

Abstract
Packaging of eukaryotic genomes into chromatin affects every process that occurs on DNA. The positioning of nucleosomes on underlying DNA plays a key role in regulation of these processes, as the nucleosome occludes underlying DNA sequences. Here, we review the literature on mapping nucleosome positions in various organisms, and discuss how nucleosome positions are established, what effect nucleosome positioning has on control of gene expression, and touch on the correlations between chromatin packaging, sequence evolution, and the evolution of gene expression programs.
Introduction
DNA molecules of eukaryotic chromosomes range from 7.8 to 520 μm in length for yeast or 1.7 to 8.5 cm for humans. Considering the size of yeast nuclei (2–6 μm in diameter) or animal cell nuclei (20–50 μm in diameter), eukaryotic organisms are faced with an information storage and packaging problem. Anyone who has had to deal with tangled rope or yarn can appreciate the importance of packaging long strings into a manageable and accessible form. DNA has to be folded in a way that allows access to a key subset of DNA sequences located throughout the molecule. So, instead of folding into one big ball like yarn or rope, DNA wraps around thousands of globular protein cores and ends up looking like “beads on a string” in electron micrographs. The “bead” or nucleosome is the smallest packaging unit of the chromatin fiber and it consists of 147bp DNA wrapped 1.65 turns around a histone octamer core, consisting of two H2A/H2B and H3/H4 heterodimers(Luger et al., 1997). Nucleosomes are then further assembled into various higher order structures that can potentially be unwrapped as needed (for review see (Luger and Hansen, 2005)).
Decades of single gene studies have confirmed the intuitive notion that depending on whether they are found in the nucleosome or in the linker DNA between nucleosomes, DNA sequences can be more or less accessible to transcription, DNA repair or DNA recombination machinery (Boeger et al., 2008; Durrin et al., 1992; Green and Almouzni, 2002; Kamakaka and Thomas, 1990; Roth and Roth, 2000). In addition to its role in DNA packaging, the arrangement of nucleosomes along a DNA sequence is therefore also a regulatory mechanism that influences gene expression and other DNA-dependent processes. Unraveling the rules and factors that determine how nucleosomes are positioned and how they influence gene activity and evolution is one of the central questions in biology today. With the implementation of high-throughput genomic scale experimental techniques we are now starting to define the general principles that guide nucleosome positioning and the ways nucleosomal organization affects DNA processes.
Nucleosome positioning over genes across many different organisms
Over the past five years, an increasing number of studies have made use of genomic technologies to map nucleosomes across the genomes of organisms ranging from budding yeast to humans (Johnson et al., 2006; Mavrich et al., 2008a; Mavrich et al., 2008b; Schones et al., 2008; Valouev et al., 2008; Whitehouse et al., 2007; Yuan et al., 2005). Briefly, crosslinked chromatin is digested to mononucleosomes using micrococcal nuclease, which preferentially digests linker DNA and leaves nucleosomal DNA intact. Protected DNA is isolated, and nucleosomal fragments are assayed either by tiling microarray or by ultra high-throughput sequencing.
These studies have provided us with a picture of chromatin structure in many organisms, most extensively yeast. First, and perhaps most surprisingly, the majority of nucleosomes in yeast are “well-positioned,” meaning that the nucleosome is positioned at the same location in all the cells in the population. Positional variability is of course a quantitative feature, but in the literature authors often refer to “well-localized” nucleosomes when the standard deviation in positioning of the nucleosome center is on the order of 10–20 bp, which corresponds to the variability in the extent of MNase digestion of nucleosome ends. Nucleosome positioning is globally more variable in multicellular organisms, but nonetheless a subset of their nucleosomes are well-positioned, and the locations of these well-positioned nucleosomes provide clues as to the mechanisms that determine nucleosome positioning (see below). The observed higher variability of nucleosome positioning in multicellular organisms is not only due to differential nucleosome positioning in different cell types, as the same level of variability has been observed in relatively homogenous cell populations such as CD4+ T cells.
Genome-wide studies also provide a view of the nucleosome positions in a typical gene (Fig. 1). Promoter regions are overall depleted of nucleosomes relative to transcribed regions. So-called “nucleosome free regions” (NFRs) are localized just upstream of the transcription start site (TSS). This pattern is found in yeast (Albert et al., 2007; Kaplan et al., 2008; Mavrich et al., 2008a; Whitehouse et al., 2007; Yuan et al., 2005), worms (Johnson et al., 2006; Valouev et al., 2008), medaka (Sasaki et al., 2009), flies (Mavrich et al., 2008b) and humans (Ozsolak et al., 2007; Schones et al., 2008). The first nucleosome downstream of the TSS (the +1 nucleosome) is strongly localized, and nucleosomes at the 5′ end of a gene are generally better-localized than nucleosomes in the middle of genes. An NFR at the 3′ end of genes with a localized nucleosome immediately upstream of it has also been identified in both yeast (Mavrich et al., 2008a; Shivaswamy et al., 2008) and flies (Mavrich et al., 2008b). Interestingly, the position of the +1 nucleosome relative to TSS seems to vary in different organisms and may reflect differences in transcriptional regulatory mechanisms, or in the core transcription machinery such as TFIIB (Jiang and Pugh, 2009; Li et al., 1994). The center of the yeast +1 nucleosome is located ~50–60 bp downstream of the TSS, such that transcription typically starts about 10 bases into the first nucleosome (Mavrich et al., 2008a; Whitehouse et al., 2007; Yuan et al., 2005). In contrast, in Drosophila the center of the +1 nucleosome is found 135bp downstream of the TSS, meaning that the upstream border of the nucleosome is located roughly 60 bp downstream of the TSS (Mavrich et al., 2008b). Interestingly, the +1 nucleosome is shifted 10 bp downstream at genes with paused RNA polymerase. A related phenomenon has also been observed in human T-cells, where the 5′ end of the +1 nucleosome is located at +40bp from the TSS in genes with elongating RNA pol II and at +10bp in inactive genes with stalled RNA Pol II, suggesting a role for RNA polymerase in shaping the chromatin landscape (see TRANS determinants, below) (Schones et al., 2008).
Figure 1. Nucleosome positioning in a typical gene.
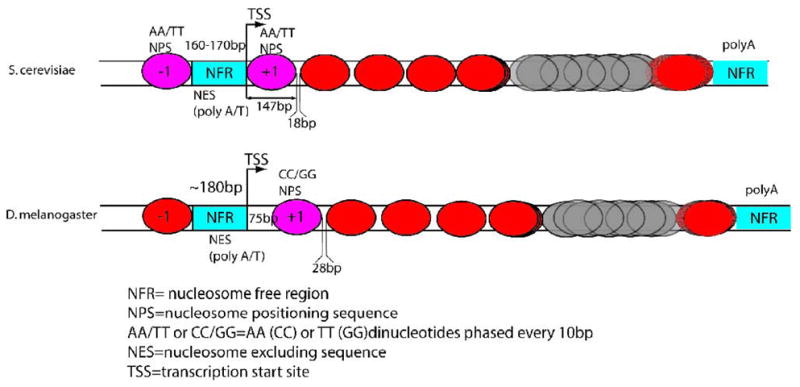
Red and grey nucleosomes are strongly and weakly positioned, respectively. Magenta nucleosomes are occasionally positioned by DNA sequence.
This canonical promoter chromatin structure (−1 nucleosome/NFR/+1 nucleosome) is found in “housekeeping” genes such as those encoding glycolysis and ribosomal proteins, and NFR width correlates somewhat with transcription levels in yeast. Conversely, stress-responsive genes, whose promoters are enriched for TATA boxes and are regulated by the SAGA complex (Basehoar et al., 2004; Huisinga and Pugh, 2004), are more likely to have a non-canonical promoter chromatin structure with a diminished NFR, often due to delocalized nucleosomes partially covering the promoter (Albert et al., 2007; Choi and Kim, 2009; Field et al., 2008; Tirosh and Barkai, 2008b). The non-canonical chromatin structure of these promoters is probably not due to transcription inactivity but is rather secondary to a lack of nucleosome-excluding sequences such as Poly(dA/dT) (see below), and may also be linked to the distinct regulatory mechanisms at work in these genes. Interestingly, Drosophila’s TATA box-containing genes also show a non-canonical nucleosome organization (Mavrich et al., 2008b), suggesting that the division of genes in two classes with distinct promoter chromatin structures - housekeeping genes and environmentally responsive genes - may be an evolutionarily-conserved feature.
