

Abstract
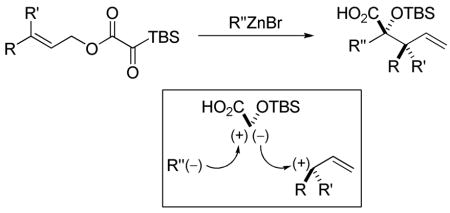
Organozinc, magnesium, and lithium nucleophiles initiate a Brook/Ireland-Claisen rearrangement sequence of allylic silyl glyoxylates resulting in formation of γ,δ-unsaturated α-silyloxy acids.
New methods to produce the glycolic acid moiety continue to be important in the creation of small molecule building blocks.1 γ,δ-Unsaturated glycolic acid derivatives represent a functional group-rich subset of substituted α-hydroxy acids with significant potential for further elaboration. The preparation of such compounds may be achieved via Ireland-Claisen rearrangement of allyl glycolate esters.2 While the standard Ireland-Claisen rearrangement is performed by sequential enolization and silylation of allylic esters, alternative approaches to the requisite silyl ketene acetal have been reported. Methods include 1,4-addition to enoates,3 silylene transfer followed by 6π-electrocyclization,4 or by Rh-catalyzed reduction of enoates.5 These methods allow for the generation of chiral products from achiral starting materials; however, a method that introduces multiple α-substituents in a single-pot process could potentially allow more rapid buildup of molecular complexity.
Silyl glyoxylates are reagents for the geminal linking of nucleophile/electrophile pairs at a glycolic acid junction.6 Since multicomponent couplings involving silyl glyoxylates are believed to proceed through a glycolate enolate intermediate, the use of an allylic ester might allow [3,3]-sigmatropic rearrangement to proceed in preference to intermolecular addition to an electrophile (Scheme 1).
Scheme 1.

Silyl Glyoxylate Reactivity
In the proposed reaction, addition of a nucleophile to the acyl silane would give a tetrahedral intermediate (Scheme 2). Proper alignment of the C–Si σ orbital with the adjacent C=O π* orbital in tetrahedral intermediate 2 would allow [1,2]-Brook rearrangement7 to proceed, forming glycolate enolate 3. At this point, the allylic ester enolate could undergo Ireland-Claisen rearrangement to afford a γ,δ-unsaturated acid.
Scheme 2.

Proposed Reaction
The Ireland-Claisen rearrangement proceeds via a well-understood transition state wherein the enolate geometry dictates the stereochemistry of the product.8 The E/Z geometry of the intermediate glycolate enolate has not been established in reactions of silyl glyoxylates; therefore, determination of enolate geometry would augment our understanding of silyl glyoxylate reactivity and provide mechanistic insight for future endeavors. Furthermore, this process would create two new C–C bonds, allowing access to densely functionalized glycolic acids. Herein, we describe experiments directed toward these ends.
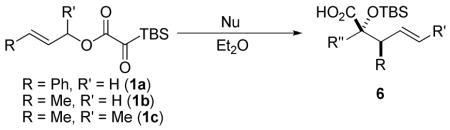
Preliminary work focused on defining a compatible nucleophilic component. Previously employed nucleophiles such as vinylmagnesium bromide and zinc acetylides were found to trigger oligomerization of the silyl glyoxylate. This result underscores the chemoselectivity displayed in previously reported three-component coupling reactions, where the nucleophile reacts selectively with the silyl glyoxylate, rather than the terminal electrophile, and the intermediate glycolate enolate reacts selectively with a terminal electrophile, rather than a second equivalent of silyl glyoxylate. The proposed Ireland-Claisen rearrangement requires a nucleophile capable of completely consuming silyl glyoxylate prior to oligomerization.
