

Abstract
Platelets have been extensively studied as hemostatic regulators, stopping uncontrolled flow of blood from an injured vessel and allowing for repair. However, multiple studies have shown that platelets can interact with bacterial proteins, particularly seen during sepsis and inflammation. Immune cells recognize pathogens through Toll-like Receptors (TLRs). These same receptors allow platelets to recognize bacterial proteins and regulate platelet immunity and function. This review examines the TLRs expressed on platelets and megakaryocytes and how these receptors affect the function of these cells. Through TLRs, platelets go beyond hemostatic regulation and play a pivotal role in inflammation and infection.
Keywords: Platelets, Toll-like Receptors, Megakaryocytes, TLR2, TLR4
Introduction
The immune system consists of two fundamental approaches to recognize and respond to harmful stimuli due to infection – innate and adaptive immunity. Innate immunity is the first line of defense against invading microorganisms and involves a series of reactions that prevent ongoing damage, isolate infective agents, and activate the repair process. Adaptive immunity involves the dynamic adaptation to new and unique epitopes on pathogens in the environment. [1] Innate immunity is primarily mediated by macrophages and neutrophils. These immune cells distinguish between pathogen and self by utilizing signals from Toll-like receptors (TLRs). Stimulation of TLRs results in NFκB and MAPK pathway activation, leading to the production of proinflammatory cytokines.
The Toll gene was first discovered as encoding for a receptor in Drosophila embryos. [2-3] Expression of TLRs is ubiquitous throughout species and has evolutionary conservation. TLR2 (with TLR1 or 6) and TLR5 primarily function at the plasma membrane, while TLRs 3, 7, 8, and 9 are reported to function intracellularly. TLR4 is found both in the plasma and intracellular spaces. Each TLR responds to a different set of ligands or pathogen associated molecular patterns (PAMPs). The most studied of the TLRs, TLR4, recognizes lipopolysaccharides (LPS) of gram-negative bacteria. The variety of TLR2 ligands is the greatest among all the TLRs and this is due to its heterodimerization with TLR1 and TLR6. TLR2 recognizes lipoproteins and lipopeptides, peptidoglycans and lipoteichoic acid of gram-positive bacteria, LPS from non-enterobacteria, lipoarabinomannan from mycobacteria, and zymosan from fungi, to name a few. The intracellular portion of these receptors consists of a Toll/Interleukin-1 Receptor (TIR) domain that binds to an adaptor molecule, MyD88. IRAK1/4 are recruited to the cell membrane, bind to MyD88, and are phosphorylated. TRAF6 then binds to and is activated by IRAK1. TRAF6 will go on to activate the NFκB and MAPK pathways. TLR signaling induces antigen presenting cell activation, pro-inflammatory cytokine production, and increased expression of co-stimulatory ligands. These events are important for induction of innate immune responses and improved acquired immunity. [4]
The interaction between infection and thrombosis has been largely studied in sepsis models, in which a systemic infection leads to the activation of the coagulation pathway, creating thrombi in the microcirculation of organs. These events lead to the consumption of platelets and coagulation proteins and multiple organ failure. Limited work has focused on the direct interaction of platelets with bacteria, leading not only to the formation of a platelet-rich clot, but also the activation of the innate immune system. In this setting, TLRs may be the link. Work done in 1977 showed that streptococcal derived lipoteichoic acid (LTA) could stimulate platelet granule release, independent of other well-characterized platelet receptors. [5] In addition, the synthetic lipopeptide, Pam3CSK4, has been shown to induce physiological platelet activation. [6] This review will focus on work that has shown the role of TLRs in both platelet production and platelet activation.
TLRs and Megakaryocytes
Megakaryocytes primarily are found in the bone marrow where they constitute 1% of the total cell population [7]. Megakaryocytes are formed through the maturation process known as megakaryopoiesis, in which the cells express thrombotic markers, increase significantly in size and DNA content (16+ N). Upon stimulation by multiple factors, including thrombopoietin, megakaryocytes adhere to the endothelium of the bone marrow sinuses, increasing mRNA and protein levels, which are shuttled to the ever elongating pseudopodia that are forming. From these structures pro-platelets bud off and flow into circulation, where they undergo further maturation into platelets, a process called thrombopoiesis. Few studies have looked into the expression and function of TLRs in megakaryocytes and whether or not these receptors could have a role in platelet production. Initial work had shown that low grade endotoxemia increases thrombopoietin levels, which resulted in an increase in reticulated platelets and increased platelet-neutrophil aggregates. [8] Murine bone marrow treated with LPS also showed an increase in the levels of thrombopoietin and cytokines, factors important for thrombopoiesis. [9] Therefore, it is possible that inflammation and infection can modulate platelet production through TLRs. Work using the Meg-01 cell line, a human megakaryoblast cell line, has shown through RT-PCR and flow cytometry that megakaryocytes only express TLR1 and TLR6. [10] Both mRNA levels are increased in a dose dependent manner over time in the presence of interferon-γ. [10] Based on work done with macrophages derived from THP-1 monocytes, it is hypothesized that the levels of TLRs increase with cell differentiation since Meg-01 cells had a lower level of TLR1 and 6 expression compared to isolated platelets. [10] TLR4 surface expression was also verified by flow cytometry and immunohistochemistry on adherent Meg-01 cells; however, in this study, it was hypothesized that any TLR4 found on platelets was a remnant from megakaryocytes. [11] Further confirmation of TLR4 on megakaryocyte cell surface was shown through flow cytometry of murine megkaryocytes isolated from fetal livers. [12] As the cells matured, as indicated by the increase in CD41 levels, the TLR4 levels also increased. [12] Additionally, TLR2 and TLR4 were both detected by flow cytometry in human megakaryocytes isolated from patients, and both receptors were increased in megakaryocytes from myelodysplatic syndrome, a disorder of the hematopoietic stem cells causing multiple lineage cytopenias. [13]
