

Abstract
Airways display robust NF-κB activation and represent targets for anti-inflammatory asthma therapies, but the functional importance of NF-κB activation in airway epithelium remains enigmatic. Therefore, transgenic mice were created in which NF-κB activation is repressed specifically in airways (CC10-IκBαsr mice). In response to inhaled Ag, transgenic mice demonstrated significantly ameliorated inflammation, reduced levels of chemokines, T cell cytokines, mucus cell metaplasia, and circulating IgE compared with littermate controls. Despite these findings, Ag-driven airways hyperresponsiveness was not attenuated in CC10-IκBαsr mice. This study clearly demonstrates that airway epithelial NF-κB activation orchestrates Ag-induced inflammation and subsequent adaptive immune responses, but does not contribute to airways hyperresponsiveness, the cardinal feature that underlies asthma.
Hallmarks of asthma include airways inflammation predominated by eosinophils, the production of mucus, Th2 cytokines (IL-4, -5, and -13), and allergen-specific IgE, as well as airways hyperresponsiveness to inhaled bronchocon-stricting agonists. The transcription factor NF-κB is considered a master regulator of inflammation and immune processes (1, 2) and has been widely implicated in the pathophysiology of asthma. NF-κB activity is tightly controlled by the inhibitory protein, IκBα, which is complexed to NF-κB dimers to promote cytoplasmic retention and low basal transcriptional activity. Upon cellular stimulation, the IκB kinase complex phosphorylates IκBα, causing its ubiquitination and degradation through the 26S proteasome pathway. IκB degradation exposes the nuclear localization sequence of NF-κB, allowing its accumulation in the nucleus, binding to DNA, and stimulation of gene transcription. Mice lacking the NF-κB subunits p50 or c-Rel develop less airway inflammation upon Ag challenge, demonstrating the causal role of NF-κB in allergic airway disease (3–5). NF-κB plays an important role in the maturation of Th cells, and the systemic failure to activate NF-κB results in a blunted immune response to Ag. However, activation of NF-κB has also been demonstrated specifically within airway epithelium in animal models of allergic airway inflammation (6, 7) and in patients with asthma (8–13). Thus, it is unclear exactly what aspects of allergic airways disease may be governed by the activation of NF-κB in these resident pulmonary epithelial cells.
To address the functional significance of airway epithelial NF-κB activation in allergic airways disease, we generated a transgenic mouse expressing an IκBα mutant, also referred to as IκBα superrepressor (IκBαsr), 3 which is resistant to phosphorylation-induced degradation. By placing this transgene under transcriptional control of the CC10 promoter, NF-κB activation was inhibited selectively in airway epithelial cells. Using the OVA model of allergic airways disease, we report here that NF-κB repression in airways significantly ameliorates inflammation, expression of NF-κB-dependent inflammatory mediators, and secondary effects on inflammatory cells, including Th cell cytokine production, Ag-specific Ig levels, and mucous cell metaplasia. Despite these findings, inhibition of NF-κB activation in airways did not dampen airways hyperresponsiveness, an important hallmark of asthma. These results demonstrate that NF-κB activation within airway epithelium is necessary to fully induce the recruitment of inflammatory cells to the airways in response to allergen challenge but also demonstrate a complex dissociation between airway inflammation and hyperresponsiveness.
Materials and Methods
CC10-IκBαsr transgenic mice
The CC10-IκBαsr transgenic mice generated as previously described (14) were backcrossed for 7–10 generations with BALB/c mice (The Jackson Laboratory, Bar Harbor, ME) and housed in the University of Vermont Animal Facility. The Institutional Animal Care and Use Committee granted approval for all studies. Mice were subjected to OVA sensitization and challenge as described elsewhere (6). Grade V OVA was obtained from Sigma-Aldrich (St. Louis, MO), and ImjectAlum was obtained from Pierce Biotechnology (Rockford, IL). Mice were euthanized by a lethal dose of pentobarbital via intraperitoneal injection.
Pulmonary function assessment
Anesthetized mice were tracheotomized and mechanically ventilated for the assessment of pulmonary function using the forced oscillation technique, as previously described (15). Mice were ventilated at a rate of 2.5 Hz, with a tidal volume of 0.2 ml and 3 cm H2O positive end-expiratory pressure (flexiVent; SCIREQ, Inc., Montreal, Canada). Data from regular ventilation was collected to establish the baseline for each animal, and inhaled doses of aerosolized methacholine (Sigma-Aldrich) in saline were then administered in successive increasing concentrations (0, 3.125, 12.5, and 50 mg/ml). Multiple linear regression was used to fit impedance spectra derived from measured pressure and volume to the constant phase model of the lung (16): Z(f) = Rn+ JωI + [(Gti+ jHti)/ωa]. Thus, we determined the following physiological properties; Newtonian resistance (Rn, a measurement of airway resistance) and elastance (Hti, a measure of parenchymal events). The peak response for each variable was determined, and the percentage change from baseline, as measured at the beginning of the protocol, was calculated.
Bronchoalveolar lavage (BAL)
BAL fluid was immediately collected from euthanized mice for the assessment of total and differential cell counts (6). Supernatants were collected for protein (BioRad, Hercules, CA) and cytokine quantitation (Eotaxin-1, IL-4, IL-5, IL-13, and IFN-γ) by ELISA according to manufacturer's instructions (R&D Systems, Minneapolis, MN).
Serum collection and Ig analysis
Following euthanasia, blood was collected in serum separator tubes, centrifuged, and serum was kept frozen at − 80°C. For Ig ELISAs, 96-well plates were coated overnight at 4°C with either 2 μg/ml OVA in 0.1 M NaHCO3 (pH 9.6) or anti-IgE (R35.72; BD Biosciences Pharmingen, San Diego, CA) in PBS (pH 7.2–7.4), washed with PBS containing 0.05% Tween 20, and blocked with 2% BSA in PBS. Plates were washed, and serum was applied at dilutions of 1:8–1:4096 and incubated overnight at 4°C. Plates were washed and incubated with biotinylated secondary Ab (Pharmingen), followed by incubation with streptavadin/peroxidase (Roche Diagnostics Corp., Indianapolis, IN) for 1 h, and detection using reagents from R&D Systems. ODs were read using a Bio-Tek Instruments PowerWaveX (Winooski, VT) plate reader at 450 nm. Data is reported as OD values (± SEM) from identical dilutions in the linear range of the readings (1:256).