CIS determinants of nucleosome positioning
The remarkably uniform and conserved nucleosomal organization of gene promoters begs the question: what determines nucleosome positions throughout the genome? Are nucleosome positions primarily “encoded” in the DNA sequence (cis factors) or are they a consequence of the regulatory activity of chromatin remodelers, transcription factors and the transcription machinery (trans factors)?
As befits a general packaging factor, the histone octamer has little sequence preference in the classical sense of having a binding motif. However, the constraint of having to wrap DNA nearly twice around a small octamer of proteins (the nucleosome’s diameter is much shorter than the ~50 nm persistence length of free DNA in solution) means that the energy required to bend a given genomic sequence does influence the binding affinity of the histone octamer (Thastrom et al., 1999). Since structural properties of DNA, such as local bendability, depend on DNA sequence, one might expect that DNA sequence will at least partially contribute to nucleosome positioning. The structure of poly-dA/dT sequences differs from the canonical double helix (Nelson et al., 1987) and is presumed to be resistant to the distortions necessary for wrapping around nucleosomes (Iyer and Struhl, 1995; Kunkel and Martinson, 1981; Segal and Widom, 2009; Sekinger et al., 2005). Conversely, sequences with AA/TT/TA dinucleotides spaced at 10 bp intervals are intrinsically bendable and thus bind the octamer with higher affinity than random sequence (Anselmi et al., 1999; Thastrom et al., 1999; Trifonov, 1980). An influential demonstration of the role for sequence in dictating chromatin structure was the report from the Struhl group that in vitro reconstitution of the HIS3 promoter into chromatin recapitulates some aspects of that promoter’s in vivo chromatin structure (Sekinger et al., 2005).
Recently, several groups have approached the question of sequence-driven nucleosome positioning computationally by trying to extract nucleosome-favoring and nucleosome-excluding DNA sequence motifs from in vivo nucleosome positions (Ioshikhes et al., 2006; Peckham et al., 2007; Segal et al., 2006; Yuan and Liu, 2008). The general strategy used in these studies was to identify DNA sequences which have strongly-positioned nucleosomes in vivo and then use these sequences as a training set to compute common DNA sequence patterns in nucleosomal DNA segments. The identified nucleosomal DNA sequence motifs were then used to predict nucleosome positions elsewhere in the genome and the predictions were compared to available nucleosome position maps.
Ioshikhes et al. (Ioshikhes et al., 1996; Ioshikhes et al., 2006) used a training set of 200 sequences from budding yeast nucleosomes. They obtained a nucleosome positioning sequence (NPS) of AA and TT dinucleotides occurring at 10 bp frequencies, mirroring each other with a gradient from the 5′ to the 3′ end of the nucleosome for AA and (of course) from 3′ to 5′ for TT dinucleotides. They then searched for correlations to this NPS profile across the yeast genome (Ioshikhes et al., 2006). NPS distribution in TATA-less genes is fairly uniform, showing the highest correlation with +1 and −1 nucleosomes and little or no correlation beyond the +3 and −2 nucleosomes. In vivo nucleosome-free regions had the strongest anticorrelation with NPS occurrence. On the other hand, TATA-containing promoters clustered into three groups distinguished by the position of the NPS/antiNPS/NPS motif relative to TSS. SAGA-regulated genes tend to have the TATA box within a predicted nucleosome, suggesting that nucleosome repositioning is likely to play a role in the regulation of these genes (see below).
Segal et al. (Segal et al., 2006) used a different training set from Ioshikhes et al., starting with 199 well-positioned S. cerevisiae nucleosomes. Here too, dinucleotide distribution analysis revealed a 10 bp periodicity of AA/TT/TA dinucleotides, with an abundance of AA/TT/TAs found towards the nucleosome edges, away from the nucleosome’s dyad axis. Similar periodicity was also observed in 177 natural chicken nucleosomes and in sequences isolated from nucleosomes reconstituted from randomly-synthesized DNA in vitro. A probabilistic model incorporating dinucleotide frequencies was generated, and used to scan the yeast genome. Consistent with the findings of Ioshikhes et al, the authors predicted that the +1 nucleosome was more likely than other nucleosomes to be positioned by pronucleosomal sequence cues. However, unlike other models, this model did not predict any contribution of nucleosome-excluding sequences to NFR formation. Curiously, AA/TT/TA frequency was the highest 10 and 20bp from the dyad in synthetic nucleosomes, in contrast to natural nucleosomes where the highest frequency was at the ends of the nucleosome (see, for example (Mavrich et al., 2008a)). The discrepancy between in vitro and in vivo nucleosomes suggests that histones in vivo do not “choose” the most favorable sequence, i.e. in vivo nucleosomes do not always occupy the most stable locations.
Overall, Segal et al. reported that their predictions of nucleosome positions, which included nucleosomes predicted from pronucleosomal sequences as well as nucleosomes positioned by in silico packing the remaining genome with nucleosomes, were accurate to within 35 bp for ~45% of in vivo nucleosomes. It is important to be aware, however, that random predictions capture >30% of nucleosome positions within this distance. Thus, it is much more accurate to say that “pronucleosomal” sequences account for significantly less than 15% (less because many in vivo positions captured by the model were nucleosomes correctly positioned based on their proximity to a sequence-directed nucleosome) of nucleosome positions observed in vivo.
In contrast to dinucleotide frequency analysis from the two studies above, Peckham et al.(Peckham et al., 2007) looked for k-mer distributions (k=1 to 6) in 1000 highest scoring and 1000 lowest scoring nucleosomes from the Yuan et al. (Yuan et al., 2005) dataset. They found that AT or GC rich k-mers turned out to be in nucleosome inhibiting or nucleosome favoring sequences, respectively. However, only a subset of nucleosome positions could be predicted using the k-mer distribution score. By the authors estimate, only 22%–25% nucleosome positions were due to a DNA sequence signal. Finally, Yuan and Liu (Yuan and Liu, 2008) computed the occurrence of periodic dinucleotide signals within nucleosomal and linker sequences and used the obtained scores to predict nucleosome positions. Peckham et al. and Yuan and Liu ‘s methods for scoring nucleosomal sequences, while different, had a similar prediction success rate and were both better than Segal et al. and Ioshikhes et al, probably because Peckham and Yuan included linker sequences in their analysis. Consistent with this, a more recent model from Segal and colleagues (Field et al., 2008) includes antinucleosomal sequences, and performs much better over nucleosome-depleted regions (see below).
Even though none of these methods could globally predict nucleosome positions, two common themes are apparent: (1) Nucleosomes clearly have some sequence preference in vitro. In vivo however, DNA sequence is probably the dominant positioning element for only a small subset of nucleosomes in the genome; most commonly the +1 and −1 nucleosomes around TSSs; (2) AT-rich sequences are good predictors of nucleosome depleted regions, which are particularly enriched for homopolymeric runs of A or T (Field et al., 2008; Yuan et al., 2005). Most of the methods were better at predicting nucleosome-depleted regions than at predicting the location of nucleosomes, suggesting that DNA sequence is most commonly used to exclude nucleosomes from specific regions (such as promoters) rather than position nucleosomes in nucleosome rich regions.
