

SUMMARY
Oncogenic fusion proteins are capable of initiating tumorigenesis but the role of their wild-type counterparts in this process is poorly understood. The mixed lineage leukemia (MLL) gene undergoes chromosomal translocations, resulting in the formation of oncogenic MLL fusion proteins (MLL-FPs). Here we show that menin recruits both wild-type (wt) MLL and MLL-AF9 to the loci of Hox genes to activate their transcription. Wild-type MLL not only catalyzes histone methylation at key target genes but also controls distinct MLL-AF9-induced histone methylation. Notably, the wt Mll allele is required for MLL-AF9-induced leukemogenesis and maintenance of MLL-AF9-transformed cells. These findings suggest an essential cooperation between an oncogene and its wild-type counterpart in MLL-AF9-induced leukemogenesis.
Keywords: MLL, mixed lineage leukemia, MLL fusion protein, MLL-AF9, menin, histone methylation, Leukemia stem cells, epigenetics
Introduction
Multiple oncogenic fusion proteins resulting from chromosomal translocations are capable of initiating tumorigenesis, but little is known about the role of the remaining wild-type allele in this process. The mixed lineage leukemia gene (MLL) is fused with one of over 60 distinct partner genes through chromosomal translocations in various human acute leukemias, resulting in the formation of multiple MLL fusion proteins (MLL-FPs) (Hess, 2004, Krivtsov, et al., 2007). MLL-FPs are capable of leukemic transformation and dysregulation of multiple Hox genes including Hoxa9. In one well-characterized example, MLL-AF10 directly interacts with Dot1L, the only known H3K79-specific methyltransferase, via the AF10 moiety and recruits Dot1L to the Hoxa9 locus to aberrantly increase H3K79 dimethylation (Okada, et al., 2005). The H3K79 methyltransferase activity of Dot1L is required for enhanced transcription of certain Hox genes and for MLL-AF10-induced bone marrow (BM) transformation. MLL-AF4 also enhances Dot1L-mediated H3K79 methylation at Hox genes (Krivtsov, et al., 2008) and the wild type counterparts of additional MLL fusion partners such as AF4 and ENL have been shown to interact with Dot1L in a large protein complex (Bitoun, et al., 2007, Mueller, et al., 2007), illustrating one common mechanism for transformation.
Wild type (wt) MLL is homologous to the Drosophila trithorax gene, a positive regulator of gene expression. Wt MLL is proteolytically cleaved into two parts, MLL-N and MLL-C by the protease Taspase 1 (Hsieh, et al., 2003). MLL-C contains a conserved SET domain (Suv3–9, Enhancer of zeste and Trithorax), which catalyzes histone H3 lysine 4 (H3K4) methylation and upregulates transcription of HOX genes in fibroblasts or epithelial cell lines (Milne, et al., 2002, Nakamura, et al., 2002). H3K4 trimethylation (H3K4m3) is associated with euchromatin and active genes and specifically recruits chromatin-remodeling proteins to stimulate gene expression (Berger, 2007, Flanagan, et al., 2005, Li, et al., 2006).
Wt MLL forms a large complex with several proteins including menin (Hughes, et al., 2004, Yokoyama, et al., 2005), a nuclear DNA-binding protein that is mutated in an inherited human endocrine tumor syndrome (La, et al., 2004). Menin interacts with the N-terminus of both MLL and MLL-FPs (Yokoyama, et al., 2005), increases H3K4 trimethylation (H3K4m3) at the Hoxa9 locus, and upregulates its transcription in MLL-FP-transformed hematopoietic cells (Chen, et al., 2006, Yokoyama, et al., 2005). Moreover, menin is required for proliferation of cells transformed by MLL-AF9 fusion protein (MA9 hereafter) (Caslini, et al., 2007, Chen, et al., 2006). However, little is known as to whether menin affects MA9-regulated H3K79 methylation and whether wt MLL is important for MA9-mediated leukemic transformation.
The potential role (or lack thereof) of wt MLL in MLL-AF9 leukemogenesis has not been addressed. On the one hand, despite a lack of the wt MLL SET domain, MA9 remains capable of initiating leukemogenesis when introduced into wt murine or human hematopoietic progenitors (Barabe, et al., 2007, Krivtsov, et al., 2006, Somervaille, et al., 2006, Wei, et al., 2008). Moreover, MLL-AF10 reduces H3K4 dimethylation at the Hoxa9 locus (Okada, et al., 2005), which is mediated at least partly by wt MLL. Further, in MLL-FP-expressing human leukemia cells, which in theory lose one of the two wt MLL alleles in chromosomal translocation, expression of wt MLL target genes such as Hoxa9 is even higher than in non-MLL-FP-leukemia cells (Armstrong, et al., 2002). These studies raise the possibility that wt MLL is not crucial for oncogenic transformation by MLL-FPs. On the other hand, wt MLL is crucial for H3K4 methylation and expression of Hox genes in fibroblasts and HeLa cells (Milne, et al., 2002, Nakamura, et al., 2002). Moreover, wt Mll excision compromises the function of hematopoietic stem cells (HSCs) and expression of 5' Hoxa genes including Hoxa9 (Jude, et al., 2007, McMahon, et al., 2007), yet these Hox genes are upregulated in an MA9-transformed leukemia stem cell (LSC)-enriched population (Krivtsov, et al., 2006), raising the possibility that wt MLL is involved in MA9-induced leukemogenesis. Therefore, whether the wt MLL allele is crucial for MA9-induced leukemogenesis remains unresolved.
A long list of oncogenic fusion proteins resulting from chromosomal translocations has been identified in various leukemias and solid cancers (Nambiar, et al., 2008). However, it is poorly understood whether the wild-type alleles influence tumorigenesis induced by the majority of the known oncogenic fusion proteins. A better understanding of the function of these wt alleles in tumorigenesis could yield insights into the mechanisms of transformation. Our earlier findings on the role of menin in proliferation and gene transcription of MA9-transformed cells prompted us to investigate the potential role of wt MLL in MA9-induced leukemogenesis.
RESULTS
Menin is required for methylation of both histone H3 lysine 4 (H3K4) and histone H3 lysine 79 (H3K79) at the Hoxa9 locus
The MLL-AF10 fusion protein has been reported to transform bone marrow (BM) by increasing Dot1L-catalyzed H3K79 methylation but repressing H3K4 methylation at the Hoxa9 locus, suggesting that H3K79-methylating Dot1L, but not H3K4-methylating wt MLL, is crucial for MLL-FP-induced leukemic transformation (Okada, et al., 2005). Although menin interacts with the N-terminus of wild-type (wt) MLL and MLL fusion proteins (Yokoyama, et al., 2005), little is known as to whether menin is crucial for H3K79 methylation at Hoxa9 in MA9-transformed bone marrow (BM) cells. To address this question, we excised the floxed Men1 gene in MA9 - transformed BM cells (AT1 cells), which harbor Men1f/f;Cre-ER, using 4-hydroxyl tamoxifen (4-OHT) to induce Cre activity (Fig. 1A, lane 2). We performed chromatin immunoprecipitation (ChIP) assays with the control and Men1-excised cells. Our results showed that Men1 excision reduced H3K79 dimethylation in two separate locations at the Hoxa9 locus, as shown by amplicons a and b (Fig. 1B). As Dot1L is the only known H3K79 methyltransferase in mammals, this finding is consistent with the notion that menin is crucial for MA9-induced Dot1L recruitment to the Hoxa9 locus and H3K79 methylation at the locus.
Figure 1. Menin is required for both H3K4 trimethylation and H3K79 dimethylation at Hoxa9 in MA9-transformed cells.
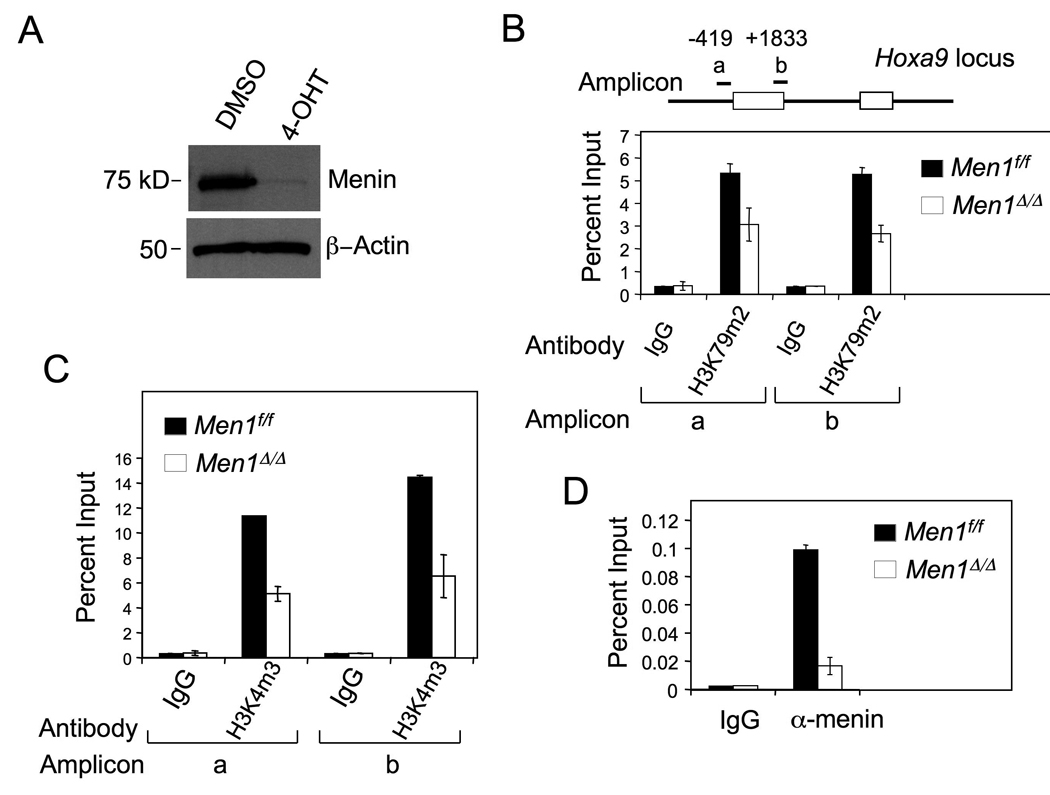
(A) Western blot for menin in control or Men1 excised MA9-transformed AT1 cells, which harbored Men1f/f;Cre-ER. The cells were treated with either control DMSO (Men1f/f) or 4-OHT (Men1Δ/Δ) to excise the floxed Men1. (B–D) ChIP assay, with two distinct amplicons, for detecting dimethylated H3K79 (B), trimethylated H3K4 (C), and menin binding (D) at Hoxa9 in Men1f/f and Men1Δ/Δ AT1 cells. Error bars denote +/− SD.