Immunostaining
Following euthanasia, lungs were instilled with PBS for 5 min at a pressure of 25 cm H2O, placed into Tissue-Tek OCT Compound (Sakura Finetek, Inc., Torrance, CA), and frozen in liquid nitrogen-chilled isopentane for the preparation of 10-μm frozen sections. Slides were fixed for 5 min with 3% paraformaldehyde in PBS, washed and permeabilized for 20 min with 1% Triton X-100 in PBS, and blocked with 10% goat serum in PBS for 1 h. Slides were then incubated at room temperature with Ab recognizing RelA (10 μg/ml, SC-372; Santa Cruz Biotechnology; Santa Cruz, CA) in 1% BSA/PBS for 3 h. Following three washes in PBS, slides were incubated for 30 min with goat anti-rabbit Alexa 647-labeled secondary Ab (Molecular Probes, Eugene, OR) in PBS and counterstained with a 1:1000 dilution of SYTOX Green (Molecular Probes) in PBS to label DNA. Slides were washed with PBS, rinsed with ddH2O, and coverslipped using Aqua Poly-Mount (Polysciences, Inc., Warrington, PA). Sections were scanned using an Olympus BX50 upright microscope configured to a Bio-Rad MRC 1024 confocal scanning laser microscope system using a 40× objective and an iris setting of 2 (depth <2 μm). SYTOX Green staining of nuclei was detected by exciting fluorescence with the 488 laser line, whereas Alexa 647 was detected following excitation with the 647 laser line.
Histopathology and morphometry
Following euthanasia and BAL, the left lung lobe was instilled with 4% paraformaldehyde in PBS (4% PFA) for 10 min at a pressure of 25 cm H2O and placed into 4% PFA at 4°C overnight for further fixation of the tissue before embedding in paraffin. Then 7-μm sections were cut, affixed to glass microscope slides, deparaffinized with xylene, and rehydrated through a series of ethanols (6) and stained with H&E, or with periodic acid schiff stains (PAS), coverslipped, and examined by light microscopy. Sections with a length:diameter ratio of <2:1 were evaluated for inflammatory cell infiltration and PAS (mucin) positivity of the airways by a pathologist blinded to the identity of the sections. Multiple airways of similar size were assessed per section, and the percentage of PAS positive airway epithelial cells were recorded.
RNase protection assay (RPA)
Lungs were removed from the mice immediately following BAL, snap frozen in liquid nitrogen, and pulverized using chilled mortars and pestles. RNA was extracted and gene expression was assessed using the RPA (Pharmingen). Ten micrograms of total lung RNA was hybridized to 32P-labeled probe sets (mCK-5c), processed according to the manufacturers protocol, hybridized components were separated on a 5% acrylamide gel, and the gel was exposed to Kodak BioMax x-ray film (Eastman Kodak, Rochester, NY) or to phosphorimaging screens and subsequently quantitated on a Molecular FX system using QuantityOne software (BioRad).
RT-PCR
Total RNA was DNase treated and reverse transcribed into cDNA using SuperscriptII, according to instructions by the manufacturer (Life Technologies, Grand Island, NY). Semiquantitative PCR was performed using PCR SuperMix (Invitrogen, Carlsbad, CA). PCR conditions were: denaturation 30 s at 94°C, annealing 30 s at 55°C, extension for 60 s at 72°C, and a final extension for 5 min at 72°C. The following intron-spanning primers were used: CCL20 (30 cycles) forward 5′-CGTCTGCTCTTCCTTGCTTT-3′, reverse 5′-TTGACAAGTCCACTGGGACA-3′; Gob-5/Clca3 (30 cycles) forward 5′-AAGCAAACCACTCCCATGAC-3′, reverse 5′-TGCGAAAGCATCAA CAAGAC-3′; β-actin (25 cycles) forward 5′-TCCTTCGTTGCCGGTC CACA-3′, reverse 5′-CGTCTCCGGAGTCCATCACA-3′. PCR products were visualized following electrophoresis in 1% aragose gels stained with ethidium bromide. Densitometry was performed using a Chemidoc XRS imager and QuantityOne software (BioRad). Real-time semiquantitative RT-PCR was performed using the Taqman system. PCRs were performed using the Taqman Universal PCR Master Mix and the ABI PRISM 7700 Sequence Detection System. The probes and primer sets used for mouse Eotaxin-1 forward 5′-AGAGCTCCACAGCTTCTATT-3′, reverse 5′-CTTACTGGT CATGATAAAGCAGCAG-3′, probe 5′-FAM-(ACGGTCACTTCCTTCAC CTCCCAGG)-BHQ-1 3′; and for HPRT forward 5′-TTTGCCGCGAGCCG-3′, reverse 5′-TAACCTGGTTCATCATCGCTAATC-3′, probe 5′ FAM-(CG ACCCGCAGTCCCAGCGTC)-BHQ-1 3′, were purchased from Biosearch Technologies (Novato, CA). Eotaxin-1 levels were normalized to HPRT, and the data are presented as average expression relative to the housekeeping gene.
Statistical analysis
Data were analyzed by two-way ANOVA, and Bonferroni correction was used for multiple comparisons.
Results
Allergen sensitized and challenged CC10-IκBαsr mice exhibit reduced airway epithelial NF-κB activation
To directly address the functional significance of airway epithelial NF-κB activation in allergic airways disease, we used a transgenic mouse expressing IκBαsr in airway epithelium by placing this transgene under transcriptional control of the CC10 promoter (CC10-IκBαsr). As we have previously described, these mice exhibit normal gross and microscopic lung anatomy and express the transgene exclusively in conducting airways, not in alveolar epithelium or macrophages (14). In an OVA-driven model of allergic airways disease, the nuclear presence of the NF-κB subunit, RelA, was readily apparent in wild-type mice, whereas its localization was exclusively cytoplasmic in CC10-IκBαsr mice, illustrating that in response to allergen challenge, NF-κB activation in airway epithelium was effectively blocked (Fig. 1). We have previously demonstrated that NF-κB rapidly translocates from the cytoplasm to the nucleus of airway epithelial cells of sensitized mice exposed to Ag, without marked translocation in alveolar epithelial cells or macrophages of the lung (6). While similar results are observed herein, NF-κB is also observed in inflammatory cells comprising the peribronchiolar legion in wild-type mice, while expression of the transgene in the CC10-IκBαsr lung also inhibits the recruitment of activated inflammatory cells to the peribronchiolar space following Ag challenge.
