

Abstract
Secondary neurodegeneration resulting from stroke is mediated by delayed proinflammatory signaling and immune cell activation. Although it remains unknown which cell surface markers signify a proinflammatory phenotype, increased isolectin binding occurs on CD11b-expressing immune cells within injured brain tissue. Several reports have confirmed the efficacy of human umbilical cord blood (HUCB) cell therapy in reducing ischemic injury in rat after middle cerebral artery occlusion (MCAO), and these effects were attributed in part to dampened neuroinflammation. The present study examined the time course of lectin binding to cells of microglia/macrophage lineage within 96 hrs after MCAO, and whether delayed HUCB cell treatment alters the migration and/or morphological characteristics of these cells throughout the period of infarct expansion. Isolectin binding was upregulated in response to injury, was maximal at 96 hrs, and colocalized with cells that expressed the putative proinflammatory markers MMP-9 and nitric oxide. Isolectin-tagged fluorescence was also significantly increased at 72 hrs and localized to greater numbers of amoeboid, CD11b-expressing cells relative to 51 hrs. Treatment with 1×106 HUCB cells significantly reduced total lectin binding at 72 hrs, as well as the total area occupied by lectin-tagged fluorescence at both 51 and 72 hrs, relative to vehicle-treated controls. This effect was accompanied by a shift in the morphology of CD11b-positive cells from amoeboid to ramified shape. These data indicate that HUCB cell therapy suppressed the recruitment of proinflammatory, isolectin-binding cells during the period of infarct expansion, thus offering a potential mechanism for the protective effects of HUCB cell therapy.
Keywords: Stroke, rat brain, microglia, macrophages, hypoxia/ischemia
Introduction
Ischemia produces an initial wave of neural injury through energy failure resulting from reduced oxygen and nutrients (Lipton 1999; Takano et al. 1996). However, neurodegeneration is enhanced by a second, delayed response involving immune cells and proinflammatory signals (Offner et al. 2006). This immune response contributes to delayed neural injury that occurs up to days after ischemia. While the brain is generally impervious to blood-borne immune cells, CNS infiltration occurs after ischemia as a result of blood brain barrier degradation (Jean et al. 1998; Matsuo et al. 1994). The inflammation elicited by the interaction of peripheral immune cells and resident microglia leads to enhanced neurodegeneration, exceeding that which results from the loss of blood flow.
Microglia are the endogenous macrophages of the brain and share many characteristics that are indistinguishable from peripheral macrophages (Streit et al. 1988). In response to insult, microglia alter their morphology from a resting, ramified form to an amoeboid cell that is associated with an activated state (Garden and Moller 2006; Streit et al. 2004). Several immune-associated markers are often used to define the state of activation, yet it is unclear which of these markers is most indicative of severe inflammation and injury after stroke. Although microglia secrete proinflammatory cytokines after insult (Allan and Rothwell 2001; Dirnagl et al. 1999), these cells have also been described as neuroprotective through their ability to release neurotrophins and other neurosupportive factors (Coull et al. 2005; da Cunha et al. 1997). Investigations using cultured microglia have provided some insight into mechanisms of proinflammatory signaling (Hall et al. 2008). Despite these data, in vivo studies are necessary to determine the complex cellular interactions in the brain after injury.
Monocytes/macrophages have been shown to infiltrate the stroke-damaged brain after 48 hours (Vendrame et al. 2005). Additionally, a recent report has shown that microglia comprise the majority of monocyte lineage cells within the injured brain, suggesting that peripheral macrophages may influence resident microglia (Tanaka et al. 2003). Thus, therapies targeting either the arrival of peripheral monocytes and/or their interactions with microglia may prove beneficial in the treatment of delayed neuronal death after stroke.
Recent studies from our laboratory showed that delayed systemic administration of human umbilical cord blood (HUCB) cells significantly reduced white matter injury (Ajmo et al. 2009), decreased infarct volume and improved behavioral recovery (Newcomb et al. 2006; Vendrame et al. 2004) in rats after middle cerebral artery occlusion (MCAO). The efficacy of HUCB cells to treat stroke at delayed time points has been attributed to combined anti-inflammatory and protective actions, and evidence indicates that this therapy reduces numbers of B cells and CD11b-expressing monocytes/macrophages in brain (Vendrame et al. 2005). Interestingly, cells that express CD11b also increase binding of isolectin IB4 in the injured brain (Matsumoto et al. 2007), suggesting that lectin binding occurs in proinflammatory cells.
The goals of the present study were two-fold. The time course of lectin binding to microglia/macrophages after MCAO was investigated with specific emphasis on whether lectin-binding cells exhibit a proinflammatory phenotype. Delayed HUCB cell therapy was also administered to determine whether HUCB cells target this population of proinflammatory cells, and thus whether the efficacy of this therapy in mitigating neural injury is associated with dampening of proinflammatory cell recruitment to the infarct.