Recently, the idea that genomic sequence directs in vivo chromatin structure has been explicitly tested genome-wide in yeast via in vitro reconstitution of genomic DNA into histone octamers. After reconstitution of nucleosomes on purified yeast genomic DNA, nucleosomes were isolated and characterized by deep sequencing, and this in vitro nucleosomal map was compared to the in vivo map (Kaplan et al., 2008). The global correlation between the two maps was 0.74, indicating that much of the nucleosomal organization in the yeast genome is indeed due to cis factors. In vivo and in vitro 3′ NFR profiles are practically superimposable, but in vivo 5′ NFRs show a higher degree of nucleosome depletion than in vitro ones, suggesting a further role for proteins in promoter nucleosome depletion. Motifs were found which differed in their in vitro and in vivo occupancy levels, including motifs for Abf1, Reb1, and Rap1, which are proteins whose binding drives nucleosome eviction from a subset of 5′ NFRs (see TRANS determinants, below).
While the correlation between in vitro and in vivo maps is quite high, the majority of this correspondence appears to be due to nucleosome exclusion in vitro at 5′ and 3′ ends of genes caused by antinucleosomal poly(dA/dT) sequences – an important analysis of pronucleosomal contributions would be to calculate the correlation coefficient between in vitro and in vivo maps only over gene bodies, discounting the dominant role for poly(dA/dT) in these maps. Indeed, strong evidence against a dominant role for pronucleosomal sequences comes from the obervation that TSS-aligned averages of chromatin profiles in vivo reveal a strongly positioned +1 nucleosome downstream of the NFR (Field et al., 2008; Kaplan et al., 2008; Mavrich et al., 2008a; Mavrich et al., 2008b; Yuan et al., 2005), whereas the corresponding in vitro average demonstrates a strong NFR but no positioned +1 nucleosome (Kaplan et al., Figure 4a). Thus, while there is clearly some statistical enrichment of intrinsically-bendable DNA that correlates with in vivo nucleosome positions, in vivo this appears to play little role in translational positioning of nucleosomes. Instead, it has been suggested (Jiang and Pugh, 2009; Mavrich et al., 2008a) that the dinucleotide periodicity detected in various computational studies contributes to rotational positioning of nucleosomes, and that instead of sequence, trans factors play the major role in positioning the center of the nucleosome to within ~5 bp. The direction of intrinsic curvature would then dictate the precise (1 bp) nucleosomal position and corresponding major groove helix exposure.
Sequence analysis of chromatin maps from other organisms return broadly similar results in terms of dinucleotide periodicity signatures, but suggest that programming nucleosome-free regions via nucleosome exclusion likely only occurs in a subset of organisms. For example, analysis of 4mers depleted from nucleosomes isolated from C. elegans revealed AAAA, among other 4mers (Valouev et al., 2008). Indeed, analysis of in vivo C. elegans data using in vitro reconstitution data from yeast genomic DNA showed a correlation of 0.6 between 5-mer nucleosome occupancy levels in vitro and nucleosome occupancy in vivo (Kaplan et al., 2008), and, as noted above, this correlation is driven largely by polyA-dependent nucleosome exclusion. Conversely, a variety of nucleosome-occupied and nucleosome-depleted sequence motifs were described by Pugh and colleagues in D. melanogaster (Mavrich et al., 2008b), but while many of the nucleosome-depleted motifs contained runs of A (such as the Hunchback motif), polyA per se was not identified. In humans, nucleosome-depleted regions occur upstream of transcribed genes (Ozsolak et al., 2007; Schones et al., 2008), but are uncommon in uninduced genes, there they instead correlate with the presence of transcriptional machinery at the promoter. These data suggest that constituitive, or “programmed”, nucleosome-free regions may play less of a role in mammalian chromatin than they do in “lower” organisms. Indeed, analysis of the presence or absence of unfavorable (defined from yeast data) 5mer sequences such as AAAAA in nucleosomes sequenced from many organisms indicates a strong role for antinucleosomal sequences in yeast and worms, but demonstrates that these sequences play a significantly less important role in chicken, fly, and human chromatin (see (Field et al., 2008), Figure 5, column 2).
Altogether, we conclude that nucleosome exclusion by poly(dA/dT) sequences at promoters and transcription termination sites acts as a major force shaping the chromatin landscape in yeast and worms. Beyond nucleosome exclusion, there is some role for intrinsically-bendable sequences with 10 bp AA/AT/TT periodicity in positioning nucleosomes (although in mammals and flies GC/CG/CC/GG periodicity appears to be better-correlated with in vivo nucleosome positioning (Kogan et al., 2006; Mavrich et al., 2008b)). For the best-studied case in yeast, we believe the results favor the interpretation that the observed dinucleotide periodicities operate more at the level of establishing rotational positioning rather than translational positioning. Yet the majority of nucleosomes in vivo in yeast are well-positioned, raising the question of what positions the majority of nucleosomes not positioned by tight-binding bendable sequences.
Early analysis of delocalized, or “fuzzy” nucleosomes, revealed that these are enriched at locations distant from promoters (Yuan et al., 2005), and in fact variability in nucleosome positioning increases with increasing distance from NFRs (Mavrich et al., 2008a). These findings are consistent with the barrier model for statistical positioning of nucleosomes, first proposed by Kornberg and Stryer (Kornberg and Stryer, 1988). According to this model, barriers located along the chromosome prevent nucleosome binding, and nucleosomes are packed between these barriers at some average spacing which varies between organisms and between cell types (Van Holde, 1989). Since there are only so many ways five nucleosomes can be placed in 850 base pairs of genome (for instance), the five nucleosomes will appear relatively well-positioned despite no strong sequence preferences for any of the 850 bp. Another implication of this idea is that nucleosomes upstream and downstream of the barrier become less and less well positioned the further away from the barrier they get, which is indeed observed in vivo.
Barriers could be nucleosome-excluding sequences like AT-rich NFRs, strongly sequence-positioned nucleosomes, or DNA binding proteins (such as transcription factors) bound to specific sequences. The barrier model predicts that in genomes with short distances between barriers, such as yeast, only a subset of nucleosome positions (or barrier regions) need to be encoded in the DNA sequence, and nucleosomes surrounding these “anchor” locations will appear well-localized due to statistical positioning. This way, even though most nucleosomes in the genome are well positioned, only a few of those positions can be predicted from DNA sequence analysis, as seen in the plethora of studies on this topic described above. Consistent with these ideas, ~60% of yeast nucleosomes are well-positioned. Conversely, many fewer nucleosomes are well-positioned in human cells (Schones et al., 2008), where the massive size of genes results in much more space between the potential barriers located at promoters, enhancers, insulators, and other regulatory regions. As in yeast, well-positioned nucleosomes occur near regulatory elements in human cells (Fu et al., 2008; Schones et al., 2008), and positioning decays with increasing distance from these putative barriers (Fu et al., 2008). Despite this mass of consistent experimental information, in silico models which expressly incorporate packing (Field et al., 2008; Segal et al., 2006) still do not correctly predict the bulk of nucleosome positions (Kaplan et al., 2008), suggesting either that packing models need to be refined, or conversely that trans positioning factors dominate global positioning. As an interesting aside, since repeat length varies between organisms and even between cell types (and is affected by chromatin assembly factors), it is easy to see how the same DNA sequence could be differently packaged in different cell types based on its distance from a barrier and the local repeat length in the cell type in question.
In summary, it is quite clear that establishment of nucleosome-free regions by stiff, antinucleosomal polyA stretches is a major driver of chromatin architecture in yeast and probably worms. Nucleosomes positioned by pro-nucleosomal, intrinsically-flexible sequences also occur in eukaryotic genomes, and play some role in positioning a small subset of nucleosomes. Finally, genome-wide mapping studies are quite consistent with the predictions of statistical positioning, though definitive tests of this idea have yet to be carried out.