We also noted that Men1 excision reduced H3K4 trimethylation at Hoxa9 (Fig. 1C), in agreement with our previous findings (Chen, et al., 2006). However, this finding is different from the proposed role of MLL-AF10 in reducing H3K4 methylation at the Hoxa9 locus (Okada, et al., 2005), which could be at least partly mediated by wt MLL. As a control, ChIP assays showed that menin bound the promoter of Hoxa9, and menin binding was abrogated in Men1Δ/Δ cells (Fig. 1D). Together, these results demonstrate that menin is crucial not only for H3K4 methylation but also for Dot1L-mediated H3K79 methylation at the Hoxa9 locus in the MA9-transformed cells. These results raise the possibility that wt MLL is crucial for MA9-induced Hox gene expression.
Menin recruits both wt MLL and MA9 to the Hoxa9 locus
To explore whether wt MLL participates in upregulating Hox gene expression in MA9-transformed cells, we first examined whether wt MLL binds the Hoxa9 locus, and if so, whether wt MLL binding to the locus depends on menin in MA9-transformed BM cells. Since MA9 lacks the MLL-C portion of wt MLL, detection of MLL-C at the Hoxa9 locus indicates that wt MLL is recruited to the locus. Hence, we chose to use an antibody that specifically recognizes only MLL-C to detect wt MLL. ChIP assays showed that MLL-C bound the Hoxa9 locus (Fig. 2A), but the MLL-C binding was abrogated when Men1 was excised (Men1Δ/Δ) in the MA9-transformed BM cells (Fig. 2A). These results indicate that menin is required for recruiting wt MLL to Hoxa9 in the MA9-transformed cells.
Figure 2. Wt MLL and MA9 are recruited to Hoxa9 in a menin-dependent manner.
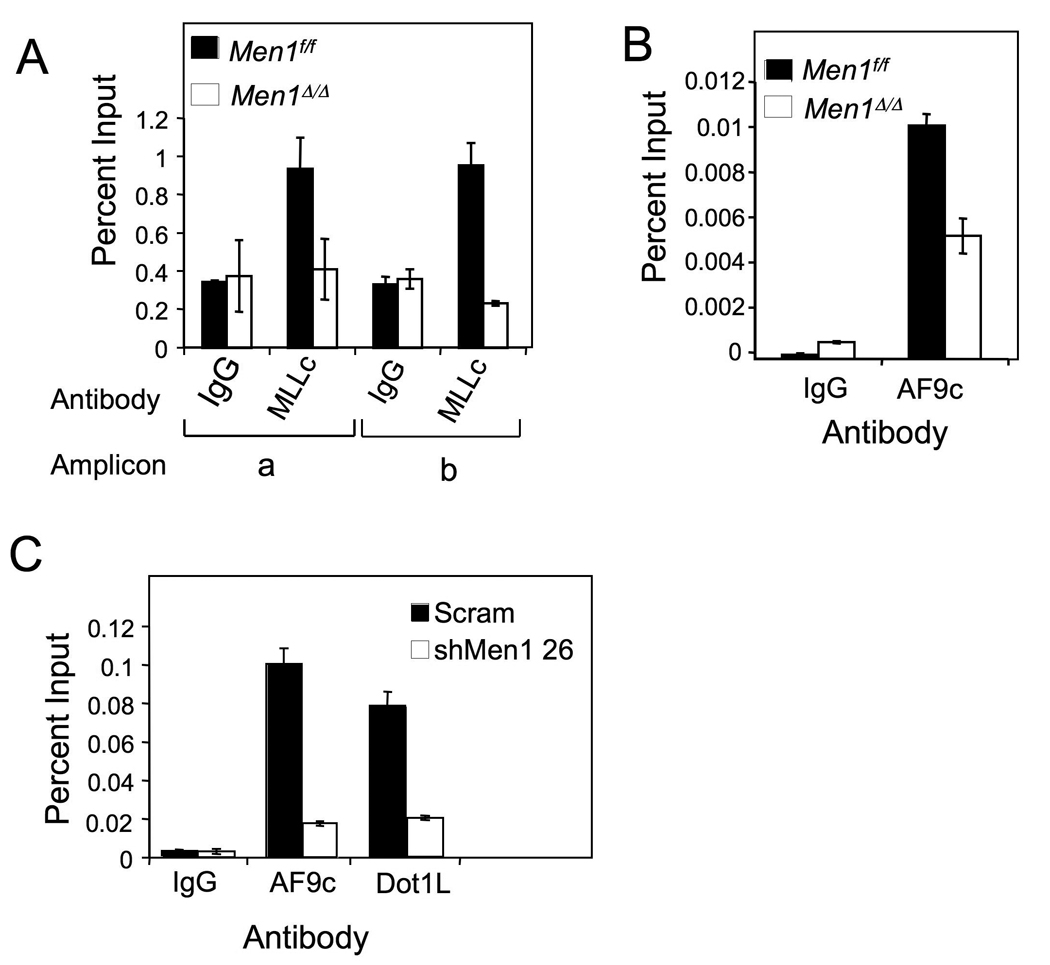
AT1 cells were treated with either DMSO (Men1f/f) or 4-OHT (Men1Δ/Δ) and processed for ChIP assay with either (A) anti-MLL-C or (B) anti-AF9 C-terminus antibodies. (C) THP-1 cells were transduced with either control scrambled or Men1 shRNA-expressing lentiviruses, and used for ChIP assay with anti-AF9 or anti-Dot1L antibodies. Error bars denote +/− SD. See also Figure S1.
As menin interacts with the N-terminus of MLL (Yokoyama, et al., 2005), we determined if menin affects recruitment of MA9 (which contains the N-terminus of MLL) to Hoxa9 by ChIP assay, using an anti-AF9 antibody that only recognizes the AF9 portion of the MA9 fusion protein (Fig. 2B). Our results indicate that MA9 bound to Hoxa9 and that Men1 excision reduced MA9 binding to the locus (Fig. 2B). As the C-terminal portion (aa 397–557) of AF9 has been reported to bind Dot1L (Zhang, et al., 2006), we tested if the AF9 part of MA9 bound Dot1L using a GST-AF9 pull-down assay, and found that the AF9 portion from MA9 bound Dot1L (Fig. S1A–C). To further evaluate whether menin affects recruitment of MA9 to the Hoxa9 locus in MA9-expressing human leukemia cells, we knocked down menin expression, using lentiviruses expressing MEN1 shRNAs, in THP-1 cells that harbor MA9 (Fig. S1D and F). MEN1 knockdown (KD) reduced the number of THP-1 cells (Fig. S1E), HOXA9 expression (Fig. S1F), and menin binding to the HOXA9 locus (Fig. S1G). Consistently, MEN1 KD also reduced the binding of both MA9 and Dot1L to the HOXA9 locus (Fig. 2C). Together with data from Figure 1, these results indicate that menin promotes recruitment of both wt MLL and MA9/Dot1L to the HOXA9 locus, thereby increasing wt MLL-mediated H3K4 methylation and Dot1L-mediated H3K79 methylation at the locus, respectively.
Wt MLL is required for proliferation and survival of MA9-transformed leukemia cells and for expression of HOX genes
It has been unresolved whether wt MLL is crucial for MLL-FP-induced leukemia. On the one hand, H3K4 methylation, which is at least partly mediated by wt MLL, is repressed in MLL-AF10-transformed cells (Okada, et al., 2005). On the other hand, wt MLL is crucial for H3K4 methylation and expression of HOX genes (Milne, et al., 2002, Nakamura, et al., 2002), some of which are crucial for BM transformation by certain MLL-FPs (Ayton, et al., 2003). Our data indicate a crucial role for menin in wt MLL recruitment to and H3K4 methylation at HOXA9 (Fig. 2A and 1C). Thus, we examined if wt MLL is important for expression of HOX and cell cycle genes and for growth of MA9-transformed bone marrow (BM) cells. We transduced AT1 cells with either control scrambled or MLL-C-targeting shRNA retroviruses (Fig. 3A, shRNA 11). The rationale for targeting only the C-terminus is to avoid affecting the mRNA encoding MA9, which lacks the MLL-C sequence. The MLL-C shRNAs, but not the scrambled vector, reduced the number of AT1 cells (Fig. 3B) and the mRNA levels of wt Mll, Hoxa9, and Ccna2, which encodes cyclin A2 (referred to as cyclin A hereafter) (Fig. 3C), and the MLL-C and cyclin A proteins (Fig. 3D).