Material and methods
Permanent Middle Cerebral Artery Occlusion Procedure
All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals with a protocol approved by the Institutional Animal Care and Use Committee at the University of South Florida. Methods used for the MCAO have been previously reported (Ajmo Jr et al. 2006). Briefly, 300–350 g Sprague Dawley rats were anesthetized, the common carotid artery was separated from the vagus nerve, and blunt dissection was performed to isolate the internal carotid artery (ICA), the external carotid artery (ECA) and the middle cerebral artery (MCA). A 40 mm monofilament was introduced into the ECA, fed distally into the ICA and advanced approximately 25 mm through the Circle of Willis to the origin of the MCA. Reductions in blood flow were recorded using a Laser Doppler Monitor (Moor Instruments, Devon, England). Only animals that showed a 60% reduction were used for this study. Following recovery, animals were randomly assigned into treatment groups (n=3 rats per group).
HUCB Cell Preparation and Delivery
For cell preparation, HUCB cells (Cambrex Corp., East Rutherford, NJ Saneron CCEL Therapeutics, Tampa, FL) were thawed rapidly at 37°C, washed in HBSS plus HEPES and centrifuged three times for 10 min at 1000 rpm. Viability was determined using the Trypan Blue dye exclusion method. Cell concentration was adjusted to 1×106 cells in 500 μl. For delivery, rats were anesthetized with 5% isofluorane in O2 at 2 L/min. The penile vein was exposed, a 30g × 1″ needle was inserted into the lumen of the vein and cells were injected. HUCB cells obtained from different donors were evenly distributed among the treatment groups.
Immunohistochemistry
Rats were perfused intracardially with normal saline followed by 4% paraformaldehyde in phosphate buffer. Brains were extracted, cryopreserved in increasing sucrose concentrations (20%, 30%), and cut into 30 μm sections using a cryostat. Sections from 0.7, −0.3, and −1.3 mm relative to Bregma were then thaw mounted onto slides and stored at −20°C until staining. The slides were thawed, rinsed with phosphate buffered saline containing 0.5 mM CaCl2 (PBS, pH 7.2), and placed in permeabilization buffer containing 10% goat serum, 3% 1M lysine, and 0.3% Triton X-100 in PBS for 1 hr at room temperature. Next, sections were incubated overnight at 4°C with primary antibody in antibody solution containing 2% goat serum and 0.3% Triton X-100. Primary antibody incubations were performed in a humidified chamber, protected from light when necessary. The next morning, slides were washed with PBS (3 × 5 min) and incubated with Alexa-Fluor® 594 goat anti-mouse secondary (1:300, Molecular Probes) in antibody solution. After final washing, slides were coverslipped with Vectashield hard set mounting media containing DAPI (Vector Laboratories, Burlingame, CA).
Primary antibodies used were mouse anti-CD11b (OX-42; 1:3,000, Serotec) and mouse anti-MMP-9 (1:1000, Chemicon). Isolectin staining was performed using 5 μl/mL Isolectin IB4 (Griffonia simplicifolia) conjugated with Alexa-Fluor 488 or 594 (Molecular Probes, Eugene, OR) that was co-incubated with primary antibody.
Fluoro-Jade Staining
The Fluoro-Jade stain identifies dead and degenerating cells, thus providing a positive quantitative marker as opposed to the absence of labeling that is observed when using Nissl stain. Fluoro-Jade was previously shown to be a more sensitive measure of neurodegeneration when compared to triphenyltetrazolium chloride (TTC) (Duckworth et al. 2005). This method was adapted from Schmued and colleagues (Schmued et al. 1997) and subsequently detailed (Duckworth et al. 2005). Sections were prepared and mounted on glass slides as described for histology. Slides were sequentially placed in 100% ethanol for 3 min, 70% ethanol for 1 min and deionized water for 1 min. Sections were oxidized for 15 min using 0.06% KMnO4 solution followed by 3 brief rinses in PBS. Slides were then immersed in a 0.001% solution of Fluoro-Jade (Histo-Chem, Jefferson, AR) in 0.1% acetic acid for 30 min, rinsed with PBS, dried for 20 min at 45°C, cleared with xylene and coverslipped using DPX medium (Electron Microscopy Sciences, Ft. Washington, PA).
Organotypic Slice Culture
Organotypic slice cultures were prepared using a method previously described (Leonardo et al. 2009). P8–P10 rat pups were decapitated, brains were removed, and intact hippocampi were dissected in cold isotonic buffer (136.89 mM NaCl, 5.37 mM KCl, 169 nM Na2HPO4, 22.04 nM KH2PO4, 27.52 nM glucose, 59.01 mM sucrose). Whole hippocampi were sliced sagitally at 400 μm thickness using a McIlwain Tissue Chopper (Mickle Laboratory Engineering Co. Ltd. Gomshall, Surrey, England). Only slices that appeared thin and translucent were selected for culture. Slices were then incubated for 90 min at 4°C. Cultures were maintained on Millicell CM (Millipore Corp., Billerica, MA) inserts and placed in 6-well plates containing Neurobasal media supplemented with B27 and 5 mM L-glutamine. Slices were cultured for 14 days in a standard tissue culture incubator at 37°C, receiving partial media changes every 3–4 days prior to experimentation. Each experiment utilized 2 pups for slice preparation. A total of 6 slices per treatment group were used for each experiment, and data were collected from 3 separate experiments.