TRANS determinants of nucleosome positioning
The modest success of sequence-driven nucleosome positioning algorithms points to the involvement of trans factors in the positioning of a significant fraction of nucleosomes. A clear example of this is the observation that in vitro reconstitution of the PHO5 promoter using only histones and DNA failed to recapitulate the in vivo nucleosome positioning, whereas an ATP-dependent activity in yeast extract was required to recreate the in vivo chromatin state, demonstrating a key role for cellular factors in the in vivo positioning of nucleosomes on this template (Korber et al., 2004). Broadly, three major classes of trans factors have been implicated in nucleosome positioning/occupancy: (1) transcription factors (Shim et al., 1998; Taylor et al., 1991; Varga-Weisz and Becker, 1995; Vettese-Dadey et al., 1994), especially the abundant multifunctional regulators known as “general regulatory factors” (GRFs), such as Abf1 and Rap1 in S.cerevisiae; (Yarragudi et al., 2004), (2) chromatin remodelers (for review see (Cairns, 2005)) and (3) RNA polymerase (Field et al., 2008; Mavrich et al., 2008b; Schones et al., 2008; Yuan et al., 2005).
In vitro reconstitution of the yeast genome into nucleosomes revealed that while most transcription factor binding sites are intrinsically depleted of nucleosomes, several motifs including sites for Reb1 and Abf1 are more nucleosome depleted in vivo than in vitro (Kaplan et al., 2008). These factors are therefore implicated in the formation of NFRs at their binding sites in vivo. Indeed, when temperature sensitive Abf1 or Reb1 mutants are shifted to the non-permissive temperature nucleosome occupancy increases at the NFRS of promoters with binding sites for Abf1 and Reb1 (Badis et al., 2008; Hartley and Madhani, 2009). The mechanism of Abf1- and Reb1-mediated nucleosome depletion is not clear. The Abf1 protein acts as a barrier that determines nucleosome positions upstream and downstream of its binding site at the RPS28A promoter, but this can occur in the absence of NFR formation – the T-rich region downstream of the Abf1 binding site is the region necessary for forming the NFR at this promoter (Lascaris et al., 2000). On the other hand, Abf1 binding can drive the removal of a well positioned nucleosome in an episomal artificial substrate in vitro (Yarragudi et al., 2004). Since Abf1 does not have an ATPase domain, it is unlikely that it can actively remove or slide nucleosomes away from its binding site. Instead, it may take advantage of some form of nucleosome unwrapping to expose its site; either turnover (Ahmad and Henikoff, 2002a; Ahmad and Henikoff, 2002b; Dion et al., 2007; Mito et al., 2007) or nucleosome unwrapping or “breathing” (Anderson et al., 2002; Anderson and Widom, 2000; Anderson and Widom, 2001; Poirier et al., 2008) are appealing candidates. The distinction between Abf1 and most other transcription factors might then lie in its relatively high abundance (Choi and Kim, 2008; Newman et al., 2006), or perhaps a high affinity for (or a slow off rate from) its binding sites. Other abundant transcription factors that have been implicated in chromatin remodeling, such as Rap1 and Reb1, may use a similar mechanism to deplete promoters of nucleosomes and thereby facilitate the binding of other transcription factors required for gene activation (Pal et al., 2004; Yarragudi et al., 2004). Alternatively, it was recently shown that NFRs where nucleosome occupancy increases in conditional mutants of REB1 or ABF1 are a subset of the NFRs where nucleosome gain is seen in conditional mutants of the RSC ATP-dependent chromatin remodeler (Hartley and Madhani, 2009), suggesting perhaps that Reb1 and Abf1 act to evict nucleosomes in vivo via recruitment of the RSC complex.
Unlike GRFs, chromatin remodelers have an ATPase domain and use the energy of ATP hydrolysis to slide nucleosomes laterally (Fazzio and Tsukiyama, 2003; Lomvardas and Thanos, 2001) or eject them from DNA (Boeger et al., 2004; Cairns, 2005), among other activities. There are several classes of chromatin remodelers, including SWI/SNF (BAF), imitation switch (ISWI), INO80, SWR1 and Mi-2/CHD. In one fascinating in vitro study, chromatin remodelers moved nucleosomes to one of four available positions on the template, but the specific location chosen was different for every different type of remodeler (Rippe et al., 2007). One can therefore imagine that only a subset of potentially nucleosome-favoring sequences in the genome will be occupied by nucleosomes at any given time, and the distribution of nucleosome occupied sites will depend on the local action of chromatin remodelers. Any given promoter could consequently adopt, through the action of different chromatin remodelers, several nucleosomal architectures that would expose or occlude different transcription factor biding sites and affect gene expression in different ways.
RSC is a yeast chromatin remodeling complex that belongs to the SWI/SNF class of remodelers and is involved in transcriptional regulation of a wide variety of genes. RSC binds to ~700 targets in the yeast genome, predominantly over Pol III genes (Damelin et al., 2002; Ng et al., 2002) and over a subset of Pol II promoters, many of which carry a specific sequence motif for the Rsc3 subunit (Badis et al., 2008; Damelin et al., 2002; Ng et al., 2002). In conditional mutants of the RSC subunit Sth1, nucleosome occupancy increases over Pol III genes and transcription ceases (Parnell et al., 2008). As for Pol II genes, RSC (like other chromatin remodelers) tends to regulate the subset of yeast genes with TATA boxes, as the common occurrence of the TATA box in the −1 nucleosome necessitates regulation by chromatin remodelers for TATA exposure (Basehoar et al., 2004). Nucleosome positioning changes upon RSC depletion at Pol II promoters are more subtle than those at Pol III promoters, but RSC-occupied promoters and NFRs do gain nucleosome occupancy upon RSC loss more than do non-RSC promoters (Badis et al., 2008; Parnell et al., 2008). The mechanism for the increase in nucleosome occupancy seems to vary between promoters and consists of a combination of nucleosome sliding as well as binding by new nucleosomes.
Crucially, some chromatin remodelers function by moving nucleosomes from their “preferred” sequence to a less favorable one. Isw2 is a yeast chromatin remodeler whose major regulatory role is as a repressor. Elegant work from the Tsukiyama lab showed that at the POT1 promoter Isw2 functions to move a nucleosome from its sequence-directed site to a less-favored site towards the NFR (Whitehouse and Tsukiyama, 2006). This single-gene study was then extended to the whole genome, where mapping of Isw2 showed it associated with tRNA genes as well as ~20% of RNA Pol II genes (Whitehouse et al., 2007). Comparison of genome-wide nucleosome positioning maps between wild type and iswΔ mutant cells revealed that ~35% of Isw2-bound targets (~400 genes) were subject to detectable Isw2-mediated chromatin remodeling. +1 nucleosomes were shifted up to 70bp (15bp average) away from the NFR region in mutant cells, suggesting that in wild-type cells Isw2 inhibits transcription by positioning nucleosomes over the TSS and the NFR, where most transcription factor binding sites are located. The same trend was also observed at the 3′ end of genes, with nucleosomes shifting away from the intergenic region towards the 3′ end of target genes in iswΔ cells. This often occurred at genes oriented tandemly (as opposed to convergently), suggesting a potential role for this repositioning in antisense transcriptional control. Indeed, surprisingly, Isw2-mediated repositioning of nucleosomes turned out to repress antisense noncoding transcription by positioning nucleosomes over cryptic transcription start sites in these intergenic regions.
We have dealt so far with trans factors that operate upstream of transcriptional by modulating nucleosome positioning and occupancy. However, as mentioned above, the very process of transcription by RNA polymerases is a major force for shaping chromatin structure (Mavrich et al., 2008b; Parnell et al., 2008; Schones et al., 2008). Generally speaking, NFR width correlates with transcription level, and this appears largely to be a result of eviction of the −1 nucleosome at actively-transcribed genes, while nucleosomes over coding regions are more delocalized at higher transcription rates. In the few studies where nucleosomes have been mapped in an organism before and after some stimulus that changes transcription is applied, it has been observed that changes in nucleosome occupancy predominate over changes in nucleosome positions (Schones et al., 2008; Shivaswamy et al., 2008).
