward ME, Tombes RM. Laminin activates CaMK-II to stabilize nascent embryonic axons. Brain Res. 2006;1092(1):59–68. doi: 10.1016/j.brainres.2006.03.099. [DOI] [PubMed] [Google Scholar]
- Faison MO, Perozzi EF, Caran N, Stewart JK, Tombes RM. Axonal localization of delta Ca2+/calmodulin-dependent protein kinase II in developing P19 neurons. Int J Dev Neurosci. 2002;20(8):585–592. doi: 10.1016/s0736-5748(02)00107-7. [DOI] [PubMed] [Google Scholar]
- Fan RS, Jacamo RO, Jiang X, Sinnett-Smith J, Rozengurt E. G protein-coupled receptor activation rapidly stimulates focal adhesion kinase phosphorylation at Ser-843. Mediation by Ca2+, calmodulin, and Ca2+/calmodulin-dependent kinase II. J Biol Chem. 2005;280(25):24212–24220. doi: 10.1074/jbc.M500716200. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39(2):283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Fleming IN, Elliott CM, Buchanan FG, Downes CP, Exton JH. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J Biol Chem. 1999;274(18):12753–12758. doi: 10.1074/jbc.274.18.12753. [DOI] [PubMed] [Google Scholar]
- Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004;6(10):977–983. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- Giannone G, Ronde P, Gaire M, Haiech J, Takeda K. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J Biol Chem. 2002;277(29):26364–26371. doi: 10.1074/jbc.M203952200. [DOI] [PubMed] [Google Scholar]
- Giannone G, Ronde P, Gaire M, Beaudouin J, Haiech J, Ellenberg J, Takeda K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J Biol Chem. 2004;279(27):28715–28723. doi: 10.1074/jbc.M404054200. [DOI] [PubMed] [Google Scholar]
- Hagel M, George EL, Kim A, Tamimi R, Opitz SL, Turner CE, Imamoto A, Thomas SM. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol Cell Biol. 2002;22(3):901–915. doi: 10.1128/MCB.22.3.901-915.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelers IH, Olivo C, Mertens AE, Pegtel DM, van der Kammen RA, Sonnenberg A, Collard JG. The Rac activator Tiam1 is required for (alpha)3(beta)1-mediated laminin-5 deposition, cell spreading, and cell migration. J Cell Biol. 2005;171(5):871–881. doi: 10.1083/jcb.200509172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CA, Crawford BD, Yan YL, Postlethwait J, Cooper MS, Hille MB. Roles for zebrafish focal adhesion kinase in notochord and somite morphogenesis. Dev Biol. 2001;240(2):474–487. doi: 10.1006/dbio.2001.0467. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Ishikawa T. Molecular pharmacology of calmodulin pathways in the cell functions. Cell Calcium. 1992;13(6–7):465–472. doi: 10.1016/0143-4160(92)90059-2. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A, Sandborg RR, Horwitz AF. Adhesion in cell migration. Curr Opin Cell Biol. 1995;7(5):697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Illario M, Cavallo AL, Monaco S, Di Vito E, Mueller F, Marzano LA, Troncone G, Fenzi G, Rossi G, Vitale M. Fibronectin-induced proliferation in thyroid cells is mediated by alphavbeta3 integrin through Ras/Raf-1/MEK/ERK and calcium/CaMKII signals. J Clin Endocrinol Metab. 2005;90(5):2865–2873. doi: 10.1210/jc.2004-1520. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Noguchi T, Matozaki T, Horikawa T, Fukunaga K, Tsuda M, Ichihashi M, Kasuga M. Roles for the protein tyrosine phosphatase SHP-2 in cytoskeletal organization, cell adhesion and cell migration revealed by overexpression of a dominant negative mutant. Oncogene. 2000;19(1):75–84. doi: 10.1038/sj.onc.1203204. [DOI] [PubMed] [Google Scholar]
- Johnson LD, Willoughby CA, Burke SH, Paik DS, Jenkins KJ, Tombes RM. Delta Ca(2+)/calmodulin-dependent protein kinase II isozyme-specific induction of neurite outgrowth in P19 embryonal carcinoma cells. J Neurochem. 2000;75(6):2380–2391. doi: 10.1046/j.1471-4159.2000.0752380.x. [DOI] [PubMed] [Google Scholar]
- Kerr BA, Otani T, Koyama E, Freeman TA, Enomoto-Iwamoto M. Small GTPase protein Rac-1 is activated with maturation and regulates cell morphology and function in chondrocytes. Exp Cell Res. 2008;314(6):1301–1312. doi: 10.1016/j.yexcr.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Moon RT. Wnt and calcium signaling: Beta-catenin-independent pathways. Cell Calcium. 2005;38(3–4):439–446. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Kuhl M, Sheldahl LC, Park M, Miller JR, Moon RT. The Wnt/ Ca2+ pathway: A new vertebrate Wnt signaling pathway takes shape. Trends Genet. 2000;16(7):279–283. doi: 10.1016/s0168-9525(00)02028-x. [DOI] [PubMed] [Google Scholar]