Oxygen Glucose Deprivation
Organotypic slices were subjected to 48 hrs of normoxia or oxygen glucose deprivation (OGD). Immediately prior to exposure, inserts were transferred into new 6-well plates containing vehicle (Dulbecco’s Modified Eagles Medium (DMEM; Mediatech, Herndon, VA) for normoxia or DMEM without glucose (Invitrogen, Carlsbad, CA) for OGD. Cultures were maintained in a standard tissue culture incubator during the normoxia exposure and a hypoxic chamber (CBS Scientific Co. Inc., Del Mar, CA) containing 1% O2, 5% CO2, balance N2, and maintained at 37°C during the OGD exposure.
DAF Imaging
Nitric oxide (NO) production in lectin – positive microglia was measured using the NO sensitive dye DAF-FM (Invitrogen, Carlsbad, CA). Organotypic slice cultures were incubated for 1 hour at 37°C in DMEM containing 5μM DAF-FM and (5μM) Isolectin B4 directly conjugated to Alexa-Fluor 594. The slices were washed in physiological saline solution (PSS) consisting of 140 mM NaCl, 5.4 mM KCl, 1.3 mM CaCl2, 1.0 mM MgCl2, 20 mM glucose, and 25 mM HEPES (pH 7.4) prior to NO measurements. DAF-FM loaded cells were visualized at 488 nm, while lectin positive cells were visualized at 594 nm. Fluorescent emission was captured using a Sensicam digital CCD camera (Cooke Corporation, Auburn Hills, MI) and recorded with Slidebook 3.0 software (Intelligent Imaging Innovations, Denver, CO). Final image analyses including generation of the merged images were performed using the Openlab (Improvision, Coventry, England) software package.
Image Analyses and Quantification
For histological staining, images were acquired using a Zeiss Axioscope 2 (model #801572) controlled by Openlab software (Improvision Ltd, Lexington, MA). Photomicrographs were captured with a Zeiss Axicam Color camera (model #412-312). All images were captured at the same exposure and digital gain settings. Isolectin quantification (n=4 animals per group) was performed using two distinct measures that included total isolectin-tagged fluorescence (arbitrary units) and total area occupied by isolectin-positive cells (mm2). Blood vessels which bound isolectin were removed from the analysis to ensure that only the fluorescence emitted from lectin-binding immune cells within the brain parenchyma was included. All data were expressed as group mean ± SEM. A value of p<0.05 was considered significant for all analyses. Significance was determined by two-way ANOVA and main effects were subjected to Bonferroni’s post-hoc tests.
Results
CD11b/isolectin-positive cells participate in the delayed immune response
Double staining for isolectin IB4 and CD11b was performed on tissues from rats subjected to MCAO or sham-MCAO and sacrificed at 48 or 96 hrs post-surgery (Fig. 1). Immunoreactivity in sham-MCAO rats localized to ramified CD11b-expressing microglia/macrophages (Fig. 1A). CD11b-positive cells did not bind isolectin, which was restricted to blood vessels (Fig. 1B). Fluoro-Jade staining was prominent 48 hrs after MCAO (Fig. 1C, inset), indicating widespread neurodegeneration throughout the corpus striatum. Immunoreactivity for CD11b was also detected in cells throughout the ipsilateral striatum at this timepoint (Fig. 1C). Isolectin localized to hypertrophic ramified cells that were also positive for CD11b (Fig. 1D). Although colocalization was detected, there were many CD11b-positive cells that did not bind isolectin. At 96 hrs, CD11b immunoreactivity was detected in cells with prominent amoeboid morphology (Fig. 1E), which is generally associated with an activated phenotype. Isolectin staining also labeled round, amoeboid cells throughout the infarct (Fig. 1F), and CD11b again colocalized with isolectin. In contrast to the 48 hr timepoint, colocalized cells appeared larger and more abundant at 96 hrs.
Figure 1. CD11b and isolectin colocalize within the striatal infarct at delayed time points.

CD11b-positive cells in sham-MCAO animals displayed a ramified shape (A) and did not bind isolectin, which only labeled blood vessels (B). Inset (C) shows Fluoro-Jade staining within the striatal infarct at 48 hrs. CD11b immunoreactivity was localized to many hypertrophic ramified cells (C) that also bound isolectin (D) 48 hrs after MCAO. At 96 hrs, colocalization of CD11b (E) and isolectin (F) was abundant in cells that displayed amoeboid morphology. Scale bars = 50 μm.