In multicellular organisms, different cell types are characterized by widely varying transcriptional activity, which is reflected in varied nucleosome positioning profiles. Perhaps this is most clearly observed in a heroic study from Zhao and colleagues, in which nucleosome positions were mapped by deep sequencing in CD4+ T cells before and after stimulation with anti-CD3 and anti CD28 antibodies (Schones et al., 2008). The +1 nucleosome of expressed genes with elongating Pol II was located at +40bp downstream of the TSS, whereas genes with stalled Pol II had their +1 nucleosome +10bp downstream of TSS. Curiously, inactive genes with no Pol II did not have well-positioned nucleosomes, although this could potentially be an artifact of insolubility, during nucleosome isolation procedures, of nucleosomes associated with repressed genes in mammals. Examination of the hundreds of genes that change expression during T cell stimulation revealed that the major change in chromatin structure at the higher expression level was, oddly, an increase in occupancy of the +1 nucleosome. We eagerly anticipate reconciliation of this result with the decreased nucleosome occupancy observed over highly-transcribed genes in yeast (Shivaswamy et al., 2008).
Nucleosome reorganization in response to stimuli is not just restricted to promoter regions. For example, the entire Drosophila Hsp70 locus displays a rapid loss of nucleosomes upon heat shock that is, surprisingly, independent of Hsp70 transcription (Petesch and Lis, 2008). The region of nucleosome depletion spans several kilobases, and is bounded by scs and scs’ insulator elements. This initial extensive nucleosome loss is necessary for the full activation of Hsp70 and is dependent on transcription factors HSF and GAF, and Poly(ADP)Ribose Polymerase (PARP), which interacts with DNA and nucleosomes (Petesch and Lis, 2008).
While to date most of what we know about RNA polymerase’s role in chromatin structure in vivo comes from studies where the transcriptional output of a cell is reprogrammed by some stimulus, it is important to note that observation of changed chromatin secondary to changed transcription does not necessarily mean that RNA polymerase itself (as opposed to remodelers, etc.) is responsible for the changes in chromatin structure. We anticipate that future studies into in vivo chromatin structure will start to untangle the contribution of RNA polymerase per se from the contributions of the many polymerase-associated factors.
Nucleosome positioning and gene regulation
Clearly, a number of factors, ranging from DNA sequence to protein complexes, affect nucleosome packaging across the genome. Of course, in order to understand chromatin’s role in biology we must also ask the converse question: how does nucleosome positioning affect cellular processes such as transcription?
Nucleosomes have classically been thought to prevent transcription factor binding to motifs located within the nucleosome, and for most transcription factors this appears to be true. In vivo, most functional transcription factor binding motifs are in nucleosome-free regions (Yuan et al., 2005), and motifs that occur in the −1 nucleosome appear to be enriched in locations where the major groove faces away from the octamer (Albert et al., 2007). However, exceptions to these broad principles tend to have interesting consequences for gene regulation.
First, as described above, some transcription factors are capable of remodeling nucleosomes in the absence of any additional factors. Elegant in vitro studies from the Widom laboratory show that the DNA located near the entry/exit point of the nucleosome transiently dissociates from the octamer, exposing any underlying binding sites (Anderson and Widom, 2000). Furthermore, binding of a transcription factor to a site near the edge of the nucleosome was also shown to enhance accessibility of other sites towards the center of the nucleosome (Polach and Widom, 1996; Vashee et al., 1998). This is particularly interesting, as nucleosome occupancy over two motifs can therefore make the binding of one factor dependent on the binding of the other even in the absence of any direct interaction between the factors on naked DNA. A recent study in human cells bears on this idea – here, a subset of NF-κB-dependent promoters were shown to be regulated normally by the subunit p65 even without its activation domain (van Essen et al., 2009). Instead of directly activating the gene, the p65 DNA-binding domain was required for other transcription factors to gain access to the promoter, thus creating a promoter that was NF-κB-dependent despite no direct role for the activation domain. While the mechanism for this was not shown to involve nucleosome eviction or unwrapping, and could in any case be explained by a recruited remodeling complex, the cooperativity inherent to having two TF motifs within the same nucleosome provides an appealing hypothesis for the underlying mechanism in this case.
Another elegant case of nucleosome positioning affecting the input function for a gene comes from the human β-interferon promoter. Here, a nucleosome positioned over the TATA box slides downstream upon activation of three different transcription factors – activation of any single factor is insufficient to activate this gene, as each of the three plays a role in recruiting a different factor required for nucleosome movement (Lomvardas and Thanos, 2002). Thanos and colleagues created a modified promoter with a tight-binding pronucleosomal sequence at the position where the nucleosome normally slides upon activation. This “slid” promoter, like a naked promoter, was responsive to any one of the three relevant transcription factors’ input. Thus, in this case the positioning of the nucleosome on the promoter changes the regulatory logic of promoter from an OR gate to an AND gate for three factors (Lomvardas and Thanos, 2002).
The general idea that transcription factors and nucleosomes compete for binding to the same DNA has global implications for regulation of gene activity. As mentioned above, in yeast and flies TATA and non-TATA genes are packaged differently, with promoters of TATA-containing genes being generally more occupied by nucleosomes. TATA-containing genes in yeast are enriched for a variety of features relative to non-TATA genes – 1) they are more likely to be sensitive to mutation of chromatin regulators (Choi and Kim, 2008; Tirosh and Barkai, 2008b), 2) they are generally stress-responsive rather than “housekeeping” (Basehoar et al., 2004), 3) they display increased transcriptional plasticity and their expression patterns diverge more rapidly during evolution (Landry et al., 2007; Tirosh et al., 2006), 4) they contain more transcription factor binding motifs (Landry et al., 2007; Tirosh et al., 2006), and 5) their expression is more variable from cell to cell within a population (Newman et al., 2006) due to transcriptional bursts rather than the continuous transcription observed in housekeeping genes (Bar-Even et al., 2006; Zenklusen et al., 2008). Several of these features may result from the increased nucleosome occupancy observed at these promoters. Most clearly, occlusion of the TATA box by a nucleosome is thought to be responsible for the requirement that many of these genes show for chromatin remodeling activity. More subtly, the presence of a nucleosome over transcription factor binding sites is proposed to result in competition between the two types of proteins, with transient opening or dissociation of nucleosomes (Anderson and Widom, 2000; Dion et al., 2007) at a promoter allowing binding of transcription factors and bursts of transcription (Choi and Kim, 2009; Field et al., 2008; Tirosh and Barkai, 2008b). The rapid off rate for most transcription factors (Hager et al., 2006; Voss and Hager, 2008) would then allow nucleosome reassembly, introducing a delay between transcription bursts. Consistent with this idea, studies of cell-to-cell variability at the PHO5 promoter implicate nucleosome dynamics as a major contributor to variability (Boeger et al., 2008; Raser and O’Shea, 2004). It is important to note here, however, that TATA boxes do not themselves cause increased promoter nucleosome occupancy. While it is true that promoters with a nucleosome immediately upstream of the TSS possess the noise characteristics described above, only half of such promoters contain a TATA box (Tirosh and Barkai, 2008b). Therefore, TATA box and increased promoter nucleosome occupancy strongly tend to co-occur, but this is not absolute, and it will be interesting to understand the constraints that led to the evolutionary co-occurrence of these two features.
Of course, not all nucleosome occlusion is subject to transcription factor invasion, as binding sites buried deep in a nucleosome are seldom exposed by breathing. Under these circumstances, binding sites found in nucleosomes will only be exposed upon nucleosome eviction. A very interesting case of this is seen at the PHO5 promoter, where multiple binding sites for the transcription factor Pho4 occur in both nucleosomal and linker locations (Venter et al., 1994). An elegant study from the O’Shea group showed that the strength of Pho4 binding sites in the linker region set the threshold of phosphate starvation (and Pho4 levels) required for activation of the promoter. On the other hand, the total number and strength of Pho4 binding sites, including those occluded by nucleosomes, contributed to the overall level of PHO5 transcription once the threshold for activation was reached (Lam et al., 2008). In this case, exposure of buried binding sites is not thought to be a direct consequence of transcription factor invasion at the nucleosome edges, but rather is secondary to recruitment of an ATP-dependent remodeling factor to evict the nucleosomes in question. But the interesting point in this case is that even cryptic nucleosome-occluded binding sites can nonetheless contribute to the regulation of the downstream gene.