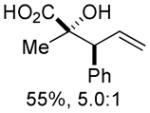
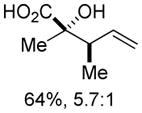
An assessment of other Grignard reagents revealed that MeMgBr had potential as an effective nucleophile as it cleanly added to cinnamyl silyl glyoxylate 1a at 0 °C. In this case, Brook rearrangement did not occur below room temperature. When the reaction was allowed to warm, only decomposition was observed. Silyl ketene acetal formation was then attempted by addition of MeMgBr at 0 °C followed by warming to room temperature and addition of TMSCl; however, this also resulted in decomposition. Given that no TMS incorporation had occurred, we turned to TMSOTf, which induced the desired Ireland-Claisen rearrangement and provided the γ,δ-unsaturated glycolic acid 7 in 55% yield (Table 1, entry 1). Silyl glyoxylates 1b and 1c reacted with similar efficiency (entries 2–3). The lithium enolate of tBuOAc also initiates the Brook/Ireland-Claisen sequence with excellent diastereoselectivity (Table 1, entry 4).
Table 1.
Sequential Brook/Ireland-Claisen Rearrangements of Silyl Glyoxylates
 | |||
|---|---|---|---|
| entry | silyl glyoxylate | Nu | yield, dr |
| 1 | 1a | MeMgBr/TMSOTf |
 7 7
|
| 2 | 1b | MeMgBr/TMSOTf |
 8 8
|
| 3 | 1c | MeMgBr/TMSOTf |
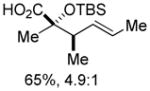 9 9
|
| 4 | 1a | 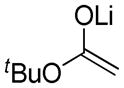 |
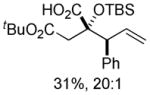 10 10
|
| 5 | 1a | ZnEt2 |
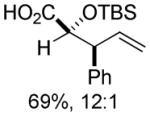 11 11
|
| 6 | 1a |
 12 12
|
|
| 7 | 1b |
 13 13
|
|
| 8 | 1c |
 14 14
|
|
| 9 | 1a | 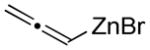 |
 15 15
|
| 10 | 1b | 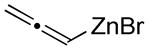 |
 16 16
|
| 11 | 1a | 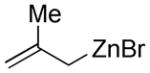 |
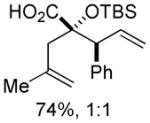 17 17
|
| 12 | 1b | 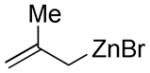 |
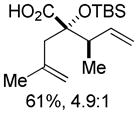 18 18
|
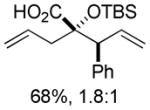
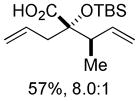
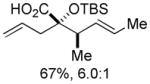
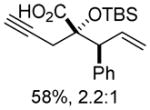
Organozinc nucleophiles were also found to be effective. ZnEt2 serves as an efficient hydride donor, triggering the sequential Brook and Ireland-Claisen rearrangements, to afford the glycolic acid in 69% yield with good diastereoselectivity (Table 1, entry 5). The reduction of α-ketoesters with ZnEt2 or EtMgBr has been previously reported; however, it is typically a minor byproduct to ethyl addition.9 Allyl zinc bromide and allenyl zinc bromide were useful triggers as well (Table 1, entries 6–10).
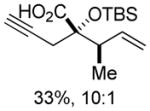
Substituted allylic zinc nucleophiles were also competent initiators. Methallyl zinc bromide provided the desired products with slightly diminished diastereoselectivity relative to allyl zinc bromide (Table 1, entries 11–12). The reaction was also tolerant of crotyl- and cinnamylzinc reagents; however, products were obtained as a complex mixture of diastereomers (not shown). Notably, none of the organozinc nucleophiles required silyl ketene acetal formation, as the zinc enolate underwent [3,3]-rearrangement spontaneously.
Extension to other organozinc bromides proved challenging. Benzylzinc bromide, propynylzinc bromide, and Reformatsky nucleophiles resulted in alkene dimer 19 (Scheme 3). This dimer was cleanly formed by addition of the glycolate enolate to a second equivalent of silyl glyoxylate. The derived alkoxide suffered Brook rearrangement and elimination. It appears that allyl- and allenylzinc bromide nucleophiles consumed the silyl glyoxylate more rapidly and therefore dimerization was not competitive.
Scheme 3.