To characterize the time course of isolectin binding, quantification of isolectin-tagged fluorescent signal was performed in tissues from a separate group of animals that were sacrificed at 3, 24, 48 or 96 hrs post-MCAO (Fig. 2). Data revealed a significant increase in fluorescent signal throughout the ipsilateral striatum at 96 hrs (p<0.02), indicating that lectin binding was most abundant at the timepoint associated with maximal injury in this model.
Figure 2. Quantification of isolectin binding in the infarcted striatum after MCAO.
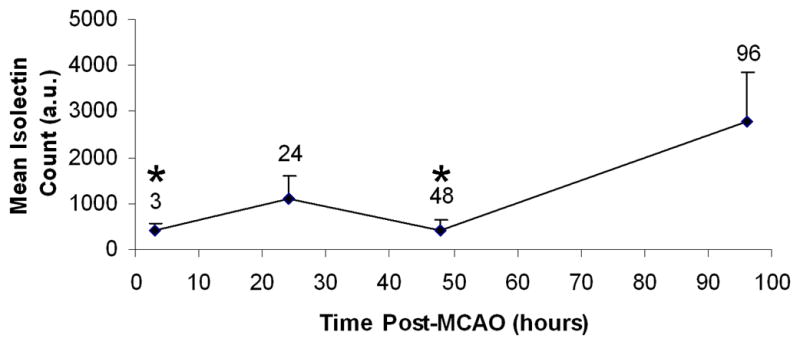
Isolectin-tagged fluorescence was quantified at 3, 24, 48 and 96 hrs after MCAO (n=3 rats per group, 3 sections per rat). Mean values were normalized to counts obtained from sham-operated rats. Total fluorescent signal did not significantly differ between the 3, 24 and 48 hr timepoints. Isolectin binding was significantly increased at 96 hrs compared to both 3 and 48 hrs (* p<0.02), while a trend toward an increase was observed at 24 hrs (p=0.06).
Isolectin-binding cells express neuroinflammatory markers in vivo and ex-vivo
Double staining experiments indicated that lectin binding to CD11b-expressing cells increased throughout the striatal infarct 96 hrs after MCAO, the timepoint when injury is most severe. However, it is unclear whether isolectin-binding cells represent a proinflammatory phenotype. Matrix metalloproteinase (MMP) activity is known to promote inflammation and exacerbate injury after stroke (Asahi et al. 2001; Brown et al. 1995; Cuadrado et al. 2008; Gu et al. 2005). Immunohistochemistry was performed using an antibody generated against MMP-9 in tissues collected 96 hrs after MCAO, and these tissues were co-incubated with isolectin IB4 (Fig. 3). MMP-9 expression was intense and ubiquitous throughout the infarcted striatum (Fig. 3A). The majority of MMP-9 immunoreactivity was detected on the surfaces of hypertrophic ramified and amoeboid cells, yet some was also associated with cellular processes. Isolectin-binding amoeboid cells were also prominent throughout the infarct (Fig. 3B) and many of these cells colocalized with MMP-9 (Fig. 3C).
Figure 3. Isolectin-binding cells express neuroinflammatory markers in vivo and ex-vivo.

MMP-9 expression was ubiquitous throughout the striatal infarct 96 hrs after MCAO (A). Isolectin binding was detected on microglia/macrophages throughout the infarct (B) and colocalized with MMP-9-expressing cells (C). Isolectin binding was also prominent in organotypic hippocampal slices exposed to OGD (D), and DAF-FM staining (E) showed that NO production occurred in isolectin-binding cells (F). Scale bars = 100 μm.
Similar to MMP-9, nitric oxide (NO) production is upregulated in microglia and is a putative inflammatory signal which promotes cell death. Because NO production cannot be measured in fixed tissues, DAF-FM staining for NO was performed in organotypic hippocampal slices exposed to 48 hrs of OGD. Since cellular connections remain intact, this model was selected as the best ex vivo model which mimics injury resulting from restricted oxygen and blood flow to the brain. Similar to data obtained from rats subjected to MCAO, organotypic slices showed abundant isolectin binding to amoeboid cells after 48 hrs exposure to OGD (Fig. 3D). DAF-FM staining was also prominent throughout the slices (Fig. 3E), demonstrating NO production in response to insult, and isolectin-binding cells that produced NO were abundant (Fig. 3F).