FIGURE 1.
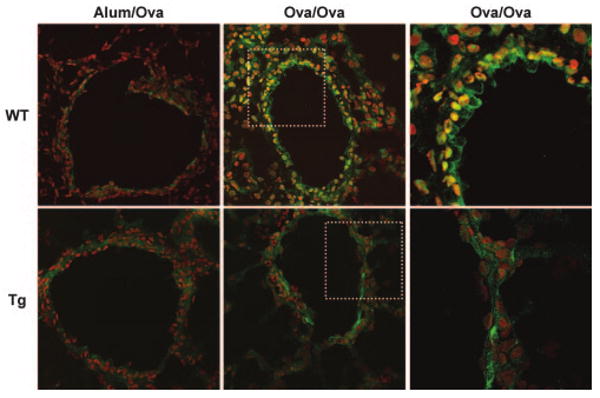
Immunolocalization of NF-κB in airway epithelial cells of allergen sensitized and challenged mice. Frozen lung sections were prepared 48 h following the third daily aerosolized allergen challenge of mock-sensitized (Alum/OVA) or OVA-sensitized (OVA/OVA) mice. Sections were stained with an Ab directed against RelA (Santa Cruz SC-372) followed by incubation with an Alexa 647-conjugated secondary Ab (pseudocolored green). A nuclear counter stain (pseudocolored red) was used to evaluate nuclear localization of RelA, in which case green and red merge to create yellow. Sections were scanned by confocal microscopy. Original magnification of left and center images, ×400. WT, wild-type; Tg, CC10-IκBαsr mice. To better illustrate the differences in nuclear NF-κB localization, the boxed areas of the center panel images were magnified using a ×2.2 optical zoom and are shown on the right. Images are representative of results from six mice per group.
Allergen-sensitized and challenged CC10-IκBαsr mice exhibit reduced airway inflammation and mucus production
We next assessed airways and tissue inflammation in response to inhaled Ag. Compared with sensitized and challenged wild-type mice, airway inflammation induced by Ag was significantly blunted in CC10-IκBαsr transgenics, based on the decreased numbers of inflammatory eosinophils and lymphocytes in BAL fluid (Fig. 2A) and peribronchiolar regions (Fig. 2B). Reductions in the number of mucin producing (PAS+) airway cells were also evident in the CC10-IκBαsr transgenics (Fig. 2C). Quantitation of the percentage of mucin-positive cells in bronchioles with a length: diameter ratio of <2:1 revealed significantly fewer cells in the transgenic (50 ± 6.3%) than in the wild-type (74 ± 7.1%) mice (p = 0.013).
FIGURE 2.
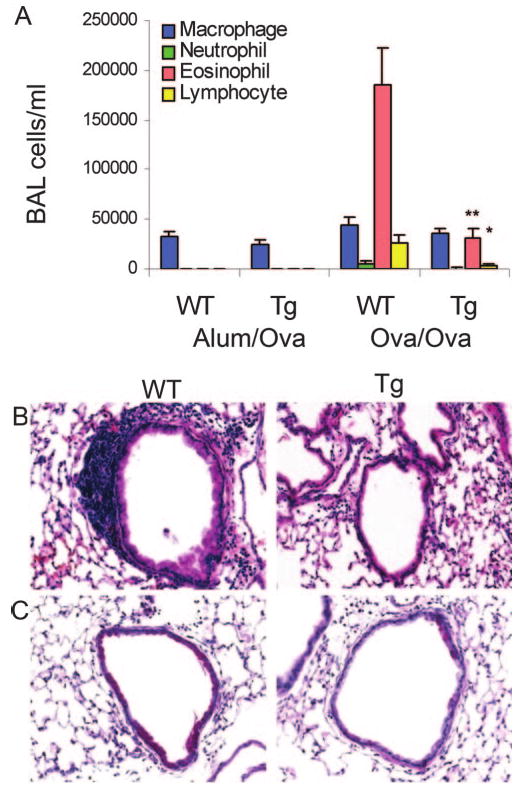
Inflammatory cell recruitment and airway remodeling following Ag sensitization and challenge. A, BAL fluid was collected and differential cell counts were performed 48 h following the third daily aerosolized OVA challenge from OVA-sensitized (OVA/OVA) or mock-sensitized (Alum/Ova) CC10-IκBαsr (Tg) or wild-type littermate control (WT) mice. Values are means (± SEM) from 8 to 16 mice per group. **, p < 0.01; *, p < 0.05 compared with values in WT OVA/OVA mice. Representative sections from paraffin-embedded lung were stained using H&E (B) and PAS reagents to visualize mucus-producing (reddish-purple) airway cells (C). Original magnification, ×400.