Dimerization Limits Nucleophile Scope
Geranyl silyl glyoxylate 20 underwent the title transformation when treated with allenyl or allylic zinc bromides, providing glycolic acid derivatives in moderate diastereoselectivity (Table 2). These results demonstrate the Brook/Ireland-Claisen sequence may show promise for the construction adjacent chiral quaternary centers.
Table 2.
Generation of Contiguous Quaternary Centers
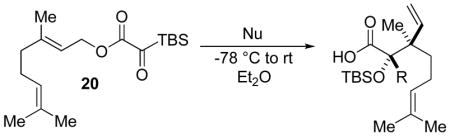 | |||
|---|---|---|---|
| entry | nucleophile | product (yield) | dr |
| 1 | 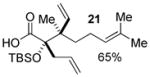 |
3.0:1 | |
| 2 | 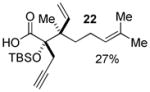 |
3.7:1 | |
| 3 | 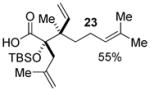 |
2.5:1 | |
Enolate geometry was inferred by examination of the relative stereochemistry of the γ,δ-unsaturated glycolic acid products. The products could be converted to cyclic products by iodolactonization or ring-closing metathesis, allowing for NOESY analysis.10 In each case, the major diastereomer was found to be a consistent with a (Z)-glycolate enolate.
The degree of diastereocontrol depended on the identity of both the nucleophile and the ester. While cinnamyl silyl glyoxylate 1a reacted with allylzinc bromide (Table 1, entry 6) and allenylzinc bromide (entry 9) with modest diastereocontrol, analogous reactions with crotyl silyl glyoxylate 1b resulted in substantially higher dr’s (entries 7,10). The formation of the chelated alkoxide 2 appears likely in light of the established tendencies for bidentate coordination in α-dicarbonyl electrophiles and the derived addition products.11 Stereoelectronic considerations would suggest subsequent formation of (Z)-enolate 3 arising from Brook rearrangement. The resulting silyl ether will dramatically weaken chelation,12 potentially permitting enolate equilibration (via the C–Zn tautomer 5) to compete with [3,3]-rearrangement from the (Z)-isomer. The rate of the sigmatropic rearrangement should be linked to steric demand at C1 and C6, where a less hindered substrate undergoes more rapid [3,3]-rearrangement;13 therefore, it is hypothesized that the more encumbered 1a undergoes enolate equilibration to a greater extent prior to sigmatropic rearrangement. The result is an erosion of the initial enolate geometric ratio and a corresponding decrease in product diastereoselectivity. This proposal is consistent with the experimental observations within the organozinc nucleophile series: the product diastereomer ratio increases as nucleophile bulk at C1 decreases (hydride > propargyl ≈ allyl > methallyl).
It is notable that MeMgBr initiates Ireland-Claisen rearrangement with diastereoselectivity independent of ester. This may be a result of silyl ketene acetal formation prior to sigmatropic rearrangement. Provided there is no enolate equilibration after TMSOTf trapping, the diastereoselectivity reflects the (Z)/(E) Mg-enolate ratio at the time of TMSOTf addition.
The high diastereoselectivity observed for the Li-enolate (Table 1, entry 4) can be explained if generation of the C–Li tautomer 5 is slow and sigmatropic rearrangement outcompetes enolate equilibration. This may be directly contrasted with a recent reaction between zinc enolates and silyl glyoxylates that do undergo facile enolate equilibration.6b
In summary, we have developed a one-pot Brook/Ireland-Claisen rearrangement sequence that generates γ,δ-unsaturated glycolic acids containing two new C–C bonds and two contiguous stereocenters. To our knowledge, this is the first report of a Brook rearrangement initiating a [3,3]-sigmatropic process. The reaction is tolerant of various reaction partners, but diastereoselectivity is linked to the identity of the metal cation and the ester. In diastereoselective cases the stereochemical outcome of the reaction is consistent with a (Z)-enolate intermediate proceeding through a chair-like transition state. The work suggests that diastereocontrol and enolate geometry in reactions of silyl glyoxylates can be tuned with the judicious selection of metal cation and that the rate of the “second stage” reaction can have an impact.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (National Institute of General Medical Sciences – GM084927) and Novartis. We thank a reviewer for a detailed analysis of the mechanism and some suggestions that were incorporated herein.