Isolectin binding and microglia/macrophage infiltration signifies the neuroinflammatory response
Rats subjected to MCAO and treated with vehicle were sacrificed at 51 or 72 hrs post-stroke to characterize the morphology and migratory capacity of lectin-binding microglia/macrophages at time points prior to maximal injury (Fig. 4). At 51 hrs, numerous cells expressing CD11b were detected in the ipsilateral striatum (Fig. 4A) as well as the peri-infarct regions seated adjacent to the corpus callosum (Fig. 4B) and lateral ventricle (Fig. 4C). The majority of these cells displayed an amoeboid morphology typical of the microglial/macrophage response to MCAO (Fig. 4A, inset). Isolectin fluorescent signal was far more ubiquitous, labeling cells that displayed a similar amoeboid shape (Fig. 4D, inset) as those which expressed the CD11b antigen. In contrast to CD11b-expressing cells, isolectin-binding cells were more concentrated in the white matter of the corpus callosum (Fig. 4E) and throughout the periventricular region, forming a line that could be traced from vessels at the base of the brain (Fig. 4F, inset). Isolectin-positive cells also showed amoeboid morphology in these regions, consistent with those observed within the striatal infarct.
Figure 4. Isolectin-binding immune cells migrate into the striatal infarct at 51 hrs.

Rats that received vehicle at 48 hrs post-MCAO showed a robust immune cell response at 51 hrs. CD11b-immunoreactivity was present on cells within the corpus striatum (A) and peri-infarct regions adjacent to the corpus callosum (B) and lateral ventricle (C). Isolectin-binding cells were also present in the striatum (D) and displayed amoeboid morphology consistent with that of CD11b-expressing cells (A,D, insets). Isolectin-tagged fluorescent cells were present in greater quantities both in and adjacent to the corpus callosum (E) as well as in the periventricular region (F), where they were densely distributed between blood vessels originating at the base of the brain (F, inset) and peri-infarct striatal tissue. Scale bars: A,D insets = 50 μm; all others = 100 μm. CC = corpus callosum. LV = lateral ventricle.
At 72 hrs (Fig. 5), the staining profiles of CD11b- and isolectin-expressing cells were indistinguishable and densely distributed throughout the striatum (Fig. 5A,D), displaying the characteristic morphology of amoeboid microglia/macrophages (Fig. 5B,E). Additionally, greater numbers of CD11b immunoreactive cells populated the peri-infarct region adjacent to the corpus callosum at 72 hrs relative to 51 hrs. A large proportion of cells in this region also bound isolectin as observed at the earlier timepoint. These data indicate that the majority of immune cells which infiltrated the striatal infarct, peri-infarct white matter and the periventricular region bound isolectin at both timepoints, whereas CD11b-expressing cells were more ubiquitous at 72 hrs relative to 51 hrs.
Figure 5. CD11b-expressing and Isolectin-binding immune cells invade the striatal infarct at 72 hrs.

Rats that received vehicle 48 hrs post-MCAO exhibited a robust neuroinflammatory response at 72 hrs. Cells displaying CD11b immunoreactivity (A) and isolectin-tagged fluorescence (D) infiltrated the striatal infarct in greater quantities relative to the 51 hr timepoint. Isolectin-binding colocalized to many CD11b-positive, amoeboid shaped immune cells (B,E). Both CD11b-expressing (C) and isolectin-binding (F) cells were also greatly elevated in peri-infarct tissues adjacent to the corpus callosum. Scale bars: A,C,D,F = 100 μm; B,E = 50 μm. CC = corpus callosum.
Intravenous administration of HUCB cells blocks the morphological change of CD11b-expressing cells and the migratory response of CD11b/isolectin-positive cells
Rats subjected to MCAO and administered 1×106 HUCB cells 48 hrs after MCAO were sacrificed at 51 or 72 hrs to determine whether this treatment reduces the number of isolectin-binding cells and CD11b-expressing immune cells (Figure 6). In tissues from rats treated with HUCB cells and sacrificed 3 hrs post-injection, dense pockets of CD11b immunoreactive and isolectin-tagged fluorescent cells were detected within the striatum (Fig. 6A,D). These cells exhibited an amoeboid shape (Fig. 6A,D, insets) similar to those observed after treatment with vehicle. CD11b-positive cells were also detected throughout the periventricular region but were less concentrated and displayed a phenotype most often associated with an inactivated or intermediate state (Fig. 6B,C). While the majority of isolectin-tagged fluorescence in this region localized to blood vessels (Fig. 6E), the few cells that bound isolectin were also ramified CD11b-expressing cells (Fig. 6C,F). Importantly, the densely distributed, isolectin-positive amoeboid cells that had infiltrated the periventricular and peri-infarct white matter regions in tissues from 51 hr vehicle-treated animals (Fig. 4E,F) were reduced at 51 hrs in rats treated with HUCB cells.
Figure 6. Delayed HUCB cell treatment blocks immune cell migration to the striatal infarct at 51 hrs.

Rats that received HUCB cell treatment 48 hrs post-MCAO showed marked reductions in immune cell migration to the striatal infarct. CD11b immunoreactivity was localized to immune cells within the striatal infarct (A) where lectin-binding cells were also found (D). These cells displayed hypertrophic ramified and amoeboid morphologies (A,D, insets) similar to vehicle-treated animals, but were far fewer in number. HUCB cell therapy greatly reduced immune cell infiltration into the periventricular region (B,E), and the few CD11b/lectin-positive cells present displayed a ramified morphology (C,F, arrows) that resembled microglia/macrophages in sham-MCAO rats. Scale bars = A,D insets, C,F = 50 μm; all others = 100 μm. LV = lateral ventricle.