Diminished chemokine and T cell cytokine expression by sensitized and challenged CC10-IκBαsr mice
Airway epithelial NF-κB activation can promote the transcriptional up-regulation of numerous chemokine genes, essential in the recruitment of inflammatory cells (14), including eosinophils as well as CD4+ T lymphocytes, which in turn secrete Th2 cytokines. Whereas mRNA levels of the chemokine genes, RANTES, IP-10, MCP-1, eotaxin-1 (Fig. 3, A and D) and CCL20 (Fig. 3B) were markedly elevated in lung tissue of wild-type mice in response to Ag, expression levels of these genes were attenuated in CC10-IκBαsr mice. Among these chemokines, RANTES and eotaxin-1 are potent eosinophil chemoattractants (17), whereas TCA-3 induces the recruitment of Th2 cells to the lung in allergic airway disease (18), and MCP-1 may contribute to Th2 polarization (19), IP-10, while contributing to the recruitment of Th1 cells, can also modulate lung inflammation and the recruitment of Th2 lymphocytes in asthma (20), and CCL20 is an NF-κB-regulated (21), epithelial-derived (22) chemokine that stimulates the recruitment of immature dendritic cells and CD4+ T cells (23). Reductions in the expression of these chemokines may impact significantly on the subsequent magnitude of the inflammatory response elicited by inhaled Ag. Additionally, reduced expression of the calcium-activated chloride channel, Gob-5 (Clca3), a gene highly expressed by mucus-producing airway epithelial cells (24), was found in the lungs of Ag-challenged CC10-IκBαsr mice compared with wild-type controls (Fig. 3B), correlating with diminished mucus metaplasia. Significant decreases in eotaxin-1, and the T cell cytokines IL-5 and IL-13, but not IL-4 or IFN-γ, were observed in the BAL fluid of CC10-IκBαsr mice (Fig. 3C). The decreases in the production of IL-13, a cytokine known to stimulate mucus production via the transcriptional up-regulation of the Muc-5/5ac gene (25), correlated with attenuated goblet cell metaplasia in CC10-IκBαsr mice (Fig. 2C), while decreases in IL-5 and eotaxin-1 correlated with decreased eosinophilic influx into the BAL fluid. Thus, airway epithelial NF-κB activation is essential for establishing the full magnitude of the inflammatory response in this model, and consequently the abundance of cytokines produced by the inflammatory lymphocytes.
FIGURE 3.
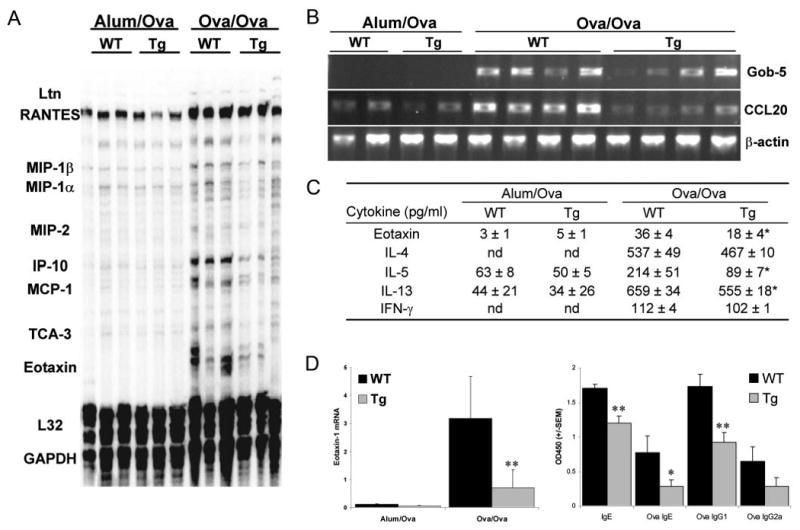
Production of chemokines, cytokines, and Ig following allergen sensitization and challenge. Lungs, BAL fluid, and serum from OVA-sensitized (OVA/OVA) or mock-sensitized (Alum/OVA) CC10-IκBαsr (Tg) mice, or transgene negative littermate control (WT) mice, were collected 48 h following the third daily aerosolized OVA challenge. RNA was extracted from lungs, reverse-transcribed, and analyzed by RPA using the commercial template mCK-5c (Pharmingen) (A), or by semiquantitative PCR (B). Data are representative of results from two separate experiments. (C) BAL fluid was analyzed by ELISA for the eosinophilic chemokine, Eotaxin-1, or for T cell cytokines. Values are means (± SEM) from 8 to 16 mice per group. nd, Not detectable. D, RNA was collected from lungs, reverse-transcribed, and analyzed for Eotaxin-1 relative to HPRT mRNA expression by semiquantitative TaqMan PCR from three to five mice per group. E, Serum was analyzed for total IgE and OVA-specific IgE, IgG1, and IgG2a by ELISA. Values are mean optical densities (± SEM) from five mice per group. **, p < 0.01; *, p < 0.05 compared with values from WT OVA/OVA mice.
Reduced IgE and Ag-specific Abs in sensitized and challenged CC10-IκBαsr mice
Circulating levels of IgE are related to asthma symptoms and decreasing IgE improves asthma, reducing the need for steroid therapy (26). Intriguingly, the circulating OVA-specific IgE, total IgE, and OVA-specific IgG1 levels, but not OVA-specific IgG2a, were significantly reduced in the CC10-IκBαsr mice compared with wildtype controls (Fig. 3E). Collectively, these findings clearly demonstrate that NF-κB activation in airway epithelium is a cardinal event in the orchestration of airways and tissue inflammation, and the expression of inflammatory mediators. We also demonstrate that NF-κB activation in airways indirectly dictates Th cell cytokine production, mucous cell metaplasia, and Ag-specific Ig levels.
CC10-IκBαsr mice are not protected from Ag-driven lung hyperresponsiveness
Asthma is defined by changes in airways physiology, notably hyperresponsiveness. We therefore assessed whether repression of NF-κB in airway epithelium would also attenuate airways hyperresponsiveness. Using mechanically ventilated mice, airway resistance (Rn) and tissue elastance (H) were calculated from data obtained with a forced oscillation technique (15, 16). Ag challenge of wild-type mice resulted in significant airway hyperresponsiveness to inhaled methacholine when compared with mock-sensitized mice (Fig. 4). CC10-IκBαsr mice displayed similar hyperresponsiveness in response to Ag as the wild-type mice, despite the significant attenuation of other variables believed to be clinically relevant to the pathophysiology of asthma. Therefore, we conclude that airway epithelial NF-κB-driven inflammation and other immune alterations are uncoupled from airways hyperresponsiveness. These findings are surprising in light of the fact that inflammatory events in the airway wall are commonly believed to be integral to the pathophysiology of asthma (27) and, as such, are current therapeutic targets (28).
FIGURE 4.