- Laabich A, Cooper NG. Neuroprotective effect of AIP on N-methyl-D-aspartate-induced cell death in retinal neurons. Brain Res Mol Brain Res. 2000;85(1–2):32–40. doi: 10.1016/s0169-328x(00)00226-6. [DOI] [PubMed] [Google Scholar]
- Lantsman K, Tombes RM. CaMK-II oligomerization potential determined using CFP/YFP FRET. Biochim Biophys Acta. 2005;1746(1):45–54. doi: 10.1016/j.bbamcr.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: A physically integrated molecular process. Cell. 1996;84(3):359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lee SY, Voronov S, Letinic K, Nairn AC, Di Paolo G, De Camilli P. Regulation of the interaction between PIPKI gamma and talin by proline-directed protein kinases. J Cell Biol. 2005;168(5):789–799. doi: 10.1083/jcb.200409028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Park ZY, Lin D, Brahmbhatt AA, Rio MC, Yates JR, III, Klemke RL. Regulation of cell migration and survival by focal adhesion targeting of Lasp-1. J Cell Biol. 2004;165(3):421–432. doi: 10.1083/jcb.200311045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg MS, Curto KA, Bilato C, Monticone RE, Crow MT. Regulation of vascular smooth muscle migration by mitogen-activated protein kinase and calcium/calmodulin-dependent protein kinase II signaling pathways. J Mol Cell Cardiol. 1998;30(11):2377–23789. doi: 10.1006/jmcc.1998.0795. [DOI] [PubMed] [Google Scholar]
- MacGillivray M, Herrera-Abreu MT, Chow CW, Shek C, Wang Q, Vachon E, Feng GS, Siminovitch KA, McCulloch CA, Downey GP. The protein tyrosine phosphatase SHP-2 regulates interleukin-1-induced ERK activation in fibroblasts. J Biol Chem. 2003;278(29):27190–27198. doi: 10.1074/jbc.M213083200. [DOI] [PubMed] [Google Scholar]
- Manes S, Mira E, Gomez-Mouton C, Zhao ZJ, Lacalle RA, Martinez AC. Concerted activity of tyrosine phosphatase SHP-2 and focal adhesion kinase in regulation of cell motility. Mol Cell Biol. 1999;19(4):3125–3135. doi: 10.1128/mcb.19.4.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PW, Maxfield FR. Transient increases in cytosolic free calcium appear to be required for the migration of adherent human neutrophils. J Cell Biol. 1990;110(1):43–52. doi: 10.1083/jcb.110.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGargill MA, Sharp LL, Bui JD, Hedrick SM, Calbo S. Active Ca2+/calmodulin-dependent protein kinase II gamma B impairs positive selection of T cells by modulating TCR signaling. J Immunol. 2005;175(2):656–664. doi: 10.4049/jimmunol.175.2.656. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: In command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Pauly RR, Bilato C, Sollott SJ, Monticone R, Kelly PT, Lakatta EG, Crow MT. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation. 1995;91(4):1107–1115. doi: 10.1161/01.cir.91.4.1107. [DOI] [PubMed] [Google Scholar]
- Pfleiderer PJ, Lu KK, Crow MT, Keller RS, Singer HA. Modulation of vascular smooth muscle cell migration by calcium/ calmodulin-dependent protein kinase II-delta 2. Am J Physiol Cell Physiol. 2004;286(6):C1238–C1245. doi: 10.1152/ajpcell.00536.2003. [DOI] [PubMed] [Google Scholar]
- Poole AW, Jones ML. A SHPing tale: Perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. 2005;17(11):1323–1332. doi: 10.1016/j.cellsig.2005.05.016. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Praetorius J, Nielsen S, Frokiaer J, Spring KR. Beta1-integrins in the primary cilium of MDCK cells potentiate fibronectin-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2004;287(5):F969–F78. doi: 10.1152/ajprenal.00096.2004. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: Integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Saxton TM, Ciruna BG, Holmyard D, Kulkarni S, Harpal K, Rossant J, Pawson T. The SH2 tyrosine phosphatase shp2 is required for mammalian limb development. Nat Genet. 2000;24(4):420–423. doi: 10.1038/74279. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15(5):2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta. 2004;1692(2–3):77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Shao H, Chou J, Baty CJ, Burke NA, Watkins SC, Stolz DB, Wells A. Spatial localization of m-calpain to the plasma membrane by phosphoinositide biphosphate binding during epidermal growth factor receptor-mediated activation. Mol Cell Biol. 2006;26(14):5481–5496. doi: 10.1128/MCB.02243-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldahl LC, Slusarski DC, Pandur P, Miller JR, Kühl M, Moon RT. Dishevelled activates Ca2+ flux. PKC, and CaMKII in vertebrate embryos. J Cell Biol. 2003;161:769–777. doi: 10.1083/jcb.200211094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIbeta functions as an F-actin targeting module that localizes CaM-KIIalpha/beta heterooligomers to dendritic spines. Neuron. 1998;21(3):593–606. doi: 10.1016/s0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181(3):968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Takahashi K. Reduced cell adhesion during mitosis by threonine phosphorylation of beta1 integrin. J Cell Physiol. 2003;197(2):297–305. doi: 10.1002/jcp.10354. [DOI] [PubMed] [Google Scholar]
- Tahinci E, Symes K. Distinct functions of Rho and Rac are required for convergent extension during Xenopus gastrulation. Dev Biol. 2003;259(2):318–335. doi: 10.1016/s0012-1606(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Takahashi K. The linkage between beta1 integrin and the actin cytoskeleton is differentially regulated by tyrosine and serine/ threonine phosphorylation of beta1 integrin in normal and cancerous human breast cells. BMC Cell Biol. 2001;2:23. doi: 10.1186/1471-2121-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombes RM, Grant S, Westin EH, Krystal G. G1 cell cycle arrest and apoptosis are induced in NIH 3T3 cells by KN-93, an inhibitor of CaMK-II (the multifunctional Ca2+/CaM kinase) Cell Growth Differ. 1995;6(9):1063–1070. [PubMed] [Google Scholar]
- Tombes RM, Mikkelsen RB, Jarvis WD, Grant S. Downregulation of delta CaM kinase II in human tumor cells. Biochim Biophys Acta. 1999;1452(1):1–11. doi: 10.1016/s0167-4889(99)00113-5. [DOI] [PubMed] [Google Scholar]
- Tombes RM, Faison MO, Turbeville JM. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene. 2003;322:17–31. doi: 10.1016/j.gene.2003.08.023. [DOI] [PubMed] [Google Scholar]
- Tran PO, Hinman LE, Unger GM, Sammak PJ. A wound-induced [Ca2+]i increase and its transcriptional activation of immediate early genes is important in the regulation of motility. Exp Cell Res. 1999;246(2):319–326. doi: 10.1006/excr.1998.4239. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Adam L, Nguyen D, Santos M, Kumar R. Differential regulation of components of the focal adhesion complex by heregulin: Role of phosphatase SHP-2. J Cell Physiol. 2002;190(2):189–199. doi: 10.1002/jcp.10054. [DOI] [PubMed] [Google Scholar]
- von Wichert G, Haimovich B, Feng GS, Sheetz MP. Force-dependent integrin-cytoskeleton linkage formation requires downregulation of focal complex dynamics by Shp2. EMBO J. 2003;22(19):5023–5035. doi: 10.1093/emboj/cdg492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang FM, Liu HQ, Liu SR, Tang SP, Yang L, Feng GS. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1beta in vivo and in vitro. Breast Cancer Res Treat. 2005;89(1):5–14. doi: 10.1007/s10549-004-1002-z. [DOI] [PubMed] [Google Scholar]
- Wang Q, Herrera Abreu MT, Siminovitch K, Downey GP, McCulloch CA. Phosphorylation of SHP-2 regulates interactions between the endoplasmic reticulum and focal adhesions to restrict interleukin-1-induced Ca2+ signaling. J Biol Chem. 2006;281(41):31093–31105. doi: 10.1074/jbc.M606392200. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin. ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6(2):154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Fassler R, Warmegard B, Johansson S. Mutational analysis of the potential phosphorylation sites in the cytoplasmic domain of integrin beta1A. Requirement for threonines 788–789 in receptor activation. J Cell Sci. 1998;111(8):1117–1126. doi: 10.1242/jcs.111.8.1117. [DOI] [PubMed] [Google Scholar]
- Yu DH, Qu CK, Henegariu O, Lu X, Feng GS. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem. 1998;273(33):21125–21131. doi: 10.1074/jbc.273.33.21125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.