Treatment with HUCB cells nearly abolished the immune cell response at 72 hrs (Fig. 7). In contrast to the earlier timepoint, no dense pockets of either CD11b- or isolectin-positive cells were present within the striatal infarct (Fig. 7A,D). Instead, immunoreactivity within this region revealed ramified or hypertrophic ramified CD11b-expressing microglia/macrophages that did not colocalize with isolectin, which labeled only blood vessels (Fig. 7B,E). Regions of peri-infarct white matter showed greatly reduced numbers of CD11b-positive cells relative to 72 hr vehicle-treated rats and were also devoid of isolectin-positive cells (Fig. 7C,F).
Figure 7. Delayed HUCB cell treatment blocks the infiltration of CD11b- and lectin-positive cells at 72 hrs.

Rats that received HUCB cell treatment 48 hrs post-MCAO showed a sparse distribution of CD11b-immunopositive microglia/macrophages within the striatum (A), and these cells displayed ramified morphology (B) characteristic of the inactivated state. Isolectin-tagged fluorescence was detected on blood vessels throughout the striatum (D) but did not label CD11b-expressing cells (E). Peri-infarct regions both in and adjacent to the corpus callosum showed reductions in the numbers of CD11b immunoreactive cells (C), and were devoid of lectin-binding cells (F). Scale bars: A,C,D,F = 100 μm; B,E = 50 μm. CC = corpus callosum.
Because the local distribution of immune cells differed between timepoints and across treatments, quantification of isolectin was performed using two distinct measures reflecting the total isolectin-tagged fluorescence present within the ipsilateral hemisphere and the actual area of the tissue that was occupied by fluorescent signal (Fig. 8). Isolectin binding was significantly increased at 72 hrs in vehicle-treated animals relative to the 51 hr timepoint, and treatment with HUCB cells significantly reduced the total fluorescent signal present at 72 hrs relative to vehicle-treated animals at both timepoints (Fig. 8A). Similar to total fluorescence, area measurements revealed a significant reduction in the total area occupied by fluorescent signal at 72 hrs in HUCBC cell-treated rats relative to vehicle-treated rats at both timepoints. Further, injection of HUCB cells also reduced the total area occupied by signal at 51 hrs (Fig. 8B), consistent with data showing that lectin-binding cells were predominantly localized to the striatum at this timepoint after HUCB cell treatment. Thus, the reduction in isolectin-bound immune cell infiltration observed 3 hrs after administration of HUCB cells relative to vehicle, as well as the alteration in morphology of CD11b-expressing cells, was enhanced at the 72 hr timepoint after treatment with HUCBC.
Figure 8. Quantification of isolectin binding.

Total isolectin-tagged fluorescent signal (A) and total area occupied by isolectin-positive cells (B) were quantified in tissues from rats treated with HUCB cells or vehicle at 48 hrs post-MCAO and sacrificed at 51 or 72 hrs. Isolectin binding was significantly elevated in vehicle-treated animals at 72 hrs relative to the 51 hr timepoint, and treatment with HUCB cells significantly reduced isolectin binding at 72 hrs relative to vehicle-treated rats at both timepoints. Treatment with HUCB cells also reduced isolectin-positive area at both timepoints relative to vehicle-treated controls. Asterisk denotes significance from 51 hr vehicle (p<0.01). Pound sign denotes significance from 72 hr vehicle (p<0.01).
Discussion
Any treatment for stroke at clinically relevant timepoints (24–48 hours) must take into account the active inflammatory response occurring in the brain. In the permanent MCAO model utilized in these studies, CD11b-expressing, amoeboid-shaped immune cells infiltrate infarcted brain tissue after MCAO at delayed time points concomitant with progressive neuroinflammation and neuronal dysfunction. Initially, as these dysfunctional neurons expressed many markers of apoptosis such as cleaved caspase-3 and TUNEL staining, it was thought that these neurons were already unsalvageable.
Recent studies by our laboratory and others have shown that this damage is reversible by therapeutics applied up to 48 hours following MCAO (Ajmo Jr et al. 2006; Newcomb et al. 2006). Interestingly, treatments that exclusively target the post-MCAO immune response, such as splenectomy, are able to reduce infarct volumes up to 85% (Ajmo et al. 2008). This reduction is similar to that observed with HUCB therapy (Newcomb et al. 2006), indicating that decreased infarct volume in response to these treatments is likely mediated by the immune system. This also implies that the necrotic core region of the infarct is smaller than previously thought, only representing approximately 15% of the infarcted tissue.