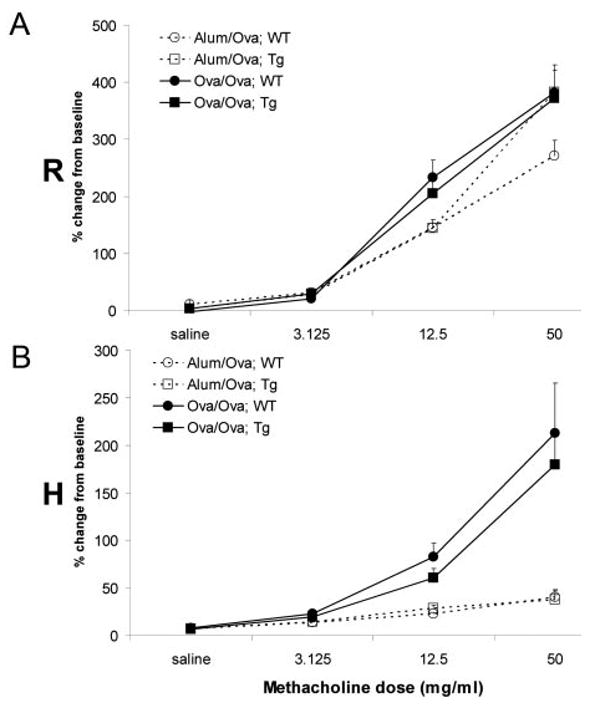
Assessment of airways hyperresponsiveness following allergen sensitization and challenge. Pulmonary hyperresponsiveness was analyzed in mock-sensitized (Alum/OVA, open symbols and broken lines) or OVA-sensitized (OVA/OVA, filled symbols and solid lines) CC10-IκBαsr (Tg, squares) or transgene negative littermate control (WT, circles) mice 48 h following the third daily aerosolized OVA challenge. Hyperresponsiveness to ascending doses of nebulized methacholine was assessed from forced oscillations and determined as Rn (airway resistance, A) and H (tissue elastance, B). As derived from the constant phase model, data are expressed as the percent change from baseline measurements (± SEM) for each of the parameters measured, are inclusive of results from three separate experiments, and comprised of 11–13 mice per group. Baseline measurements before methacholine were found not to be significantly different between any of the groups (data not shown).
Discussion
Stimulation of airway epithelial cells by inhaled materials evokes responses that may contribute to the development and maintenance of chronic pulmonary inflammation through the activation of NF-κB (8). Previous studies have demonstrated that NF-κB plays a causal role in allergic airways inflammation and suggest that inhibition of NF-κB serves as a therapeutic target for the treatment of acute or chronic pulmonary inflammatory disease states (29). However, these studies failed to consider the anatomical location of NF-κB activation within the lung. NF-κB activation has been demonstrated in lung tissue (30–33), and specifically within airway epithelium, in animal models of allergic airway inflammation (6, 7, 34) and in patients with asthma (8–13). Yet, conclusive evidence implicating the importance of NF-κB activity within the airways in the etiology of allergic airways disease was lacking.
The studies presented herein are novel and important because of the selective targeting of NF-κB repression in the epithelial cells of conducting airways, without affecting other cells of the lung, nor the alveolar compartment (6). We demonstrate here that repression of NF-κB in CC10-IκBαsr-expressing mice is sufficient to blunt the inflammatory response observed in the OVA model of airways inflammation. Marked reductions in airway and tissue neutrophils, eosinophils, and lymphocytes were observed in response to Ag in CC10-IκBαsr mice compared with wild type. As expected, the reduced inflammatory response to Ag in transgenic mice was accompanied by reduced expression of chemokines, including eotaxin-1 and CCL20, which are important in recruitment and activation of inflammatory cells such as eosinophils and CD4+ T lymphocytes, respectively. Many resident cell types of the lung are capable of secreting eosinophil and lymphocyte chemoattractants in response to stimulation. We have previously observed rapid and selective activation of NK-κB in airway epithelium of Ag-sensitized and -challenged mice (6) and report herein that inhibiting NF-κB activation, via phosphorylation of serines 32 and 36 of IκBα, selectively in these cells reduces the overall magnitude of the inflammatory response to Ag challenge. Combined, these findings support our notion of the central role of airway epithelial NF-κB activation in allergic airway inflammation, but do not minimize the importance of additional signaling pathways, additional cell types, and the complex interactions between them in dictating the pathophysiology of allergic airway disease.
Surprisingly, the expression of IL-5 and IL-13, cytokines derived from Th2 lymphocytes and total and Ag-specific Ig production were also attenuated following repression of NF-κB in the airways, suggesting that NF-κB-dependent gene products generated by airway epithelium are essential for maximal recruitment and stimulation of T and B cells in this model of allergic airways disease. Although the capacity of airway epithelial cells to process and present Ag, resulting in proliferation of OVA-sensitized T lymphocytes, has been demonstrated in vitro (35), the identity of the molecules responsible for lymphocyte stimulation remain unknown. B7 family (36) and MHC class II (37) molecules are expressed by lung epithelial cells and are putative candidates. In addition, cell-surface molecules such as VCAM-1 and ICAM-1, necessary for leukocyte recruitment and their local activation, are inducibly expressed by airway epithelial cells in response to stimulation with TNF-α or IL-1β, cytokines that signal through activation of NF-κB (38). It remains unknown from our work whether in an immunized mouse the airway epithelium can directly recognize Ag as a result of up-regulation of relevant receptors on the epithelium, or that a product derived from mast cells or dendritic cells is responsible for activating NF-κB in airway epithelial cells. Ag stimulation of bronchial brushings from asthmatics rapidly induced NF-κB activation (10), giving additional credence to the notion that airway epithelium, in a proper immunologically primed context, can directly recognize Ag. Our prior demonstration of rapid and widespread activation of NF-κB in airways following Ag challenge in sensitized mice (6), coupled to studies demonstrating that mast cell-deficient mice display robust inflammation (39), suggest a direct role of airway epithelium in Ag recognition, although additional studies are clearly needed.
One of the T cell cytokines that was attenuated in CC10-IκBαsr mice was IL-13, an important stimulant of mucus production via the transcriptional up-regulation of the Muc-5/5ac gene (25). While IL-13 does not require epithelial NF-κB activation to induce goblet cell metaplasia (40), evaluation of mucus production in CC10-IκBαsr-expressing mice demonstrated lessened PAS reactivity, illustrating that repression of NF-κB is an important strategy to attenuate mucus production and potentially hypersecretion, likely through the reduced recruitment of IL-13-producing Th2 cells to an appropriate lung compartment for stimulation subsequent to allergen inhalation. The reduced number of lymphocytes recovered in the BAL fluid supports this supposition. Alternatively, the possibility exists that Th2 cells are in the correct anatomic location but lack sufficient NF-κB-dependent co-stimulatory signals from the airway epithelium for optimal responsiveness. However, our current strategy failed to completely prevent mucus production, suggesting that other pathways or cell types contribute to mucus metaplasia in the mouse model of allergic airways disease, or that the residual inflammatory response and IL-13 secretion that remains is sufficient to drive mucus production.