Figure 3. Wt Mll is required for growth of MA9-transformed leukemia cells and expression of Hoxa9 and CCNA2.
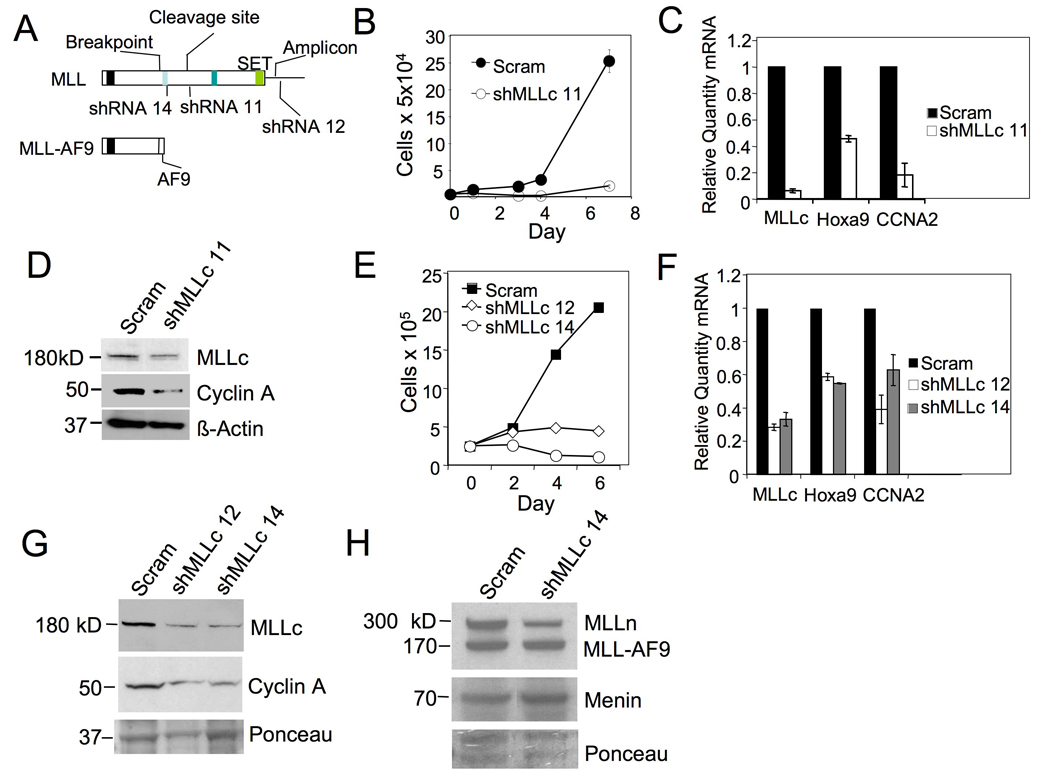
(A) A diagram for the structure of wt MLL, MA9 fusion protein, and shRNAs targeting various parts of MLL-C but not MA9. AT1 cells were transduced with either vector or MLL-C shRNA 11 retroviruses and monitored for cell number (+/− SD) (B), wt Mll, Hoxa9, and cyclin A (CCNA2) mRNAs (C), and the protein levels of MLL-C and cyclin A (+/− SD) (D). THP-1 cells were transduced with either control scrambled shRNA lentiviruses (Scram) or MLL-C shRNAs. The resulting cells were monitored for change in number (+/− SD) (E), the mRNA levels of wt Mll, Hoxa9, and CCNA2 (+/− SD) (F), and the protein levels of MLL-C and cyclin A (G), MLL-N, MLL-AF9 (MA9), and menin (H). See also Figure S2.
To determine if wt MLL is also crucial for proliferation of human MA9-expressing leukemia cells, we knocked down wt MLL in THP-1 cells by transducing lentiviruses expressing shRNAs, that targeted the C-terminus of human and murine MLL (Fig. 3A). Two independent MLL-C shRNAs, but not the control scrambled shRNA, reduced the number of THP-1 cells (Fig. 3E). We also detected an increased percentage of dead cells from wt MLL knockdown (KD) THP-1 cells (Fig. S2). As expected, wt MLL expression in the shRNA-transduced cells was reduced, as shown by qRT-PCR and Western blotting (Fig. 3F–G). The protein level of MLL-N was also reduced, however, the menin and MA9 levels were not affected (Fig. 3H). The expression of HOXA9 and CCNA2 was also reduced in wt MLL KD cells (Fig. 3F–G). Collectively, these results indicate that wt MLL upregulates expression of HOXA9 and CCNA2 as well as promoting proliferation and survival of the human leukemia cells.
To further confirm the impact of wt Mll on primary MA9-transformed bone marrow (BM) cells, we used a genetically tractable mouse model to specifically excise the wt Mll gene. We bred the Mllf/f mice (Jude, et al., 2007) with ubc9-Cre-ER mice (Ruzankina, et al., 2007), and demonstrated efficient Mll excision that was induced by 4-hydroxyl tamoxifen (4-OHT) in splenocytes from the Mllf/f;Cre-ER mice (Fig. 4A–B). Mll excision after BM cells were transformed with MA9-expressing retroviruses reduced the number of the MA9-transformed BM cells (Fig. 4C) and the expression of Hox genes in these cells (Fig. 4D), while 4-OHT did not affect proliferation of MA9-transformed cells that did not express Cre-ER (Fig. S3A–B). Together, these experiments demonstrate that wt MLL is important for proliferation/ survival of MA9-transformed leukemia cells and for high level expression of HOX genes in these cells.
Fig. 4. Wt Mll excision reduces the number of MA9-transformed cells and Hox gene expression.
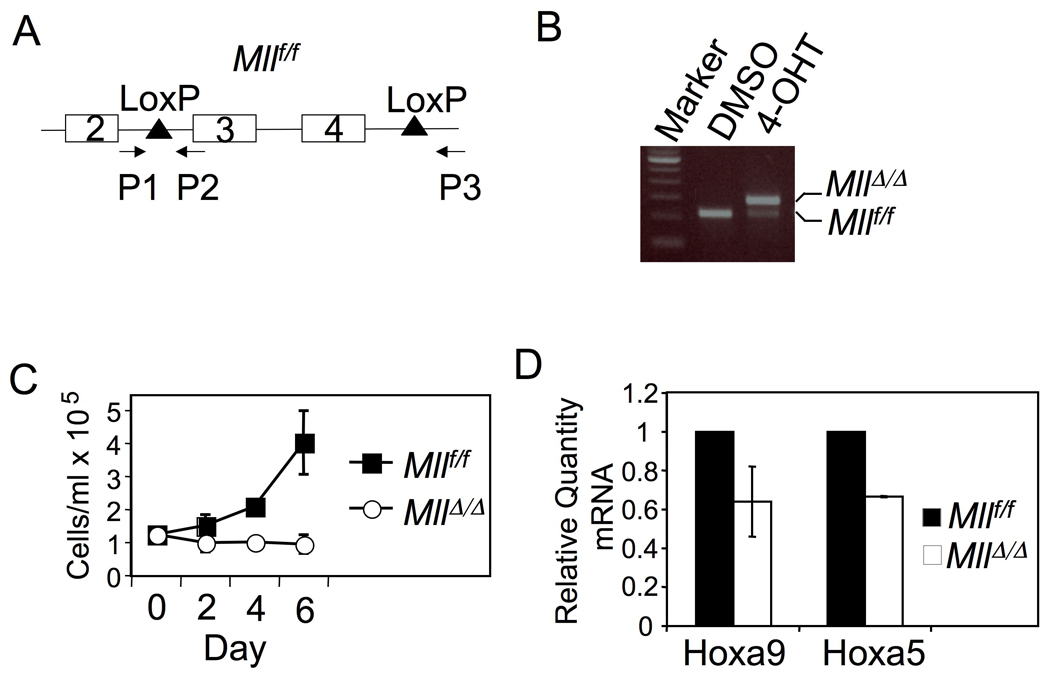
(A) A diagram for the floxed Mll and the primers used to detect the intact or excised Mll. (B) 4-OHT induced excision of the floxed Mll. Spleen cells from a Mllf/f;Cre-ER mouse were cultured with either DMSO or 4-OHT, followed by isolation of the genomic DNA and PCR amplification. (C) A growth curve for MA9-transformed BM cells with either Mllf/f or MllΔ/Δ (+/− SD, cells seeded in triplicate) (D) Quantification of Hoxa9 and Hoxa5 mRNAs in either Mllf/f or MllΔ/Δ MA9-transformed cells (+/− SD). See also Figure S3.
Wt MLL is crucial for maximal methylation of both H3K4 and H3K79 at the target genes
The SET domain of wt MLL methylates H3K4, yet is deleted from MA9 fusion protein (Milne, et al., 2002). On the other hand, loss of menin, an MLL-interacting protein, reduced H3K4 methylation at Hoxa9 in MA9-transformed cells (Fig. 1C). It has been unclear whether the remaining wt MLL allele in human MA9-expressing leukemia cells is still crucial for H3K4 methylation. We performed ChIP assays with control and wt MLL KD THP-1 cells. Wt MLL KD reduced MLL-C binding to HOXA9 (Fig. 5A), indicating that menin recruits wt MLL to its targets.
Figure 5. Wt MLL is crucial for maximal methylation of both H3K4 and H3K79 at key target genes.