Many of the CD11b-expressing, amoeboid cells also increase binding of isolectin IB4, a plant lectin that binds to sugars which are upregulated on microglia/macrophage cell membranes in response to injury. Data here showed that treatment with HUCB cells 48 hrs after MCAO altered the migration and lectin-binding profile of infiltrating immune cells, and these cells represented a proinflammatory phenotype.
In non-treated animals, amoeboid cells containing elevated levels of isolectin binding were observed within the infarct at 48 hrs and predominated at 96 hrs after MCAO. These cells infiltrate the site of injury rapidly by traveling via collateral vasculature and through nearby white matter tracts. MMP-9, a well-known contributor to ischemic pathology (Asahi et al. 2001; Brown et al. 1995; Cuadrado et al. 2008; Gu et al. 2005), was also highly expressed on isolectin-positive cells within the infarct. This protease degrades basement membrane constituents, causing blood brain barrier leakage and increasing leukocyte invasion from the vasculature. Further experiments were conducted to determine whether isolectin-binding cells upregulate the putative proinflammatory marker NO. Because NO staining must be performed in live slices, organotypic slice culture was utilized as an ex vivo system to assess NO production. Results showed that isolectin-positive cells upregulated NO upon exposure to OGD. These data support the notion that isolectin binding to cell surfaces is elevated in response to ischemic insult, and these cells facilitate proinflammatory signaling that serves as a critical component of the enhanced inflammatory response.
Isolectin binding was further investigated in tissues from animals treated with HUCB cells or vehicle at 48 hrs post-MCAO and sacrificed at 51 or 72 hrs, thus representing a window between the onset of stroke injury and the pinnacle of ischemic damage. At 51 hrs, both treatment groups exhibited an immune cell response throughout the ipsilateral hemisphere. Clusters of isolectin-positive cells could be traced along migratory paths, from collateral blood vessels at the base of the brain and from the white matter of the corpus callosum, into the corpus striatum. Systemic injection of 1×106 HUCB cells reduced the magnitude of this response and mediated a morphological shift in CD11b-positive cells from amoeboid to ramified shape just 3 hrs after the injection.
Interestingly, CD11b immunoreactivity and isolectin-tagged fluorescence did not colocalize completely. This finding was most evident at 51 hrs, and although quantification revealed no significant differences in total isolectin-tagged fluorescence between treatments at this timepoint, the total area occupied by isolectin-binding cells was significantly reduced in HUCB cell-treated rats relative to those treated with vehicle. These data were consistent with the isolectin-binding distribution profile, which showed dense clusters of isolectin-positive cells within the striatum that were rarely present within peri-infarct regions. Importantly, cells that bound isolectin were virtually absent 72 hrs after HUCB cell therapy. Thus, the binding of isolectin precedes complete infiltration and activation of proinflammatory cascades that result in enhanced neural injury.
HUCB cell therapy has previously been shown to protect the rat brain from focal ischemia when administered 48 hrs (as in the present study), but not 72 hrs, post-MCAO (Newcomb et al. 2006). Data here provide a potential mechanism for these results in the context of the temporal immune cell response. Because HUCB cells block the migration of proinflammatory cells to the injured site, this therapy would only be expected to provide efficacy at time points which precede the massive infiltration of proinflammatory cells, and in turn, heightened proinflammatory signaling. Indeed, we show that lectin binding to immune cell surfaces increased approximately 66% in vehicle-treated animals from 51 to 72 hrs, and while lectin-positive cells were present in high numbers even after HUCB cell treatment at 51 hrs, this therapy reduced the numbers of lectin-binding cells in peri-infarct regions where immune cells traffic to the striatum. Importantly, CD11b-positive, isolectin-binding microglia and/or peripheral macrophages displaying amoeboid shape were first observed within the infarct at 48 hours, just prior to HUCB cell injection. Thus, the timing of treatments aimed at targeting the immune cell response is critical in achieving efficacy.
To date, there is much controversy as to whether activated immune cells signify a protective or destructive phenotype after stroke. The expression of proinflammatory markers after ischemic insult and the time course of isolectin binding suggest a destructive role. Further, the neuroprotective HUCB cell therapy was efficacious in blocking the migratory response of amoeboid CD11b-positive lectin-binding cells, providing additional evidence that these cells contribute to neural injury. Further investigations into the precise role of isolectin-binding cells, and the manner in which HUCB cells mediate inflammatory signaling in these cells, will be critical in developing alternate therapies which mimic the actions of HUCB cell therapy but are safer, more selective, and easier to deliver.
Acknowledgments
This work was supported in part by the National Institutes of Health (NINDS RO1-NS052839) and the American Heart Association (0715096B).