Despite the significant attenuation of parameters believed to be clinically relevant to the pathophysiology of asthma, the airflow alterations that characterize airways hyperresponsiveness were not affected in CC10-IκBαsr-expressing mice. These findings are puzzling in light of the fact that the inflammatory state is currently believed to be integral to the asthma disease process. It is possible that the residual inflammation and accompanying secretion of mediators observed in the CC10-IκBαsr mice, or residual NF-κB activation that still might occur in the airway epithelium, albeit not readily detectable, are sufficient to fully drive the airways hyperresponsiveness. This implies that anti-inflammatory therapies would have to be highly effective to correct changes in lung function. It is important to note that the rat CC10 promoter targets expression of the transgene to conducting airways, while not directing transgene expression to the alveolar compartment (14). Alveolar epithelial NF-κB activation is unlikely to play a significant role in airflow alterations, given our previous observations that alveolar NF-κB activation and inflammatory gene expression are minimal (6). Our results show that NF-κB driven inflammation is uncoupled from airway hyperresponsiveness, as has been previously reported in human subjects (41). This notion is further supported by studies documenting that marked changes in allergen-induced airways physiology can occur in the absence of airway inflammation (42, 43). We have documented that in the mouse the location of inflammation, whether peribronchiolar or alveolar, is critical in driving airways hyperresponsiveness and showed that the alveolar or peripheral processes dominate this response (15). A recent study published by our laboratory has demonstrated that fibrin deposition in the lung is a major determinant of airway hyperresponsiveness, and that manipulation of the fibrinolytic cascade effectively modulates airway hyperresponsiveness (44). Because fibrin deposition is unlikely to be modulated by airway epithelial NF-κB activation, this would explain why airway hyperresponsiveness is retained in the CC10-IκBαsr mice, despite reductions in many other features of asthma. The current study further extends the previous work by implicating an alveolar-based process consistent with associated transbronchial biopsy studies in humans (45). However, it must be kept in mind that the mouse model employed herein is acute and while germane to the majority of patients in the onset of mild asthma exacerbations may not represent the patient with more severe or other forms of the syndrome (46–48).
In summary, this study demonstrates that NF-κB activation in airways drives a majority of the Ag-induced inflammation and is a significant contributor to adaptive immune responses and structural remodeling of the airway wall. In contrast, NF-κB activation in airways does not appear to play a prominent role in the genesis of airways hyperresponsiveness, a cardinal feature thought to underlie allergic airways disease. Alternative signaling pathways activated in airways, residual NF-κB activity, or the residual inflammatory cells present in the CC10-IκBαsr mouse lungs may be responsible for the observed airways hyperresponsiveness. Alternatively, events localized more distally within the alveolar compartment, such as leakage of macromolecules from the microvas-culature, alveolar injury, or surfactant dysfunction (49–51), may dominate in the genesis of airways hyperresponsiveness. Thus, avenues that effectively target these mechanisms and treatment modalities directed to the distal regions of the lung will be essential to minimize airways hyperresponsiveness.
Acknowledgments
We thank Mercedes M. Rincon for invaluable guidance with the generation of the transgenic mice, Ralph C. Budd and Scott S. Wagers for critically reading the manuscript, and Jean Claude Sirard for helpful discussions.
Footnotes
This work was supported by the National Institutes of Health (NIH) Program Project Award PO1-HL-67004, the Vermont Genetics Network through the NIH Grant 1 P20 RR16462 from the Biomedical Research Infrastructure Network program of the National Center for Research Resources (NCRR), NIH Transition to Independent Positions Award K22 ES011652, and NIH/NCRR Center for Biomedical Research Excellence P20 RR15557.
Abbreviations used in this paper: IκBαsr, IkappaB alpha superrepressor; CC10, clara cell 10-kDa protein; BAL, bronchoalveolar lavage; RPA, ribonuclease protection assay; PAS, periodic acid schiff stain; PFA, paraformaldehyde.