Lest we get carried away with that idea that nucleosomes simply serve to conditionally hide factor binding sites, we also note that nucleosomes are sometimes believed to activate transcription, as well. Perhaps best-studied is the human U6 snRNA promoter (Stunkel et al., 1997), where the presence of a nucleosome between two transcription factor binding sites for Oct1 and SNAPc brings the binding sites into physical proximity by wrapping up the intervening DNA. This facilitates a physical interaction between the two transcription factors which is necessary for transcription initiation (Zhao et al., 2001). Despite the observation that hundreds of genes are downregulated in yeast upon depletion of histones (Wyrick et al., 1999), suggesting a broader role for nucleosomes in gene activation, the mechanistic basis for gene activation by nucleosomes has been understudied to date.
Relationship between nucleosome positioning, gene expression variation, and DNA sequence evolution
Phenotypic diversity in related species arises from coding DNA sequence variation and gene expression divergence (ED). Curiously, DNA sequence variation in coding regions of genes does not correlate with gene expression divergence (Tirosh and Barkai, 2008a), suggesting that the structural and functional properties of proteins, and their expression patterns, evolve under different selective constraints. ED can result from promoter sequence divergence (cis effects) or variation in transcription regulators (trans effects). According to a linkage analysis study of segregants from a cross between two S. cerevisiae strains (Brem et al., 2002), promoter sequence divergence accounts for ~30% of the observed variability in gene expression (Choi and Kim, 2008). The remaining 70% are due to trans effects, most of which involve chromatin modifiers thanks to their highly pleiotriopic effects on gene expression (Choi and Kim, 2008; Lee et al., 2006).
While in the above example promoter divergence affects a minority of expression changes between two strains of the same species, the converse appears to be true between species (Tirosh et al., 2009). For example, analysis of the transcriptional response to mating pheromone in S.cerevisiae, S.paradoxus and S.mikatae revealed that most of the differential gene expression was correlated with the loss of transcription factor binding sites in orthologous gene promoters (Tirosh et al., 2008). However, there was still a significant number of genes that displayed gene expression variation, despite transcription factor binding site conservation. Interestingly, in these genes, it was the sequence at the sites flanking transcription factor binding motifs that had diverged in the three species, and this sequence divergence correlated with changes in predicted nucleosome occupancy. In other words, differences in flanking sequences have turned the region of transcription factor binding into a “pronucleosomal” sequence, thus increasing the likelihood of nucleosome binding and inhibition of TF binding at these promoters (Borneman et al., 2007). Gene expression variability in general (including stochastic variation in cell populations, variation in response to environmental cues and interspecies/interstrain variation) correlates with the presence of “pronucleosomal” sequences in the region upstream of the TSS (Choi and Kim, 2009). It seems therefore that there are two widespread ways for promoter sequence variation to affect gene expression divergence: (1) through changes in transcription factor binding motifs and (2) through changes in nucleosome favoring and/or nucleosome excluding sequences. Changes in nucleosome architecture at gene promoters either through trans changes in the activity of chromatin remodelers (Choi and Kim, 2008; Lee et al., 2006) or cis changes in nucleosome excluding/favoring sequences (Tirosh et al., 2008) are therefore clearly a significant factor in the evolution of gene expression profiles.
Nucleosomes could also affect the evolution of DNA sequence, as the DNA repair machinery appears to have preferential access to linker DNA over nucleosomal DNA (Shim et al., 2007; Suter et al., 1997). Indeed, recent studies of sequence evolution in multiple organisms have uncovered a fascinating role for nucleosomes in sequence variation. Specifically, the pattern of sequence variation, when superimposed on the pattern of nucleosomes in vivo, shows a correlation with nucleosome positioning in yeast (Warnecke et al., 2008; Washietl et al., 2008). Frequencies of sequence polymorphisms are highest near the dyad axis, and fall continuously towards the ends of the nucleosome, reaching a minimum in linker sequence. This effect was also observed in medaka fish (Sasaki et al., 2009), and the authors here further noted that short insertions and deletions were most common in linker DNA. Interestingly, a recent study of human polymorphism data identified skew in codon utilization for roughly 80 base pairs on either side of SNPs (Hodgkinson et al., 2009). While the authors did not suggest chromatin as a potential contributor to this pattern, it is interesting to note that 80 bp is roughly one-half nucleosome. Detailed nucleosome maps, such as those determined in human T cells (Schones et al., 2008), will need to be determined for germ cells in humans in order for this hypothesis to be directly tested.
What accounts for this sequence “shadow” of nucleosomes? Most likely, the limited access of nucleosomal DNA to DNA repair proteins is the cause of the increased mutation rate within nucleosomes relative to linkers. Another possibility is that mutation rate is the same in nucleosomal and linker DNA, but purifying selection operates to maintain nucleosome-disfavoring codons in linker regions. Evidence for the former is abundant, while a recent study suggested some evidence for the latter in yeast (Warnecke et al., 2008). Since nucleosomes can move or change occupancy when environmental conditions change, the nucleosomal shadow on genomic sequence evolution could conceivably be modulated by long-term growth in different conditions. Perhaps eventually this idea will be tested via genome sequencing of many members of the same species which occupy different ecological niches, or populations undergoing long term laboratory evolution in differing growth conditions.
Conclusion
The packaging of eukaryotic genomes into nucleosomes has profound impacts on every DNA-based process studied. The recent use of genomic technologies to study chromatin has allowed researchers to more deeply explore longstanding ideas about the relationship between sequence, chromatin structure, and regulation of cellular processes. DNA sequence plays a major role in establishing nucleosome-free regions in yeast, but sequence-based models have modest success in predicting the positions of nucleosomes outside of NFRs. A plethora of trans-acting factors also modulate nucleosome positioning and occupancy, and global analyses of discrepancies between in vitro and in vivo nucleosome maps are providing a rapid means of identifying these factors. Binding site occlusion by nucleosomes has a wide range of effects on the regulatory logic of promoters, ranging from changing the Boolean input functions for promoters, to effects on cell-to-cell variability, to tuning the signaling response thresholds of different genes. In addition to gene regulation, chromatin architecture is a significant contributor to phenotypic diversity through its effects on gene expression variation and DNA sequence evolution. Studies on many fronts in the chromatin field will continue to expand our understanding of transcriptional control, systems biology, and evolution.