Research support: National Institute of Neurological Disorders and Stroke, grant # RO1-NS052839 (AEW, KRP); American Heart Association, grant # 0715096B (AAH)
References
- Ajmo CT, Jr, Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, Cuevas J, Willing AE, Pennypacker KR. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp Neurol. 2009;218(1):47–55. doi: 10.1016/j.expneurol.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajmo CT, Jr, Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. 2008;86(10):2227–2234. doi: 10.1002/jnr.21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajmo C, Jr, Vernon D, Collier L, Pennypacker K, Cuevas J. Sigma receptor activation reduces infarct size at 24 hours after permanent middle cerebral artery occlusion in rats. Cur Neurovascular Res. 2006;3(2):89–98. doi: 10.2174/156720206776875849. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2(10):734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Asahi M, Wang X, Mori T, Sumii T, Jung J, Moskowitz M, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21(19):7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Kornfeld M, Mun-Bryce S, Sibbit R, Rosenberg G. Comparison of magnetic resonance imaging and histology in collagenase-induced hemorrhage in the rat. J Neuroimaging. 1995;5:23–33. doi: 10.1111/jon19955123. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438(7070):1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cuadrado E, Ortega L, Hernandez-Guillamon M, Penalba A, Fernandez-Cadenas I, Rosell A, Montaner J. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol. 2008;84(1):207–214. doi: 10.1189/jlb.0907606. [DOI] [PubMed] [Google Scholar]
- da Cunha A, Jefferson JJ, Tyor WR, Glass JD, Jannotta FS, Cottrell JR, Resau JH. Transforming growth factor-beta1 in adult human microglia and its stimulated production by interleukin-1. J Interferon Cytokine Res. 1997;17(11):655–664. doi: 10.1089/jir.1997.17.655. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz M. Pathobiology of ischemic stroke: an integrated view. TINS. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR. Temporary focal ischemia in the mouse: technical aspects and patterns of Fluoro-Jade evident neurodegeneration. Brain Res. 2005;1042(1):29–36. doi: 10.1016/j.brainres.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1(2):127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25(27):6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AA, Herrera Y, Ajmo CT, Jr, Cuevas J, Pennypacker KR. Sigma receptors suppress multiple aspects of microglial activation. Glia. 2008 doi: 10.1002/glia.20802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43(6):1382–1396. doi: 10.1097/00006123-199812000-00076. discussion 1396–1387. [DOI] [PubMed] [Google Scholar]
- Leonardo CC, Hall AA, Collier LA, Gottschall PE, Pennypacker KR. Inhibition of gelatinase activity reduces neural injury in an ex vivo model of hypoxia-ischemia. Neuroscience. 2009;160(4):755–766. doi: 10.1016/j.neuroscience.2009.02.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Kumon Y, Watanabe H, Ohnishi T, Shudou M, Ii C, Takahashi H, Imai Y, Tanaka J. Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions. J Neurosci Res. 2007 doi: 10.1002/jnr.21198. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Onodera H, Shiga Y, Nakamura M, Ninomiya M, Kihara T, Kogure K. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat. Effects of neutrophil depletion. Stroke. 1994;25(7):1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- Newcomb JD, Ajmo CT, Sanberg CD, Sanberg PR, Pennypacker KR, Willing AE. Timing of cord blood treatment after experimental stroke determine therapeutic efficacy. Cell Transplant. 2006;15(3):213–223. doi: 10.3727/000000006783982043. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26(5):654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Schmued L, Albertson C, Slikker W. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Streit W, Graeber M, Kreutzberg G. Functional plasticity of microglia: A review. Glia. 1988;1:301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1(1):14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano K, Latour LL, Formato JE, Carano RA, Helmer KG, Hasegawa Y, Sotak CH, Fisher M. The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann Neurol. 1996;39(3):308–318. doi: 10.1002/ana.410390307. [DOI] [PubMed] [Google Scholar]
- Tanaka R, Komine-Kobayashi M, Mochizuki H, Yamada M, Furuya T, Migita M, Shimada T, Mizuno Y, Urabe T. Migration of enhanced green fluorescent protein expressing bone marrow-derived microglia/macrophage into the mouse brain following permanent focal ischemia. Neuroscience. 2003;117(3):531–539. doi: 10.1016/s0306-4522(02)00954-5. [DOI] [PubMed] [Google Scholar]
- Vendrame M, Cassady CJ, Newcomb J, Butler T, Pennypacker KR, Zigova T, Davis Sanberg C, Sanberg PR, AEW Infusion of human umbilical cord blood cells in a rat model of stroke dose-dependently rescues behavioral deficits and reduces infarct volume. Stroke. 2004;35:2390–2395. doi: 10.1161/01.STR.0000141681.06735.9b. [DOI] [PubMed] [Google Scholar]
- Vendrame M, Gemma C, De Mesquita D, Collier L, Bickford PC, Sanberg CD, Sanberg PR, Pennypacker KR, Willing AE. Anti-inflammatory effects of human cord blood cells in a rat model of stroke. Stem Cells Dev. 2005;14:595–604. doi: 10.1089/scd.2005.14.595. [DOI] [PubMed] [Google Scholar]