References
- 1.Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh S, May MJ, Kopp EB. NF-κB and rel proteins: Evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 3.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-κB in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2:45. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 4.Donovan CE, Mark DA, He HZ, Liou HC, Kobzik L, Wang Y, De Sanctis GT, Perkins DL, Finn PW. NF-κB/Rel transcription factors: c-Rel promotes airway hyperresponsiveness and allergic pulmonary inflammation. J Immunol. 1999;163:6827. [PubMed] [Google Scholar]
- 5.Yang BL, Cohn L, Zhang DH, Homer R, Ray A, Ray O. Essential role of nuclear factor κB in the induction of eosinophilia in allergic airway inflammation. J Exp Med. 1998;188:1739. doi: 10.1084/jem.188.9.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poynter M, Irvin C, Janssen-Heniniger Y. Rapid activation of nuclear factor-κB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol. 2002;160:1325. doi: 10.1016/s0002-9440(10)62559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bureau F, Bonizzi G, Kirschvink N, Delhalle S, Desmecht D, Merville MP, Bours V, Lekeux P. Correlation between nuclear factor-κB activity in bronchial brushing samples and lung dysfunction in an animal model of asthma. Am J Respir Crit Care Med. 2000;161:1314. doi: 10.1164/ajrccm.161.4.9907010. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton LM, Davies DE, Wilson SJ, Kimber I, Dearman RJ, Holgate ST. The bronchial epithelium in asthma—Much more than a passive barrier. Monaldi Arch Chest Dis. 2001;56:48. [PubMed] [Google Scholar]
- 9.Zhao S, Qi Y, Liu X, Jiang Q, Liu S, Jiang Y, Jiang Z. Activation of NF-κB in bronchial epithelial cells from children with asthma. Chin Med J. 2001;114:909. [PubMed] [Google Scholar]
- 10.Stacey MA, Sun G, Vassalli G, Marini M, Bellini A, Mattoli S. The allergen Der p1 induces NF-κB activation through interference with IκBα function in asthmatic bronchial epithelial cells. Biochem Biophys Res Commun. 1997;236:522. doi: 10.1006/bbrc.1997.6997. [DOI] [PubMed] [Google Scholar]
- 11.Vignola AM, Chiappara G, Siena L, Bruno A, Gagliardo R, Merendino AM, Polla BS, Arrigo AP, Bonsignore G, Bousquet J, Chanez P. Proliferation and activation of bronchial epithelial cells in corticosteroid-dependent asthma. J Allergy Clin Immunol. 2001;108:738. doi: 10.1067/mai.2001.119160. [DOI] [PubMed] [Google Scholar]
- 12.Hart L, Lim S, Adcock I, Barnes J, Chung KF. Effects of inhaled corticosteroid therapy on expression and DNA-binding activity of nuclear factor κB in asthma. Am J Respir Crit Care Med. 2000;161:224. doi: 10.1164/ajrccm.161.1.9809019. [DOI] [PubMed] [Google Scholar]
- 13.Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF. Activation and localization of transcription factor, nuclear factor-κB, in asthma. Am J Respir Crit Care Med. 1998;158:1585. doi: 10.1164/ajrccm.158.5.9706116. [DOI] [PubMed] [Google Scholar]
- 14.Poynter M, Irvin C, Janssen-Heininger Y. A prominent role for airway epithelial NF-κB activation in lipopolysaccharide-induced airway inflammation. J Immunol. 2003;170:6257. doi: 10.4049/jimmunol.170.12.6257. [DOI] [PubMed] [Google Scholar]
- 15.Tomioka S, Bates JH, Irvin CG. Airway and tissue mechanics in a murine model of asthma: Alveolar capsule vs. forced oscillations. J Appl Physiol. 2002;93:263. doi: 10.1152/japplphysiol.01129.2001. [DOI] [PubMed] [Google Scholar]
- 16.Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol. 1992;72:168. doi: 10.1152/jappl.1992.72.1.168. [DOI] [PubMed] [Google Scholar]
- 17.Lampinen M, Carlson M, Hakansson LD, Venge P. Cytokine-regulated accumulation of eosinophils in inflammatory disease. Allergy. 2004;59:793. doi: 10.1111/j.1398-9995.2004.00469.x. [DOI] [PubMed] [Google Scholar]
- 18.Chensue SW, Lukacs NW, Yang TY, Shang X, Frait KA, Kunkel SL, Kung T, Wiekowski MT, Hedrick JA, Cook DN, Zingoni A, Narula SK, Zlotnik A, Barrat FJ, O'Garra A, Napolitano M, Lira SA. Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J Exp Med. 2001;193:573. doi: 10.1084/jem.193.5.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rose CE, Jr, Sung SS, Fu SM. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation. 2003;10:273. doi: 10.1038/sj.mn.7800193. [DOI] [PubMed] [Google Scholar]
- 20.Medoff BD, Sauty A, Tager AM, Maclean JA, Smith RN, Mathew A, Dufour JH, Luster AD. IFN-γ-inducible protein 10 (CXCL10) contributes to airway hyperreactivity and airway inflammation in a mouse model of asthma. J Immunol. 2002;168:5278. doi: 10.4049/jimmunol.168.10.5278. [DOI] [PubMed] [Google Scholar]
- 21.Fujiie S, Hieshima K, Izawa D, Nakayama T, Fujisawa R, Ohyanagi H, Yoshie O. Proinflammatory cytokines induce liver and activation-regulated chemokine/macrophage inflammatory protein-3α/CCL20 in mucosal epithelial cells through NF-κB [correction of NK-κB] Int Immunol. 2001;13:1255. doi: 10.1093/intimm/13.10.1255. [DOI] [PubMed] [Google Scholar]
- 22.Starner TD, Barker CK, Jia HP, Kang Y, McCray PB., Jr CCL20 is an inducible product of human airway epithelia with innate immune properties. Am J Respir Cell Mol Biol. 2003;29:627. doi: 10.1165/rcmb.2002-0272OC. [DOI] [PubMed] [Google Scholar]
- 23.Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 24.Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, Shirafuji H, Fujisawa Y, Nishimura O, Fujino M. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc Natl Acad Sci USA. 2001;98:5175. doi: 10.1073/pnas.081510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuhdi Alimam M, Piazza FM, Selby DM, Letwin N, Huang L, Rose MC. Muc-5/5ac mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways. Am J Respir Cell Mol Biol. 2000;22:253. doi: 10.1165/ajrcmb.22.3.3768. [DOI] [PubMed] [Google Scholar]
- 26.Burrows B, Sears MR, Flannery EM, Herbison GP, Holdaway MD. Relationships of bronchial responsiveness assessed by methacholine to serum IgE, lung function, symptoms, and diagnoses in 11-year-old New Zealand children. J Allergy Clin Immunol. 1992;90:376. doi: 10.1016/s0091-6749(05)80018-1. [DOI] [PubMed] [Google Scholar]