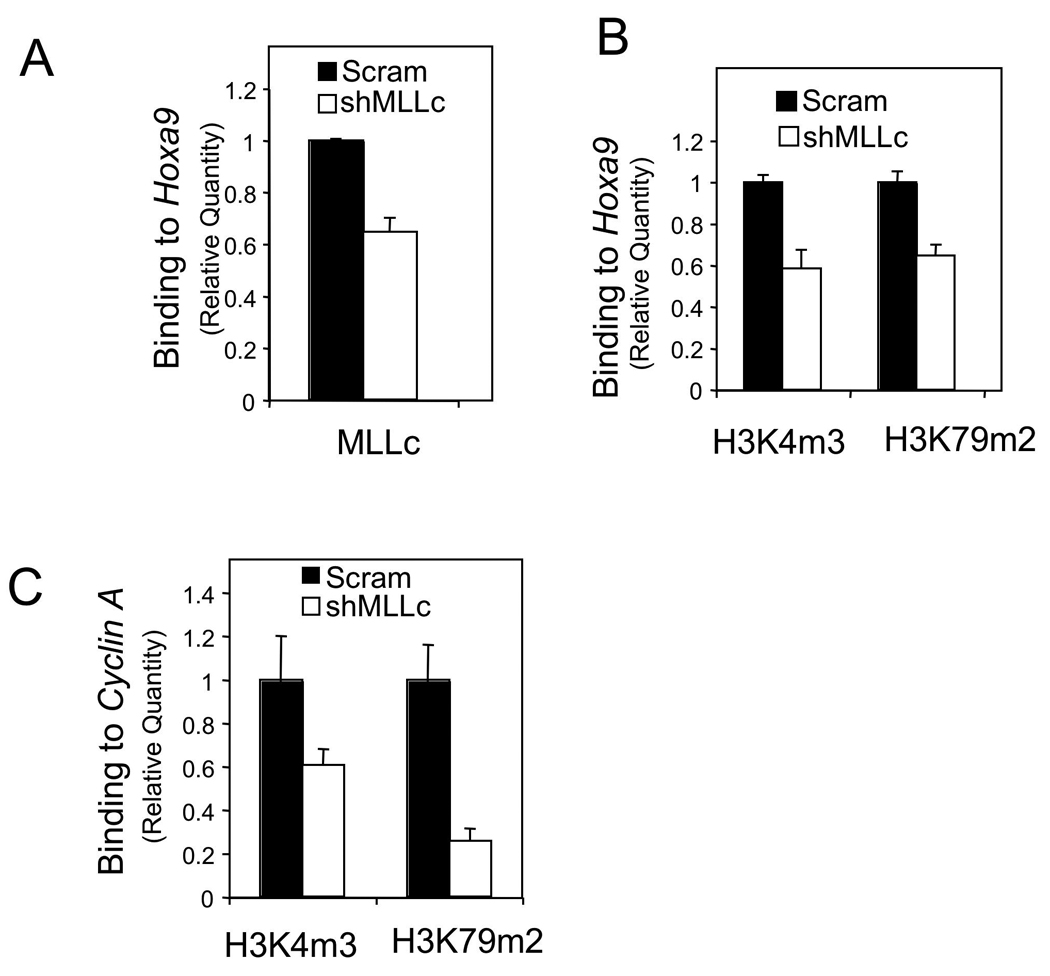
THP-1 cells were transduced with either control scrambled shRNA or MLL-C shRNA 14 lentiviruses to knock down wt MLL and evaluated using ChIP assay for MLL-C binding to Hoxa9 (A) and for histone H3K4m3 and H3K79m2 at Hoxa9 (B) and CCNA2 (C). The specificity of the anti-H3K4m3 antibody and the anti-H3K79m2 were confirmed using specifically modified peptides and Western blot. Error bars denote +/− SD. See also Figure S4.
We have shown that menin is crucial for both H3K4 and H3K79 methylation at the Hoxa9 locus (Fig. 1) through recruiting wt MLL and MA9/Dot1L, respectively (Fig. 2). Wild-type counterparts of several MLL fusion partners, such as AF4, AF9, and ENL, interact with each other as well as with Dot1L in a transcription-activating complex (Bitoun, et al., 2007, Mueller, et al., 2007). However, as wt MLL is not known to directly interact with the wild type-proteins of its fusion partners, it remains elusive whether wt MLL is important for MA9/Dot1L-mediated H3K79 methylation. Notably, H3K79 methylation was also reduced at the HOXA9 locus in these wt MLL KD cells (Fig. 5B), indicating a role for wt MLL in MA9/Dot1L-mediated H3K79 methylation. Similarly, wt MLL is also required for maximal methylation of both H3K4 and H3K79 at the CCNA2 locus (Fig. 5C), reinforcing the role of wt MLL in both H3K4 and H3K79 methylation. Collectively, these results suggest that wt MLL is crucial for maximal expression of both hematopoietic stem cell (HSC) enriched HOX genes and pro-proliferating cell cycle genes such as CCNA2, likely reflecting a dual role in regulating the self-renewal and proliferation of MA9-transformed LSCs.
Ablation or knockdown of wt MLL reduces colony formation of MA9-transduced bone marrow
Regulation of H3K79 methylation by wt MLL raised an intriguing possibility that wt MLL is important for transformation of bone marrow (BM) by MA9 or maintenance of MA9-transformed cells. We examined the impact of wt MLL knockdown on colony formation of MA9-transduced BM using a colony formation assay. Plating of MLL-ENL-transduced BM in a semi-solid medium three consecutive rounds leads to immortalization and transformation of the hematopoietic progenitors (Lavau, et al., 1997). To examine the impact of wt MLL KD on MA9-induced BM transformation, we transduced either the control scrambled shRNA or each of the two MLL-C shRNAs into the MA9-transduced BM cells after the second plating, followed by puromycin selection (Fig. 6A). The titers of these distinct shRNA and control lentiviruses were comparable and wt Mll KD was efficient in mouse cells (data not shown). At the fourth plating, numerous colonies appeared from the control cells (Fig. 6B, Scram). However, wt MLL KD with each of the MLL-C shRNAs reduced colony formation from the MA9-transduced BM (Fig. 6B–C).
Figure 6. Wt MLL knockdown suppresses colony formation of MA9-transduced BM.
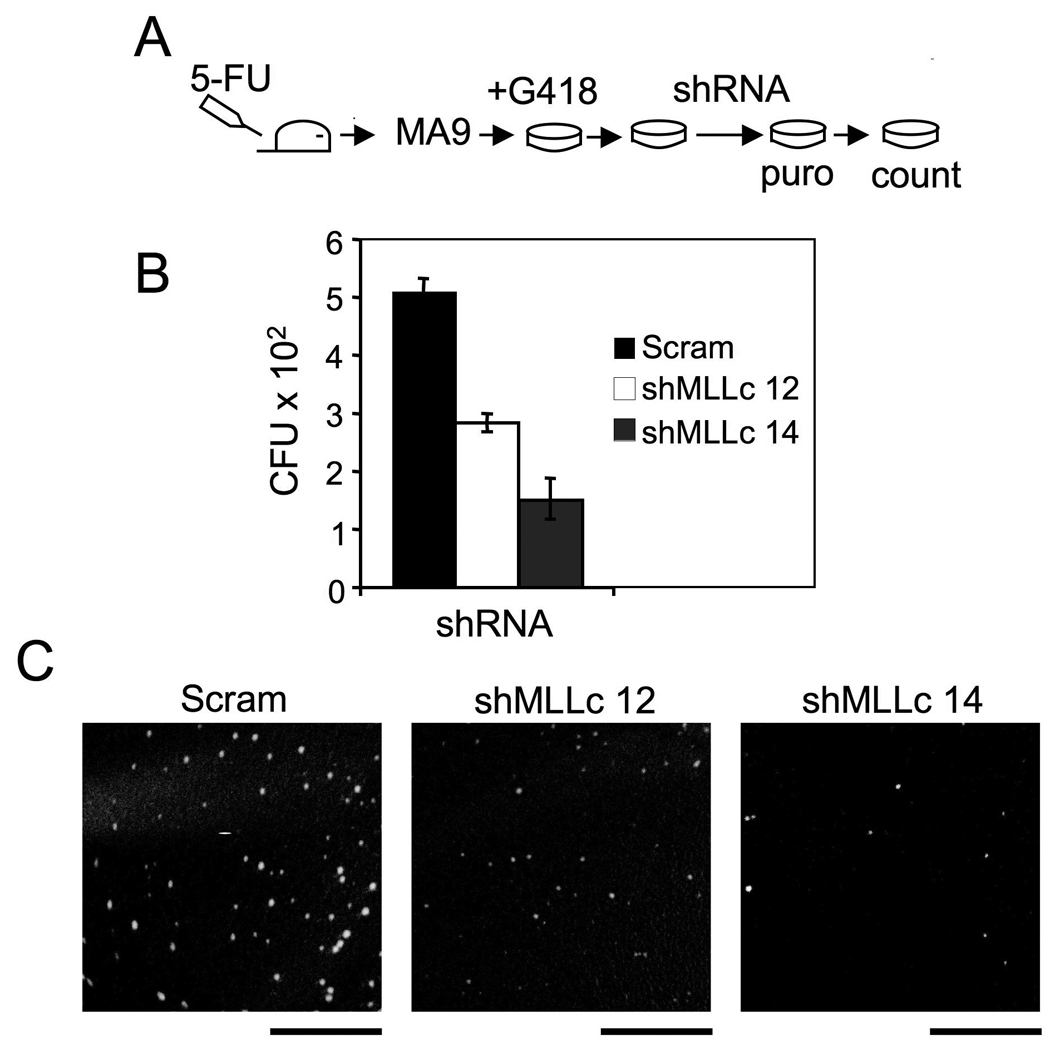
(A) Procedure for the colony formation assay. Bone marrow (BM) cells from a C57B6 mouse were transduced with pMSCV-MA9 retroviruses, and replated in triplicate weekly in methylcellulose medium with G418. After the second plating, surviving MA9 cells were transduced with each of the MLL-C shRNAs (12 and 14) or scrambled vector. (B) A summary of colony numbers for control or Mll shRNA-transduced BM. (C) Representative colonies from the culture plates (Scale bars 5mM). Error bars denote +/− SD.