Acknowledgments
OJR is supported in part by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund, and by grants from NIGMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad K, Henikoff S. Epigenetic consequences of nucleosome dynamics. Cell. 2002a;111:281–4. doi: 10.1016/s0092-8674(02)01081-4. [DOI] [PubMed] [Google Scholar]
- Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell. 2002b;9:1191–200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–6. doi: 10.1038/nature05632. [DOI] [PubMed] [Google Scholar]
- Anderson JD, Thastrom A, Widom J. Spontaneous access of proteins to buried nucleosomal DNA target sites occurs via a mechanism that is distinct from nucleosome translocation. Mol Cell Biol. 2002;22:7147–57. doi: 10.1128/MCB.22.20.7147-7157.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J Mol Biol. 2000;296:979–87. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- Anderson JD, Widom J. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol Cell Biol. 2001;21:3830–9. doi: 10.1128/MCB.21.11.3830-3839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmi C, Bocchinfuso G, De Santis P, Savino M, Scipioni A. Dual role of DNA intrinsic curvature and flexibility in determining nucleosome stability. J Mol Biol. 1999;286:1293–301. doi: 10.1006/jmbi.1998.2575. [DOI] [PubMed] [Google Scholar]
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, Gebbia M, Talukder S, Yang A, Mnaimneh S, Terterov D, Coburn D, Li Yeo A, Yeo ZX, Clarke ND, Lieb JD, Ansari AZ, Nislow C, Hughes TR. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell. 2008;32:878–87. doi: 10.1016/j.molcel.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Even A, Paulsson J, Maheshri N, Carmi M, O’Shea E, Pilpel Y, Barkai N. Noise in protein expression scales with natural protein abundance. Nat Genet. 2006;38:636–43. doi: 10.1038/ng1807. [DOI] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Kornberg RD. Nucleosome retention and the stochastic nature of promoter chromatin remodeling for transcription. Cell. 2008;133:716–26. doi: 10.1016/j.cell.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol Cell. 2004;14:667–73. doi: 10.1016/j.molcel.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Borneman AR, Gianoulis TA, Zhang ZD, Yu H, Rozowsky J, Seringhaus MR, Wang LY, Gerstein M, Snyder M. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–9. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–5. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Cairns BR. Chromatin remodeling complexes: strength in diversity, precision through specialization. Curr Opin Genet Dev. 2005;15:185–90. doi: 10.1016/j.gde.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Choi JK, Kim YJ. Epigenetic regulation and the variability of gene expression. Nat Genet. 2008;40:141–7. doi: 10.1038/ng.2007.58. [DOI] [PubMed] [Google Scholar]
- Choi JK, Kim YJ. Intrinsic variability of gene expression encoded in nucleosome positioning sequences. Nat Genet. 2009;41:498–503. doi: 10.1038/ng.319. [DOI] [PubMed] [Google Scholar]
- Damelin M, Simon I, Moy TI, Wilson B, Komili S, Tempst P, Roth FP, Young RA, Cairns BR, Silver PA. The genome-wide localization of Rsc9, a component of the RSC chromatin-remodeling complex, changes in response to stress. Mol Cell. 2002;9:563–73. doi: 10.1016/s1097-2765(02)00475-6. [DOI] [PubMed] [Google Scholar]
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315:1405–8. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- Durrin LK, Mann RK, Grunstein M. Nucleosome loss activates CUP1 and HIS3 promoters to fully induced levels in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:1621–9. doi: 10.1128/mcb.12.4.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Tsukiyama T. Chromatin remodeling in vivo: evidence for a nucleosome sliding mechanism. Mol Cell. 2003;12:1333–40. doi: 10.1016/s1097-2765(03)00436-2. [DOI] [PubMed] [Google Scholar]
- Field Y, Kaplan N, Fondufe-Mittendorf Y, Moore IK, Sharon E, Lubling Y, Widom J, Segal E. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sinha M, Peterson CL, Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CM, Almouzni G. When repair meets chromatin. First in series on chromatin dynamics. EMBO Rep. 2002;3:28–33. doi: 10.1093/embo-reports/kvf005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, Elbi C, Johnson TA, Voss T, Nagaich AK, Schiltz RL, Qiu Y, John S. Chromatin dynamics and the evolution of alternate promoter states. Chromosome Res. 2006;14:107–16. doi: 10.1007/s10577-006-1030-0. [DOI] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137:445–58. doi: 10.1016/j.cell.2009.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, Ladoukakis E, Eyre-Walker A. Cryptic variation in the human mutation rate. PLoS Biol. 2009;7:e1000027. doi: 10.1371/journal.pbio.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell. 2004;13:573–85. doi: 10.1016/s1097-2765(04)00087-5. [DOI] [PubMed] [Google Scholar]
- Ioshikhes I, Bolshoy A, Derenshteyn K, Borodovsky M, Trifonov EN. Nucleosome DNA sequence pattern revealed by multiple alignment of experimentally mapped sequences. J Mol Biol. 1996;262:129–39. doi: 10.1006/jmbi.1996.0503. [DOI] [PubMed] [Google Scholar]
- Ioshikhes IP, Albert I, Zanton SJ, Pugh BF. Nucleosome positions predicted through comparative genomics. Nat Genet. 2006;38:1210–5. doi: 10.1038/ng1878. [DOI] [PubMed] [Google Scholar]
- Iyer V, Struhl K. Poly(dA:dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995;14:2570–9. doi: 10.1002/j.1460-2075.1995.tb07255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–72. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ. Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin. Genome Res. 2006;16:1505–16. doi: 10.1101/gr.5560806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakaka RT, Thomas JO. Chromatin structure of transcriptionally competent and repressed genes. EMBO J. 1990;9:3997–4006. doi: 10.1002/j.1460-2075.1990.tb07621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Leproust EM, Hughes TR, Lieb JD, Widom J, Segal E. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2008 doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan SB, Kato M, Kiyama R, Trifonov EN. Sequence structure of human nucleosome DNA. J Biomol Struct Dyn. 2006;24:43–8. doi: 10.1080/07391102.2006.10507097. [DOI] [PubMed] [Google Scholar]
- Korber P, Luckenbach T, Blaschke D, Horz W. Evidence for histone eviction in trans upon induction of the yeast PHO5 promoter. Mol Cell Biol. 2004;24:10965–74. doi: 10.1128/MCB.24.24.10965-10974.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Stryer L. Statistical distributions of nucleosomes: nonrandom locations by a stochastic mechanism. Nucleic Acids Res. 1988;16:6677–90. doi: 10.1093/nar/16.14.6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel GR, Martinson HG. Nucleosomes will not form on double-stranded RNa or over poly(dA).poly(dT) tracts in recombinant DNA. Nucleic Acids Res. 1981;9:6869–88. doi: 10.1093/nar/9.24.6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FH, Steger DJ, O’Shea EK. Chromatin decouples promoter threshold from dynamic range. Nature. 2008;453:246–50. doi: 10.1038/nature06867.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007;317:118–21. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- Lascaris RF, Groot E, Hoen PB, Mager WH, Planta RJ. Different roles for abf1p and a T-rich promoter element in nucleosome organization of the yeast RPS28A gene. Nucleic Acids Res. 2000;28:1390–6. doi: 10.1093/nar/28.6.1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Pe’er D, Dudley AM, Church GM, Koller D. Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. Proc Natl Acad Sci U S A. 2006;103:14062–7. doi: 10.1073/pnas.0601852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Flanagan PM, Tschochner H, Kornberg RD. RNA polymerase II initiation factor interactions and transcription start site selection. Science. 1994;263:805–7. doi: 10.1126/science.8303296. [DOI] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D. Nucleosome sliding via TBP DNA binding in vivo. Cell. 2001;106:685–96. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell. 2002;110:261–71. doi: 10.1016/s0092-8674(02)00822-x. [DOI] [PubMed] [Google Scholar]
- Luger K, Hansen JC. Nucleosome and chromatin fiber dynamics. Curr Opin Struct Biol. 2005;15:188–96. doi: 10.1016/j.sbi.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008a;18:1073–83. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, Gilmour DS, Albert I, Pugh BF. Nucleosome organization in the Drosophila genome. Nature. 2008b;453:358–62. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y, Henikoff JG, Henikoff S. Histone replacement marks the boundaries of cis-regulatory domains. Science. 2007;315:1408–11. doi: 10.1126/science.1134004. [DOI] [PubMed] [Google Scholar]