- 27.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 28.Barnes PJ. Therapeutic strategies for allergic diseases. Nature. 1999;402:B31. doi: 10.1038/35037026. [DOI] [PubMed] [Google Scholar]
- 29.Lee JI, Burckart GJ. Nuclear factor κB: Important transcription factor and therapeutic target. J Clin Pharmacol. 1998;38:981. doi: 10.1177/009127009803801101. [DOI] [PubMed] [Google Scholar]
- 30.Kang JL, Lee HW, Lee HS, Pack IS, Chong Y, Castranova V, Koh Y. Genistein prevents nuclear factor-κB activation and acute lung injury induced by lipopolysaccharide. Am J Respir Crit Care Med. 2001;164:2206. doi: 10.1164/ajrccm.164.12.2104017. [DOI] [PubMed] [Google Scholar]
- 31.Held HD, Boettcher S, Hamann L, Uhlig S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-κB and is blocked by steroids. Am J Respir Crit Care Med. 2001;163:711. doi: 10.1164/ajrccm.163.3.2003001. [DOI] [PubMed] [Google Scholar]
- 32.Blackwell TS, Lancaster LH, Blackwell TR, Venkatakrishnan A, Christman JW. Differential NF-κB activation after intratracheal endotoxin. Am J Physiol. 1999;277:L823. doi: 10.1152/ajplung.1999.277.4.L823. [DOI] [PubMed] [Google Scholar]
- 33.Walley KR, McDonald TE, Higashimoto Y, Hayashi S. Modulation of proinflammatory cytokines by nitric oxide in murine acute lung injury. Am J Respir Crit Care Med. 1999;160:698. doi: 10.1164/ajrccm.160.2.9809081. [DOI] [PubMed] [Google Scholar]
- 34.Bureau F, Delhalle S, Bonizzi G, Fievez L, Dogne S, Kirschvink N, Vanderplasschen A, Merville MP, Bours V, Lekeux P. Mechanisms of persistent NF-κB activity in the bronchi of an animal model of asthma. J Immunol. 2000;165:5822. doi: 10.4049/jimmunol.165.10.5822. [DOI] [PubMed] [Google Scholar]
- 35.Suda T, Sato A, Sugiura W, Chida K. Induction of MHC class II antigens on rat bronchial epithelial cells by interferon-γ and its effect on antigen presentation. Lung. 1995;173:127. doi: 10.1007/BF02981472. [DOI] [PubMed] [Google Scholar]
- 36.Kurosawa S, Myers AC, Chen L, Wang S, Ni J, Plitt JR, Heller NM, Bochner BS, Schleimer RP. Expression of the costimulatory molecule B7–H2 (inducible costimulator ligand) by human airway epithelial cells. Am J Respir Cell Mol Biol. 2003;28:563. doi: 10.1165/rcmb.2002-0199OC. [DOI] [PubMed] [Google Scholar]
- 37.Schwiebert LM, Schleimer RP, Radka SF, Ono SJ. Modulation of MHC class II expression in human cells by dexamethasone. Cell Immunol. 1995;165:12. doi: 10.1006/cimm.1995.1181. [DOI] [PubMed] [Google Scholar]
- 38.Atsuta J, Sterbinsky SA, Plitt J, Schwiebert LM, Bochner BS, Schleimer RP. Phenotyping and cytokine regulation of the BEAS-2B human bronchial epithelial cell: Demonstration of inducible expression of the adhesion molecules VCAM-1 and ICAM-1. Am J Respir Cell Mol Biol. 1997;17:571. doi: 10.1165/ajrcmb.17.5.2685. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, Gelfand EW. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med. 1997;186:449. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whittaker L, Niu N, Temann UA, Stoddard A, Flavell RA, Ray A, Homer RJ, Cohn L. Interleukin-13 mediates a fundamental pathway for airway epithelial mucus induced by CD4 T cells and interleukin-9. Am J Respir Cell Mol Biol. 2002;27:593. doi: 10.1165/rcmb.4838. [DOI] [PubMed] [Google Scholar]
- 41.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, Brusasco V. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Respir Crit Care Med. 1998;157:4. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 42.Brewer JP, Kisselgof AB, Martin TR. Genetic variability in pulmonary physiological, cellular, and antibody responses to antigen in mice. Am J Respir Crit Care Med. 1999;160:1150. doi: 10.1164/ajrccm.160.4.9806034. [DOI] [PubMed] [Google Scholar]
- 43.Ewart SL, Kuperman D, Schadt E, Tankersley C, Grupe A, Shubitowski DM, Peltz G, Wills-Karp M. Quantitative trait loci controlling allergen-induced airway hyperresponsiveness in inbred mice. Am J Respir Cell Mol Biol. 2000;23:537. doi: 10.1165/ajrcmb.23.4.4199. [DOI] [PubMed] [Google Scholar]
- 44.Wagers SS, Norton RJ, Rinaldi LM, Bates JH, Sobel BE, Irvin CG. Extravascular fibrin, plasminogen activator, plasminogen activator inhibitors, and airway hyperresponsiveness. J Clin Invest. 2004;114:104. doi: 10.1172/JCI19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kraft M, Djukanovic R, Wilson S, Holgate ST, Martin RJ. Alveolar tissue inflammation in asthma. Am J Respir Crit Care Med. 1996;154:1505. doi: 10.1164/ajrccm.154.5.8912772. [DOI] [PubMed] [Google Scholar]
- 46.Epstein MM. Do mouse models of allergic asthma mimic clinical disease? Int Arch Allergy Immunol. 2004;133:84. doi: 10.1159/000076131. [DOI] [PubMed] [Google Scholar]
- 47.Kips JC, Anderson GP, Fredberg JJ, Herz U, Inman MD, Jordana M, Kemeny DM, Lotvall J, Pauwels RA, Plopper CG, Schmidt D, Sterk PJ, Van Oosterhout AJ, Vargaftig BB, Chung KF. Murine models of asthma. Eur Respir J. 2003;22:374. doi: 10.1183/09031936.03.00026403. [DOI] [PubMed] [Google Scholar]
- 48.Kumar RK, Foster PS. Modeling allergic asthma in mice: Pitfalls and opportunities. Am J Respir Cell Mol Biol. 2002;27:267. doi: 10.1165/rcmb.F248. [DOI] [PubMed] [Google Scholar]
- 49.Kaminsky DA, Bates JH, Irvin CG. Effects of cool, dry air stimulation on peripheral lung mechanics in asthma. Am J Respir Crit Care Med. 2000;162:179. doi: 10.1164/ajrccm.162.1.9806079. [DOI] [PubMed] [Google Scholar]
- 50.Wagers SS, Bouder TG, Kaminsky DA, Irvin CG. The invaluable pressure-volume curve. Chest. 2000;117:578. doi: 10.1378/chest.117.2.578. [DOI] [PubMed] [Google Scholar]
- 51.Kaminsky DA, Wenzel SE, Carcano C, Gurka D, Feldsien D, Irvin CG. Hyperpnea-induced changes in parenchymal lung mechanics in normal subjects and in asthmatics. Am J Respir Crit Care Med. 1997;155:1260. doi: 10.1164/ajrccm.155.4.9105064. [DOI] [PubMed] [Google Scholar]