To further confirm the role of wt MLL in MA9-induced colony formation, we used Mllf/f;Cre-ER BM to excise wt Mll after transformation. BM from control Mllf/f or Mllf/f;Cre-ER mice was transduced with MA9 retroviruses and serially replated on a semi-solid medium (Fig. 7A). Mll excision induced by 4-OHT significantly reduced the number of colonies from the MA9-transformed Mllf/f;Cre-ER BM (Fig. 7B). As a control, 4-OHT failed to reduce colony formation from MA9 Mllf/f BM (Fig. 7C). Tamoxifen (4-OHT)-induced excision of the floxed Mll in MA9-transformed BM from the Mllf/f;Cre-ER mice was confirmed by genomic PCR (Fig. 7D, lane 2). These results indicate that the wt Mll alleles are required for maintenance of MA9- transformed cells.
Figure 7. Wt Mll is required for colony formation of MA9-induced BM.
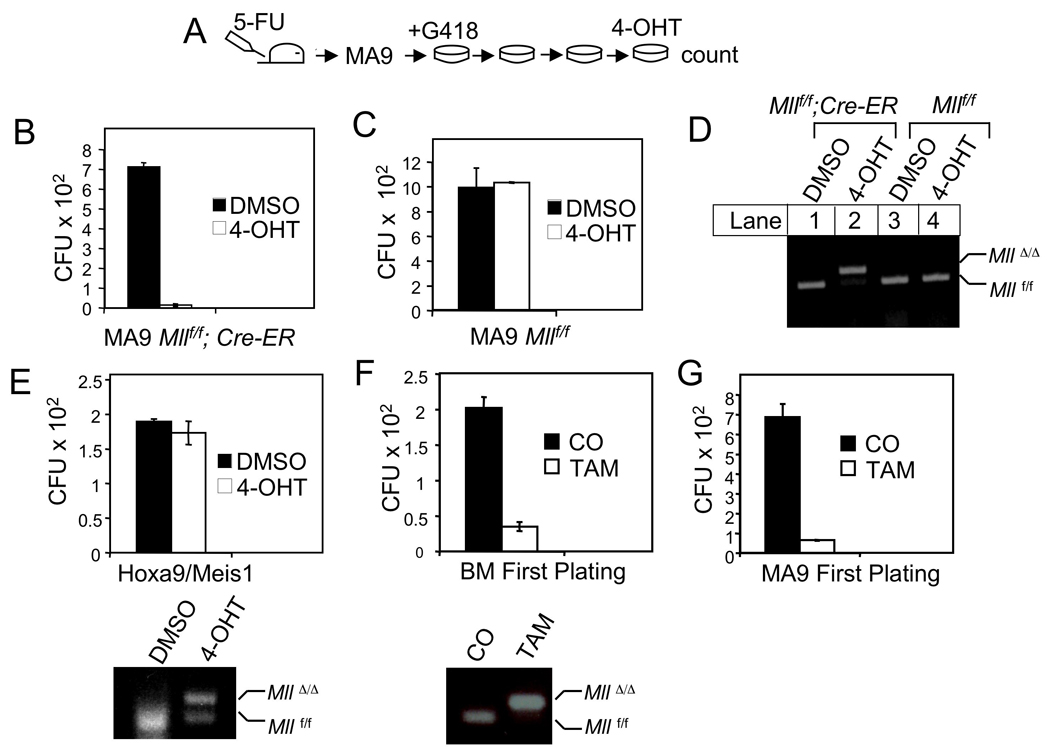
(A) A flowchart for procedures of MA9-induced transformation and 4-OHT-induced Mll excision. (B) Mll excision reduced the number of colonies from MA9-transduced BM from the Mllf/f;Cre-ER mice. (C) 4-OHT failed to reduce colony formation of MA9 retrovirus-transduced BM from the Mllf/f mice. (D) Genotyping with genomic DNA showed that 4-OHT induced Mll excision in MA9-transformed cells with the Mllf/f;Cre-ER genotype (lane 2) but failed to induce Mll excision in MA9-transformed BM cells with Mllf/f but without the Cre-ER transgene (lane 4). (E) Wt Mll excision failed to reduce Hoxa9/Meis1-induced BM colony formation (Top). 4-OHT-induced wt Mll excision in Hoxa9/Meis1-transformed BM (Bottom). (F) Wt Mll excision reduced colony formation from normal BM. BM from corn oil (CO) or TAM-treated Mllf/f;Cre-ER mice was plated on methylcellulose medium and the colony number was scored at the first plating (Top). Wt Mll was excised in BM from TAM-treated, but not from corn oil-fed, Mllf/f;Cre-ER mice (Bottom). (G) BM from corn oil or TAM-treated Mllf/f;Cre-ER mice was first transduced with MA9, followed by plating on methylcellulose medium, and the colony number was scored at the first plating. Error bars denote +/− SD.
Hoxa9 and Meis1 have previously been shown to have the ability to transform primary BM (Kroon, et al., 1998). Wt Mll excision did not inhibit BM colony formation induced by Hoxa9/Meis1 (Fig. 7E). These results indicate that wt Mll is not required for colony formation induced by Hoxa9/Meis1, likely because both Hoxa9 and Meis1 are direct MLL targets and act downstream of MLL (Guenther, et al., 2005, Nakamura, et al., 2002). This finding is consistent with the notion that wt MLL is essential for maintenance of MA9-transformed cells at least partly through upregulating certain Hox genes.
We further examined the effect of pre-existing wt Mll excision on colony formation from normal BM as well as MA9-transduced BM. BM from MLLf/f; Cre-ER mice that were treated with tamoxifen (TAM) displayed effective excision of the floxed Mll allele (Fig. 7F, bottom) and reduction of colony formation (Fig. 7F, top). Moreover, BM with wt Mll or with previous deletion of wt Mll was transduced with retroviruses expressing MA9, and plated on semi-solid medium. We found that previous deletion of wt Mll reduced colony formation at the first plating (Fig. 7G). These results, coupled with other results from the colony formation assay, indicate that wt Mll is crucial for survival and/or proliferation of BM progenitors and the maintenance of MA9-transformed cells, but not necessarily for MA9-induced transformation. From a standpoint of leukemia therapy, inhibiting the maintenance of MA9-transformed cells is more important than inhibiting MA9-induced transformation, because failure in maintaining MLL-FP-transformed cells could lead to eradication of the leukemia cells.
Mll excision in MA9-transformed cells inhibits the development of MA9-induced leukemia in mice
We next determined the role of wt MLL in MA9-induced leukemogenesis in mice using a xenotransplantation model. Either scrambled vector or MLL-C shRNA-transduced THP-1 cells were injected into NOD-SCID mice. Six weeks after transplantation, histological examination revealed that wt MLL KD reduced leukemic infiltration in long bones and spleens (Fig. S4A). The spleens from mice injected with the control cells were significantly larger than those of the mice injected with the wt MLL KD cells (Fig. S4B–C, P<0.05). These results suggest that wt MLL KD reduced engraftment of the human leukemia cells.
To confirm these results, we established a mouse leukemia model in which the floxed Mll can be excised in a temporally controllable manner. BM from the Mllf/f;Cre-ER mice was transduced with MA9 retroviruses. These transduced primary BM cells were transplanted into lethally irradiated C57B6 × B6.SJL F1 mice (Fig. 8A). Flow cytometry analysis of peripheral blood demonstrated successful engraftment of MA9-transduced donor BM (CD45.2+ only) and co-transplanted wild-type BM (CD45.1+/2+) (Fig. S5A). The percentage of cells expressing CD11b or CD11b/Gr-1 (myeloid markers) was much higher within the MA9-transduced BM (Fig. S5C–D) than that from the co-transplanted normal BM (Fig. S5B). Overt acute leukemia developed because obvious leukemia cell infiltrations were detectable in various organs including the femur, liver, and spleen (Fig. S5H–J).
Figure 8. Wt Mll is required for MA9-induced leukemogenesis in mice.