- Nelson HC, Finch JT, Luisi BF, Klug A. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature. 1987;330:221–6. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–6. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Genome-wide location and regulated recruitment of the RSC nucleosome-remodeling complex. Genes Dev. 2002;16:806–19. doi: 10.1101/gad.978902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Song JS, Liu XS, Fisher DE. High-throughput mapping of the chromatin structure of human promoters. Nat Biotechnol. 2007;25:244–8. doi: 10.1038/nbt1279. [DOI] [PubMed] [Google Scholar]
- Pal S, Cantor AB, Johnson KD, Moran TB, Boyer ME, Orkin SH, Bresnick EH. Coregulator-dependent facilitation of chromatin occupancy by GATA-1. Proc Natl Acad Sci U S A. 2004;101:980–5. doi: 10.1073/pnas.0307612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell TJ, Huff JT, Cairns BR. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J. 2008;27:100–10. doi: 10.1038/sj.emboj.7601946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham HE, Thurman RE, Fu Y, Stamatoyannopoulos JA, Noble WS, Struhl K, Weng Z. Nucleosome positioning signals in genomic DNA. Genome Res. 2007;17:1170–7. doi: 10.1101/gr.6101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petesch SJ, Lis JT. Rapid, transcription-independent loss of nucleosomes over a large chromatin domain at Hsp70 loci. Cell. 2008;134:74–84. doi: 10.1016/j.cell.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J Mol Biol. 2008;379:772–86. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Widom J. A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol. 1996;258:800–12. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–4. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippe K, Schrader A, Riede P, Strohner R, Lehmann E, Langst G. DNA sequence- and conformation-directed positioning of nucleosomes by chromatin-remodeling complexes. Proc Natl Acad Sci U S A. 2007;104:15635–40. doi: 10.1073/pnas.0702430104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Roth SY. Unequal access: regulating V(D)J recombination through chromatin remodeling. Cell. 2000;103:699–702. doi: 10.1016/s0092-8674(00)00173-2. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Mello CC, Shimada A, Nakatani Y, Hashimoto S, Ogawa M, Matsushima K, Gu SG, Kasahara M, Ahsan B, Sasaki A, Saito T, Suzuki Y, Sugano S, Kohara Y, Takeda H, Fire A, Morishita S. Chromatin-associated periodicity in genetic variation downstream of transcriptional start sites. Science. 2009;323:401–4. doi: 10.1126/science.1163183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–98. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–8. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol. 2009;19:65–71. doi: 10.1016/j.sbi.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekinger EA, Moqtaderi Z, Struhl K. Intrinsic histone-DNA interactions and low nucleosome density are important for preferential accessibility of promoter regions in yeast. Mol Cell. 2005;18:735–48. doi: 10.1016/j.molcel.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Shim EY, Hong SJ, Oum JH, Yanez Y, Zhang Y, Lee SE. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol. 2007;27:1602–13. doi: 10.1128/MCB.01956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Woodcock C, Zaret KS. Nucleosome positioning by the winged helix transcription factor HNF3. Genes Dev. 1998;12:5–10. doi: 10.1101/gad.12.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, Iyer VR. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6:e65. doi: 10.1371/journal.pbio.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunkel W, Kober I, Seifart KH. A nucleosome positioned in the distal promoter region activates transcription of the human U6 gene. Mol Cell Biol. 1997;17:4397–405. doi: 10.1128/mcb.17.8.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Livingstone-Zatchej M, Thoma F. Chromatin structure modulates DNA repair by photolyase in vivo. EMBO J. 1997;16:2150–60. doi: 10.1093/emboj/16.8.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IC, Workman JL, Schuetz TJ, Kingston RE. Facilitated binding of GAL4 and heat shock factor to nucleosomal templates: differential function of DNA-binding domains. Genes Dev. 1991;5:1285–98. doi: 10.1101/gad.5.7.1285. [DOI] [PubMed] [Google Scholar]
- Thastrom A, Lowary PT, Widlund HR, Cao H, Kubista M, Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J Mol Biol. 1999;288:213–29. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Barkai N. Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 2008a;24:109–13. doi: 10.1016/j.tig.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res. 2008b;18:1084–91. doi: 10.1101/gr.076059.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science. 2009;324:659–62. doi: 10.1126/science.1169766. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Bezalel D, Kaganovich M, Barkai N. On the relation between promoter divergence and gene expression evolution. Mol Syst Biol. 2008;4:159. doi: 10.1038/msb4100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Carmi M, Barkai N. A genetic signature of interspecies variations in gene expression. Nat Genet. 2006;38:830–4. doi: 10.1038/ng1819. [DOI] [PubMed] [Google Scholar]
- Trifonov EN. Sequence-dependent deformational anisotropy of chromatin DNA. Nucleic Acids Res. 1980;8:4041–53. doi: 10.1093/nar/8.17.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Ichikawa J, Tonthat T, Stuart J, Ranade S, Peckham H, Zeng K, Malek JA, Costa G, McKernan K, Sidow A, Fire A, Johnson SM. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–63. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Essen D, Engist B, Natoli G, Saccani S. Two modes of transcriptional activation at native promoters by NF-kappaB p65. PLoS Biol. 2009;7:e73. doi: 10.1371/journal.pbio.1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Holde KE. Chromatin. Springer-Verlag; New York: 1989. [Google Scholar]
- Varga-Weisz PD, Becker PB. Transcription factor-mediated chromatin remodelling: mechanisms and models. FEBS Lett. 1995;369:118–21. doi: 10.1016/0014-5793(95)00549-o. [DOI] [PubMed] [Google Scholar]
- Vashee S, Melcher K, Ding WV, Johnston SA, Kodadek T. Evidence for two modes of cooperative DNA binding in vivo that do not involve direct protein-protein interactions. Curr Biol. 1998;8:452–8. doi: 10.1016/s0960-9822(98)70179-4. [DOI] [PubMed] [Google Scholar]
- Venter U, Svaren J, Schmitz J, Schmid A, Horz W. A nucleosome precludes binding of the transcription factor Pho4 in vivo to a critical target site in the PHO5 promoter. EMBO J. 1994;13:4848–55. doi: 10.1002/j.1460-2075.1994.tb06811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vettese-Dadey M, Walter P, Chen H, Juan LJ, Workman JL. Role of the histone amino termini in facilitated binding of a transcription factor, GAL4-AH, to nucleosome cores. Mol Cell Biol. 1994;14:970–81. doi: 10.1128/mcb.14.2.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Hager GL. Visualizing chromatin dynamics in intact cells. Biochim Biophys Acta. 2008;1783:2044–51. doi: 10.1016/j.bbamcr.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke T, Batada NN, Hurst LD. The impact of the nucleosome code on protein-coding sequence evolution in yeast. PLoS Genet. 2008;4:e1000250. doi: 10.1371/journal.pgen.1000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Machne R, Goldman N. Evolutionary footprints of nucleosome positions in yeast. Trends Genet. 2008;24:583–7. doi: 10.1016/j.tig.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. Chromatin remodelling at promoters suppresses antisense transcription. Nature. 2007;450:1031–5. doi: 10.1038/nature06391. [DOI] [PubMed] [Google Scholar]
- Whitehouse I, Tsukiyama T. Antagonistic forces that position nucleosomes in vivo. Nat Struct Mol Biol. 2006;13:633–40. doi: 10.1038/nsmb1111. [DOI] [PubMed] [Google Scholar]
- Wyrick JJ, Holstege FC, Jennings EG, Causton HC, Shore D, Grunstein M, Lander ES, Young RA. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–21. doi: 10.1038/46567. [DOI] [PubMed] [Google Scholar]
- Yarragudi A, Miyake T, Li R, Morse RH. Comparison of ABF1 and RAP1 in chromatin opening and transactivator potentiation in the budding yeast Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:9152–64. doi: 10.1128/MCB.24.20.9152-9164.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan GC, Liu JS. Genomic sequence is highly predictive of local nucleosome depletion. PLoS Comput Biol. 2008;4:e13. doi: 10.1371/journal.pcbi.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–30. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Zenklusen D, Larson DR, Singer RH. Single-RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol. 2008;15:1263–71. doi: 10.1038/nsmb.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Pendergrast PS, Hernandez N. A positioned nucleosome on the human U6 promoter allows recruitment of SNAPc by the Oct-1 POU domain. Mol Cell. 2001;7:539–49. doi: 10.1016/s1097-2765(01)00201-5. [DOI] [PubMed] [Google Scholar]