Footnotes
Supporting Information Available Experimental procedures and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ley SV, Sheppard TD, Myers RM, Chorghade MS. Bull Chem Soc Jpn. 2007;80:1451–1472. [Google Scholar]
- 2.(a) Ager DJ. Tetrahedron Lett. 1982;23:3419–3420. [Google Scholar]; (b) Bartlett PA, Tanzella DA, Barstow JF. J Org Chem. 1982;47:3941–3945. [Google Scholar]; (c) Burke SD, Fobare WF, Pacofsky GJ. J Org Chem. 1983;48:5221–5228. [Google Scholar]; (d) Sato T, Tanaka A, Tajima K, Fujisawa T. Chem Lett. 1987:1979–1980. [Google Scholar]; (e) Fujiwara K, Goto A, Sato D, Kawai H, Suzuki T. Tetrahedron Lett. 2005;46:3465–3468. [Google Scholar]; (f) Gould TJ, Balestra M, Wittman MD, Gary JA, Rossano LT, Kallmerten J. J Org Chem. 1987;52:3889–3901. [Google Scholar]
- 3.Chai Y, Hong SP, Lindsay HA, McFarland C, McIntosh MC. Tetrahedron. 2002;58:2905–2928. [Google Scholar]
- 4.(a) Calad SC, Woerpel KA. J Am Chem Soc. 2005;127:2046–2047. doi: 10.1021/ja042893r. [DOI] [PubMed] [Google Scholar]; (b) Howard BE, Woerpel KA. Org Lett. 2007;9:4651–4653. doi: 10.1021/ol702148x. [DOI] [PubMed] [Google Scholar]; (c) Howard BE, Woerpel KA. Tetrahedron. 2009;65:6447–6453. doi: 10.1016/j.tet.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller SP, Morken JP. Org Lett. 2002;4:2743–2745. doi: 10.1021/ol026273b. [DOI] [PubMed] [Google Scholar]
- 6.(a) Nicewicz DA, Johnson JS. J Am Chem Soc. 2005;127:6170–6171. doi: 10.1021/ja043884l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Greszler SN, Johnson JS. Angew Chem Int Ed. 2009;48:3689–3691. doi: 10.1002/anie.200900215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brook AG. Acc Chem Res. 1974;7:77–84. [Google Scholar]
- 8.(a) Ireland RE, Mueller RH, Willard AK. J Am Chem Soc. 1976;98:2868–2877. [Google Scholar]; (b) Hiersemann M, Nubbemeyer U, editors. The Claisen Rearrangement: Methods and Applications. Wiley-VCH: Weinheim, Germany; 2007. [Google Scholar]
- 9.(a) Hameury T, Guillemont J, Van Hijfte L, Bellosta V, Cossy J. Org Lett. 2009;11:2397. doi: 10.1021/ol900494g. [DOI] [PubMed] [Google Scholar]; (b) Fennie MW, DiMauro EF, O’Brien EM, Annamalai V, Kozlowski MC. Tetrahedron. 2005;61:6249–6265. [Google Scholar]
- 10.See the Supporting Information for experimental details and NOESY data.
- 11.(a) Evans DA, Burgey CS, Kozlowski MC, Tregay SW. J Am Chem Soc. 1999;121:686–699. [Google Scholar]; (b) Masuyama Y, Tsunoda T, Kurusu Y. Chem Lett. 1989:1647–1650. [Google Scholar]
- 12.Keck GE, Boden EP. Tetrahedron Lett. 1984;25:265–268. [Google Scholar]
- 13.(a) Hiersemann M, Abraham L. Org Lett. 2001;3:49–52. doi: 10.1021/ol006760w. [DOI] [PubMed] [Google Scholar]; (b) Wilcox CS, Babston RE. J Am Chem Soc. 1986;108:6636–6642. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.