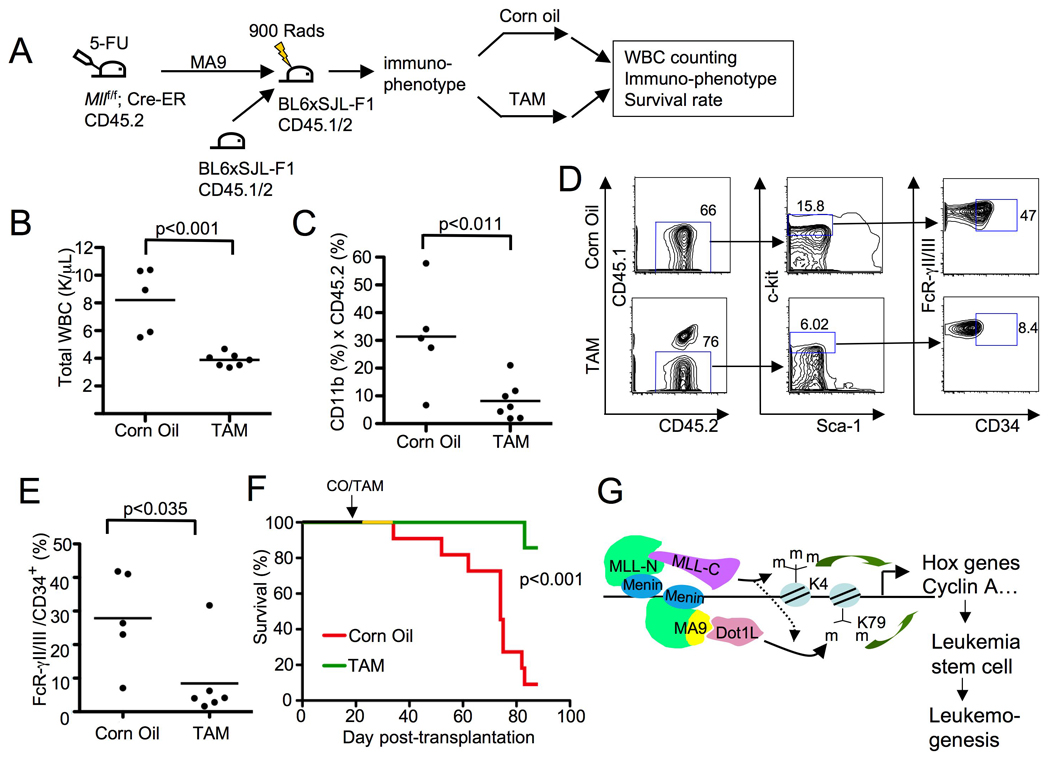
(A) A diagram for MA9-induced leukemogenesis and wt Mll excision in mice. TAM feeding was done 3 weeks after BM transplantation. (B) The total peripheral blood white blood cells (WBCs) in mice transplanted with MA9-transduced Mllf/f;Cre-ER BM were measured five weeks after corn oil or TAM feeding. (C) Flow cytometry analysis of peripheral blood CD11b+ MA9-transformed donor cells from transplanted mice, five weeks after the mice were fed with corn oil or TAM. (D) Flow cytometry analysis of CD45.2+ splenocytes from either terminally ill and corn oil-fed mice or TAM-fed mice to detect the percentage of c-kit high cells that were FcRγII/III+/CD34+. (E) A summary of the percentage of c-kit high cells that were FcRγII/III+/CD34+ from the corn oil or TAM-fed mice. (F) TAM-induced Mll excision in transplanted MA9-transduced BM in recipient mice increased the survival rate of the mice. Kaplan Meier curve for mice (C57B6/B6-SJL F1) transplanted with MA9-transduced Mllf/f;Cre-ER BM that were fed with either control corn oil (n=11) or TAM (n=7), 3 weeks after MA9 BM transplantation. (G) A model for menin, wt MLL, and MA9/Dot1L tripartite complex-controlled regulation of coupled yet distinct histone methylations, gene transcription, MA9-induced LSCs, and leukemogenesis. See also Figure S5.
To examine the impact of wt Mll excision on the development of MA9-induced acute myeloid leukemia (AML), the MA9 transduced BM-transplanted mice were fed with either control corn oil or TAM to excise the floxed Mll from MA9-transduced cells and monitored for peripheral white blood cell (WBC) number, immunophenotypes of the WBCs, and survival rate of the recipient mice (Fig. 8A). The number of total peripheral WBCs and the percentage of MA9-transduced BM-derived CD11b+ myeloid cells were significantly lower in TAM-treated mice than in the corn oil-fed control mice (Fig. 8B–C, p<0.001 and 0.011, respectively). As a control, effective Mll excision in peripheral WBCs in TAM-treated mice was observed (Fig S5K, lanes 7–13).
In splenocytes from terminally ill corn oil-fed mice or TAM-fed control mice, wt Mll excision reduced the percentage of the cells bearing the markers for L-GMP (Fig. 8D–E), namely c-kit+/Sca-1-/FcRgII/III+/CD11b+/CD34+, which have been reported to be enriched in MA9-induced leukemia stem cells (LSCs) (Krivtsov, et al., 2006). Wt Mll excision from the MA9-expressing cells also significantly increased survival rate of the recipient mice, based on Kaplan Meier analysis (Fig. 8F, p<0.001). Wt Mll excision also modestly reduced the viability of cells from CD45.2+ splenocytes (Fig. S5L). Together, these results suggest that wt Mll is crucial for the development of MA9-induced leukemia partly through enhancing leukemia stem cell proliferation and/or survival.
Discussion
The wt Mll allele, the common precursor of all MLL-FPs, controls MA9-induced leukemogenesis
Oncogenic fusion proteins resulting from chromosomal translocations represent a major molecular lesion in leukemia and certain solid tumors (Nambiar, et al., 2008). Studies about these malignant diseases are often centered on the fusion proteins with little attention paid to the role of the cognate wt alleles in tumorigenesis. A prevalent model suggests that MLL-FPs induce leukemogenesis by aberrantly upregulating expression of certain 5'-Hoxa genes (Yokoyama, et al., 2008, Yokoyama, et al., 2005), partly through enhancing Dot1L-mediated H3K79 while repressing H3K4 methylation (Okada, et al., 2005). The SET domain in wt MLL was not thought to be crucial for MLL-FP-triggered leukemogenesis, as MLL-AF10 even suppresses H3K4 dimethylation (Okada, et al., 2005), which could be at least partly mediated by wt MLL. Therefore, it has been unclear and unresolved whether wt MLL plays a role in MLL-FP-mediated leukemic transformation (Popovic, et al., 2005).
We have shown that wt MLL is crucial for maximal levels of H3K4 trimethylation at the loci of HOXA9 and CCNA2 and for MA9-induced leukemogenesis. Although MLL-AF10 suppresses H3K4 dimethylation, as wt MLL is able to convert dimethylated H3K4 (H3K4m2) to trimethylated H3K4 (H3K4m3), the repression of H3K4 dimethylation (H3K4m2) by MLL-AF10 could actually result from increased wt MLL-mediated conversion of H3K4m2 to H3K4m3. This explanation is consistent with the observation that MLL-ENL induces H3K79 methylation, but does not suppress H3K4 trimethylation at Hoxa9 (Milne, et al., 2005). Additionally, in MLL-AF4-expressing human leukemia cells, both methylated H3K4 and H3K79 are colocalized at large chromatin domains, including the domain harboring the HOXA7 and HOXA9 loci (Guenther, et al., 2008).
Wt MLL may control MA9-induced leukemogenesis by facilitating the expression of HOX genes and other self-renewal genes and supporting LSC maintenance. Although wt MLL is crucial for preventing HSCs from abnormal entry into cell cycle, it may be particularly important for promoting proliferation and survival of MA9 LSCs (Fig. 8D–E), a function distinct from that in HSCs (Jude, et al., 2007, McMahon, et al., 2007). Supporting this explanation, the expression of Hoxa9 is reactivated in MA9-transformed LSCs (Krivtsov, et al., 2006), and wt MLL is crucial for growth and survival of MA9-transformed leukemia cells (Fig. 3–Fig 4, Fig. S3, and Fig. S5L), and for maintenance of MA9 LSCs (Fig. 8D–E). Therefore, both growth and survival may represent mechanisms by which wt MLL influences MA9 transformed cells. However, our data using Hoxa9/Meis1-mediated BM transformation suggest that this requirement for wt MLL can be replaced by ectopic expression of Hoxa9/Meis1, likely because they are direct targets of MLL, therefore in this case act downstream of MLL (Fig. 7E).
There are over 60 distinct MLL-FPs and further work remains to determine whether our findings are generally applicable to other MLL-FPs. Given that MLL-AF10 and MLL-AF4 increase H3K79 methylation at target loci (Krivtsov, et al., 2008, Okada, et al., 2005) and wt MLL is also necessary for maximal H3K79 methylation (Fig. 5), it is likely that many MLL-FPs require wt MLL for leukemogenesis. On the other hand, deletion of the wt Mll allele does not change expression of Hoxa9, but reduces the colony forming activity (CFU-GEMM) of mouse fetal liver cells expressing MLL-PTD (partial tandem duplication) (Dorrance, et al., 2008). MLL-PTD still retains the C-terminal SET domain, yet wt MLL is silenced in patients' leukemia cells expressing MLL-PTD (Whitman, et al., 2005), raising the possibility that MLL-PTD-initiated leukemia arises by a distinct mechanism.
The mechanisms for wt MLL-dependent epigenetic regulation and MLL-AF9-induced leukemogenesis
We have uncovered a crucial role for the wt Mll allele in promoting MA9-induced leukemogenesis. MA9 is insufficient for maintaining the MA9-transformed leukemia cells without the wt Mll allele. Rather, it depends on co-expression of the wt Mll allele, which we have shown to upregulate not only H3K4 methylation but also MA9/Dot1L-mediated H3K79 methylation at Hoxa9. Consistent with this model, Men1 depletion reduced recruitment of wt MLL to the Hoxa9 locus and diminished not only H3K4 methylation but also H3K79 methylation at the locus.
These findings have revealed a link between wt MLL and MA9 in enhancing stem cell-related Hox genes and certain cell cycle genes in MA9-transformed leukemia cells. These results are consistent with a model whereby menin, wt MLL, and MA9 converge at the loci of Hoxa9 and certain cell cycle genes such as CCNA2. Menin may recruit both wt MLL and MA9 at these target genes to epigenetically promote their transcription, leading to maintenance of LSCs and the development of MA9-induced leukemia (Fig. 8G). MLL is also associated with the cyclin E locus in mouse embryonic fibroblasts (Takeda, et al., 2006). Wt MLL is degraded in an SCF (Skp2) and APC (cdc2)-dependent manner at distinct phases of the cell cycle, but the degradation of tested mutant MLL-FPs is inhibited (Liu, et al., 2007). It is unclear whether menin and MLL-FPs affect wt MLL degradation.
Unlike the previous report that MLL-AF10 suppresses H3K4 dimethylation (Okada, et al., 2005), our results indicate that wt MLL controls not only H3K4 trimethylation but also H3K79 dimethylation, two distinct positive histone H3 modifications, at HOXA9 and CCNA2 loci in MA9-transformed cells. H3K4m3 and H3K79m2 co-exist at the loci of multiple active genes (Guccione, et al., 2006, Steger, et al., 2008). It is likely that menin recruits both wt MLL and MLL-FP/Dot1L, which can trimethylate H3K4 and dimethylate H3K79, respectively, because menin physically interacts with the N-terminus of both wt MLL and MLL-FPs (Yokoyama, et al., 2005). A combination of H3K4m3 and H3K79m2 may cooperatively activate transcription of certain HOX genes and cell cycle genes, triggering leukemic transformation and supporting the maintenance of LSCs (Fig. 8G).
In agreement with this model, MLL-AF4 binds chromatin regions that are also rich in H3K4m3 and H3K79m2 in human leukemia cells (Guenther, et al., 2008). As H3K4m3 is a mark of transcriptional initiation and H3K79-methylating Dot1L is a component of a transcriptional elongation complex, wt MLL and menin may be crucial for regulating the crucial steps in enhancing expression of HOX genes and other targets that are important for MLL-FP-induced leukemogenesis. The protein LEDGF also interacts with both menin and the N-terminus of MLL to facilitate their recruitment to chromatin (Yokoyama, et al., 2008), but it is unclear whether the function of this protein is regulated by menin or wt MLL.
It remains unclear how wt MLL controls H3K79 dimethylation. Several wild-type counterparts of MLL fusion partners, such as AF4, AF9 and ENL, form a transcriptional elongating complex containing RNA polymerase II transcription elongation factor b (P-TEFb) and Dot1L to increase gene expression (Bitoun, et al., 2007, Mueller, et al., 2007, Zeisig, et al., 2005). It is unknown whether wt MLL is also in this complex and crucial for the function of this transcription elongation complex or Dot1L-mediated H3K79 methylation. We have found that wt MLL influences H3K79 dimethylation at the Hoxa9 gene, and it is possible that the SET-domain and/or SET-mediated H3K4 methylation are important for the role of wt MLL in MA9-induced leukemogenesis. In this regard, methylated H3K4 serves as a docking site to recruit various transcription-activating proteins such as WDR5 and BPTF (Wysocka, et al., 2005, Wysocka, et al., 2006). As menin influences wt MLL recruitment and H3K4 trimethylation as well as Dot1L recruitment (Fig. 2A and C), it cannot be ruled out that H3K4m3-binding proteins may also affect the recruitment or activity of Dot1L and subsequent H3K79 methylation.
Contrary to wt MLL-dependent H3K79 dimethylation at HOXA9, H3K4 trimethylation does not appear to rely on Dot1L-mediated H3K79 methylation. MLL-ENL enhances H3K79 dimethylation at Hoxa9, but is dispensable for H3K4 trimethylation at the locus (Milne, et al., 2005). Moreover, Dot1L ablation from cells abrogates H3K79 dimethylation but does not affect H3K4 trimethylation (Steger, et al., 2008), suggesting a uni-directional order of H3K4 and H3K79 methylation that is controlled by wt MLL (Fig. 8G).
Wt MLL as a potential target for leukemia therapy
Our findings demonstrate that wt MLL is crucial for maximal expression of HOXA9 and MA9-induced leukemogenesis. One may argue that if the wt MLL allele is required for MLL-FP-induced leukemogenesis, then what is the benefit for the leukemia cells to cripple one of the two wt MLL alleles to generate an MLL-FP? One likely explanation is that, for instance, MA9 or MA9-induced H3K79 dimethylation cooperates with wt MLL or wt MLL-mediated H3K4 trimethylation in activation of HOX genes and epigenetic chromatin reprogramming, offsetting the loss of one of the two wt MLL alleles. This is consistent with the findings that wt MLL and MA9 target genes, HOX genes, are expressed at a higher level in MLL-FP-expressing human leukemia cells than in non-MLL-FP-expressing human leukemia cells (Armstrong, et al., 2002). Moreover, wt MLL may also regulate genes controlling proliferation and self-renewal in MA9-transformed leukemia cells or LSCs.
The necessity for wt MLL in multiple processes of leukemogenesis, including MA9/Dot1L-mediated H3K79 dimethylation at HOX genes and maintenance of MA9-expressing LSCs, could render MA9-harboring leukemia cells selectively sensitive to inhibition of wt MLL. Despite our results that wt Mll was also crucial for colony formation from normal BM progenitors in culture, wt Mll excision in adult mice is well tolerated and does not adversely affect homeostatic hematopoiesis (McMahon, et al., 2007). MLL-FP-expressing leukemia cells may become particularly "addicted" to wt MLL for expression of stem cell-related HOX genes and certain cell growth/survival-related genes. This "addiction" may be attributable to the reduced cellular wt MLL levels in human leukemia cells that in theory result from disruption of one wt MLL allele in chromosomal translocation. In agreement with this interpretation, in murine HSCs harboring endogenous knockin MA9, the amount of wt MLL is only half that found in wt HSCs (Chen, et al., 2008). Moreover, wt MLL may also be more crucial to the MLL-FP-expressing leukemia cells because these cells rely on wt MLL for MLL-FP-induced H3K79 dimethylation and gene transcription (Fig. 8G). However, it cannot be ruled out that wt MLL is also crucial for transformation induced by non-MLL-FPs.
Collectively, our findings support development of strategies to treat MLL-FP-induced leukemia, in part by targeting the pathway of menin and wt MLL, the common wt allele of over 60 MLL-FPs. Wt MLL may well serve as a "non-oncogene addiction" target for treating MLL-FP-expressing leukemia (Solimini, et al., 2007). These studies raise the possibility for developing lead compounds that specifically inhibit wt MLL, its interacting proteins, processing enzymes, or its methyltransferase activity to treat MLL-FP-induced acute leukemia.
Experimental procedures
Plasmids and cell culture
Various plasmids were as previously described: pMX-GFP, pMX-puro, pMX-puro-menin, pMSCV-MLL-AF9 (MA9), pMSCVpgk-Hoxa9-GFP, pMSCVpacMeis1A, and pMSCV-GFP (Chen, et al., 2006, Jin, et al., 2003). Retroviral or lentiviral constructs expressing shRNAs were obtained from Open Biosystems (Huntsville, AL): retroviral constructs for mouse shRNAs, pshRNA 11-MLL-C (mouse, 5'-gctggcctcccataatttat-3'); lentiviral constructs expressing MLL-C shRNAs-12 and 14 (cgcggtattatcctaatttaa and cgccttcacttgaccataatt), which were highly conserved between the mouse and human wt MLL mRNAs. pLKO.1 vector (Scram) was from (Sigma, St. Louis, MO). Lentiviral packaging plasmids, pMD2G and pAX2G, were purchased from Addgene (Cambridge, MA).
HEK 293T cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% Pen/Strep. AT1 cells were generated from BM cells of Men1f/f;Cre-ER mice by transduction with retroviruses expressing MA9 and cultured in medium with 10 ng/ml IL3 (Chen, et al., 2006). In MA9-transformed BM cells, Men1f/f or Mllf/f was excised by treating the cells with 4-OH tamoxifen (4-OHT, 200 nM). THP-1 cells were maintained in RPMI-1640 containing 10% FBS and 1% Pen/Strep supplemented with 0.05mM 2-mercaptoethanol.
Packaging of recombinant retroviruses and lentiviruses and transduction of cells
pMX-GFP, pMX-puro, and pshRNAs were co-transfected with psi-2 helper plasmid into 293T cells using the calcium chloride precipitation method, as previously described (Jin, et al., 2003). To package lentiviruses, lenti-GFP, scrambled pLKO.1 vector or specific pshRNAs were cotransfected into 293T cells with pAX2G and pMD2G, using Fugene-mediated transfection. The resulting recombinant retro- or lenti-viruses were collected to transduce cells by spinoculation, followed by selection in 2 µg/ml puromycin for 2 days.
RNA extraction and quantitative RT-PCR
Total RNA was extracted from the cultured cells with Trizol and an RNeasy extraction kit (Qiagen), and used as template (1 µg) to synthesize cDNA for quantitative RT-PCR (qRT-PCR) analysis in a 7500 fast real-time PCR system (Applied Biosystems). Relative mRNA levels of a specific gene were calculated by δ–δ-CT values calibrated with either GAPDH or beta-actin, using SYBR green dye for detection (Qiagen, QuantiTect SYBR Green Mastermix). The sequences of primers for qRT-PCR are listed in The Supplemental Experimental Procedures.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed as previously described, using a Quick ChIP kit (Imgenex, San Diego, CA) (Chen, et al., 2006, Yan, et al., 2006). Briefly, cells (106) were fixed with 1% formaldehyde and lysed in ChIP lysis buffer with protease inhibitors. The genomic DNA was incubated with either control IgG or a specific primary antibody at 4°C overnight, and collected with protein G agarose beads. The protein-DNA complexes were eluted from the beads and incubated at 65°C overnight to reverse the protein-DNA crosslinking. Quantity of the precipitated DNA was determined with qPCR (Qiagen, QuantiTect SYBR Green Mastermix) and normalized with the input genomic DNA. The sequences of the primer pairs for ChIP assay are listed in the Supplemental Experimental Procedures.
Mice and bone marrow transformation
All laboratory mice were maintained on a 12-h light-dark cycle in the animal facility at the University of Pennsylvania. All experiments on mice in our research protocol were approved by Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and were performed in accordance with relevant institutional and national guidelines and regulations.. Mllf/f mice (in C57B6-SJL background) (Jude, et al., 2007) were bred with ubc9-Cre-ER mice (in C57B6 background) (Schnepp, et al., 2006), and BM from the mice was isolated and transformed, as we previously described (Chen, et al., 2006). Briefly, C57B6 mice or Mllf/f;Cre-ER mice (6–8 weeks old) were injected with 5-fluorouracil, and BM cells were collected from femurs and prestimulated with a cocktail of cytokines and growth factors, as previously described (Chen, et al., 2006). The cells were transduced twice with pMSCV-MA9 retroviruses by spinoculation and replated weekly in MethoCult GF M3434 medium (StemCell Technologies) with 1mg/ml G418. After the second plating, surviving cells from C57B6 or Mllf/f;Cre-ER mice were transduced twice with either scrambled control retroviruses or lentiviruses or their counterparts expressing one of the MLL-C shRNAs. The transduced cells (2 ×104) were seeded in a 35-mm Petri dish with methylcellulose-based medium containing 2µg/ml puromycin and scored for colonies with >50 cells one week after plating. To excise the floxed Mll, MA9-transformed BM with Mllf/f;Cre-ER were treated with either DMSO or 4-OHT (400 nM) at the fourth plating. For in vivo Mll excision prior to the first plating, Mllf/f;Cre-ER mice were treated with corn oil or 200 mg/kg body weight TAM to excise the floxed Mll, as we previously described (Schnepp, et al., 2006), then isolated bone marrow was either plated directly in methylcellulose, or transduced with MA9, followed by plating.
Leukemia induction and wt Mll excision from leukemic cells in mice
The Mllf/f mice were backcrossed with C57B6sjl mice (CD45.1+) for over nine generations (Jude, et al., 2007), and the mice were then bred with transgenic mice expressing ubc9-Cre-ER (in C57B6 background, CD45.2+) (Ruzankina, et al., 2007). The Mllf/f ;Cre-ER mice were intercrossed to maintain the Mllf/f ;Cre-ER genotype with the CD45.2+ marker. BM cells from these mice were transduced with MA9-retroviruses, and transplanted retro-orbitally into C57B6 × C57B6-SJL F1 female mice (CD45.1+/2+, 8 weeks old, 106 cells per mouse, Taconic), together with 2.5×105 BM cells from an F1 mouse. The recipient mice were irradiated with 900 rad, prior to transplantation. The mice were fed with either control corn oil or tamoxifen (TAM) (Sigma, St. Louis, MO) at a dose of 200 mg/kg body weight to excise the floxed Mll. Organs from control and leukemic mice were isolated, fixed, and processed for H & E staining and analyzed under microscope.
Flow cytometry analysis and antibodies
Cells from peripheral blood, bone marrow or spleen were harvested for analysis of immunophenotypes. After blocking unspecific binding with unlabeled rat+mouse IgG (Sigma), cells were stained on ice in PBS + 4% FCS and analyzed on LSR II, FACSCalibur or FACSAria (Becton Dickinson). Files were analyzed in FlowJo (Tree Star, San Carlos, CA). Antibodies for flow cytometry analysis, immunoblotting, and ChIP assay were described in supplemental Materials and Methods. All the biochemical experiments, including the ChIP assay, qRT-PCR, and cell proliferation, were repeated at least twice with consistent results.
Statistical analysis
Microsoft Excel and GraphPad Prism software was used for statistical analysis. Student's t test was used to determine the significance of the results unless otherwise indicated. Kaplan-Meier statistical analysis was performed using the log rank test.
Highlights
Menin recruits both wild type MLL protein and MLL-AF9 to loci of HOX genes.
Wt MLL controls both H3K4 methylation and MLL-AF9 induced H3K79 methylation.
Wt MLL allele is required for MLL-AF9 induced leukemogenesis.
Wt MLL is crucial for maintenance of MLL-AF9-transformed leukemia cells.
SIGNIFICANCE
The potential role of wt MLL in the development of mixed lineage leukemia, which is highly aggressive and often refractory to therapy, has been elusive. We demonstrate a crucial role for wt MLL, the common wild-type precursor of over 60 distinct MLL-FPs, in MLL-AF9-induced leukemogenesis. Wt MLL influences MLL-AF9-induced histone methylation and gene expression as well as growth and survival of MLL-AF9-transformed leukemia cells. These findings underscore the importance of wt MLL in the development of MLL-AF9-induced acute leukemia.
Supplementary Material
Acknowledgements
This work was in part supported by NIH grants (R01 CA113962 to XH), a Leukemia and Lymphoma Society SCOR grant (XH), and a T32 training grant CA09140 (AT). We thank Dr. J. Alan Diehl for his critical reading of the manuscript and Drs. M. Celeste Simon, Warren Pear, Martin Carroll and Wei Tong for stimulating discussions. We appreciate the discussions with other members in our laboratory. We thank Dr. Gwenn Danet-Desnoyers and his associates for assistance with leukemia cell engraftment in NOG mice, and Ms. Hong Wei at the Histology Core Facility for histological studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabe F, Kennedy JA, Hope KJ, Dick JE. Modeling the initiation and progression of human acute leukemia in mice. Science. 2007;316:600–604. doi: 10.1126/science.1139851. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- Caslini C, Yang Z, El-Osta M, Milne TA, Slany RK, Hess JL. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007;67:7275–7283. doi: 10.1158/0008-5472.CAN-06-2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, Lund EA, Sam TN, Kersey JH. Malignant Transformation Initiated by Mll-AF9: Gene Dosage and Critical Target Cells. Cancer Cell. 2008;13:432–440. doi: 10.1016/j.ccr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A. 2006;103:1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrance AM, Liu S, Chong A, Pulley B, Nemer D, Guimond M, Yuan W, Chang D, Whitman SP, Marcucci G, et al. The Mll partial tandem duplication: differential, tissue-specific activity in the presence or absence of the wild-type allele. Blood. 2008;112:2508–2511. doi: 10.1182/blood-2008-01-134338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall' Olio V, Zardo G, Nervi C, Bernard L, Amati B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–770. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, Croce CM, Nakamura T, Canaani E, Young RA. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22:3403–3408. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med. 2004;10:500–507. doi: 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Jin S, Mao H, Schnepp RW, Sykes SM, Silva AC, D'Andrea AD, Hua X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63:4204–4210. [PubMed] [Google Scholar]
- Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. Embo J. 1998;17:3714–3725. doi: 10.1093/emboj/17.13.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La P, Silva AC, Hou Z, Wang H, Schnepp RW, Yan N, Shi Y, Hua X. Direct binding of DNA by tumor suppressor menin. J Biol Chem. 2004;279:49045–49054. doi: 10.1074/jbc.M409358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavau C, Szilvassy SJ, Slany R, Cleary ML. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. Embo J. 1997;16:4226–4237. doi: 10.1093/emboj/16.14.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Cheng EH, Hsieh JJ. Bimodal degradation of MLL by SCFSkp2 and APCCdc20 assures cell cycle execution: a critical regulatory circuit lost in leukemogenic MLL fusions. Genes Dev. 2007;21:2385–2398. doi: 10.1101/gad.1574507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon KA, Hiew SY, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, Kioussis D, Williams O, Brady HJ. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1:338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005;65:11367–11374. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Nambiar M, Kari V, Raghavan SC. Chromosomal translocations in cancer. Biochim Biophys Acta. 2008;1786:139–152. doi: 10.1016/j.bbcan.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Popovic R, Zeleznik-Le NJ. MLL: how complex does it get? J Cell Biochem. 2005;95:234–242. doi: 10.1002/jcb.20430. [DOI] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp RW, Chen YX, Wang H, Cash T, Silva A, Diehl JA, Brown E, Hua X. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–5715. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–988. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–268. doi: 10.1016/j.ccr.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Chen DY, Westergard TD, Fisher JK, Rubens JA, Sasagawa S, Kan JT, Korsmeyer SJ, Cheng EH, Hsieh JJ. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 2006;20:2397–2409. doi: 10.1101/gad.1449406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, Zheng Y, Cancelas JA, Gu Y, Jansen M, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitman SP, Liu S, Vukosavljevic T, Rush LJ, Yu L, Liu C, Klisovic MI, Maharry K, Guimond M, Strout MP, et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood. 2005;106:345–352. doi: 10.1182/blood-2005-01-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Yan J, Chen YX, Desmond A, Silva A, Yang Y, Wang H, Hua X. Cdx4 and menin co-regulate hoxa9 expression in hematopoietic cells. PLoS ONE. 2006;1:e47. doi: 10.1371/journal.pone.0000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The Menin Tumor Suppressor Protein Is an Essential Oncogenic Cofactor for MLL-Associated Leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar MP, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene. 2005;24:5525–5532. doi: 10.1038/sj.onc.1208699. [DOI] [PubMed] [Google Scholar]
- Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCalpha in an aldosterone-sensitive manner. J Biol Chem. 2006;281:18059–18068. doi: 10.1074/jbc.M601903200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.