As for the role of TLRs in megakaryocytes, in 2 different studies, TLR4-/- mice were shown to have a decreased circulating [12, 14] and reticulated [14] platelet count compared to WT, suggesting TLR4 may have a role in thrombopoiesis. After a single sublethal dosage of LPS, circulating platelets levels significantly increase compared to untreated mice; however, circulating platelets in TLR4-/- mice were still lower than WT. [14]
TLRS and Platelets
There is growing research examining the non-hemostatic role of platelets, specifically related to inflammation and infection. Platelets can have an early role in immune surveillance and the transfer of pathogen information to other innate immune cells. Platelets can also modify adaptive immune responses by localizing at sites of bacterial invasion, aggregating around bacteria, and promoting clearance. Coagulation abnormalities are common in severe sepsis and it is possible that the presence of TLRs on platelets could be the link between disseminated intravascular coagulation and sepsis. [15-16] While growing evidence has clearly established the relevance of platelets in inflammation, there is much circumstantial evidence that they are also relevant in the setting of infection. An early study showed that LPS, at high doses, actually inhibited platelet activation. [17] However, a much earlier study showed that E. coli endotoxin in dogs caused a sharp fall in circulating platelet numbers and changes in platelet morphology, suggesting platelet activation. [18] More recently, platelets were shown to bind and internalize pathogens and release microcidal proteins.[19-20] In anaphylaxis-like shock (ALS) induced by mannan, a component of fungal cell surfaces, severity increases when platelets have been depleted, suggestive that platelets play a role in the recognition and clearance of this pathogen. [16] These results are contrary to LPS- induced ALS, in which platelet depletion attenuated the disease state. [16]
Other bacteria have been shown to influence platelet function in a variety of settings. Interaction of Staphyloccus aureaus with circulating platelets has been implicated as a virulent mechanism in the induction of endocarditis. Previous investigations related to the activation of platelets in the setting of infective endocarditis have demonstrated both fibrinogen-dependent [21-22] and -independent pathways may trigger platelet aggregation, [23] as may engagement of glycoprotein Ib on the platelet surface. [24-25] Serotype polysaccharides from Streptococcus mutans have also been shown to induce platelet aggregation. [26] Salmonella typhimurium porin was shown to not induce platelet aggregation but enhance ADP- and thrombin-induced activation. [27] In the setting of infection, strokes are associated with increased platelet-leukocyte interactions. [28] Related to cardiovascular disease, Chlamydia pneumoniae has been shown to adhere to platelets, stimulate P-selectin expression, and trigger aggregation. [29-30] The pro-aggregatory effects of Porphyromonas gingivalis have also been established in mouse [31] and human platelets in vitro. [32-35] Thus, in the same way that TLRs are the sentinel receptors of the immune system, platelets may be a sentinel cell in the blood by modifying the acute response to infection and injury. As seen by these studies, not only are platelet thrombotic pathways activated, but platelet-white cell interactions are stimulated in the setting of infection.
Many studies have identified TLRs on platelets, but unlike megakaryocytes, there is more work being done to understand the physiological relevance of these immunological receptors. In addition, there is still some controversy. In one study, platelets were shown to not bind to LPS, nor express TLR4 or CD14. [36] It was determined that LPS primed platelets by binding to monocyte TLR4 and CD14 and triggered the release of platelet activating factor (PAF) and oxygen radicals, which increases agonist-induced aggregation and heterotypic aggregate formation with monocytes. [36] Other studies, however, have shown that TLRs are present on platelets, summarized in Figure 1. Through RT-PCR and immunoblotting, platelets were shown to express TLR1 and 6. [10] Further, tissue sections of coronary thrombi from patients with acute coronary syndrome show a colocalization of staining for CD41 (platelet marker) and TLR1 and TLR6. [10] Through flow cytometry, platelets were shown to express TLR2 and 4, but not TLR1. [11] Treatment with LPS (TLR4 ligand) and Pam3CSK4 (TLR2 ligand) did not activate platelets nor augment agonist-induced platelet activation in this study. [11] Another study found that platelets do express TLR1, 2, 4, 6, 8, and 9 on the platelet surface. Intracellularly, there were higher levels of TLR2, 4, and 9. Upon activation, platelet TLR2 [37] and 9 [37-38] increased on the cell surface, while intracellularly, TLR2, 4, and 9 levels increased. [37] Taken in total, the data is highly suggestive of multiple TLRs in platelets.
Figure 1.
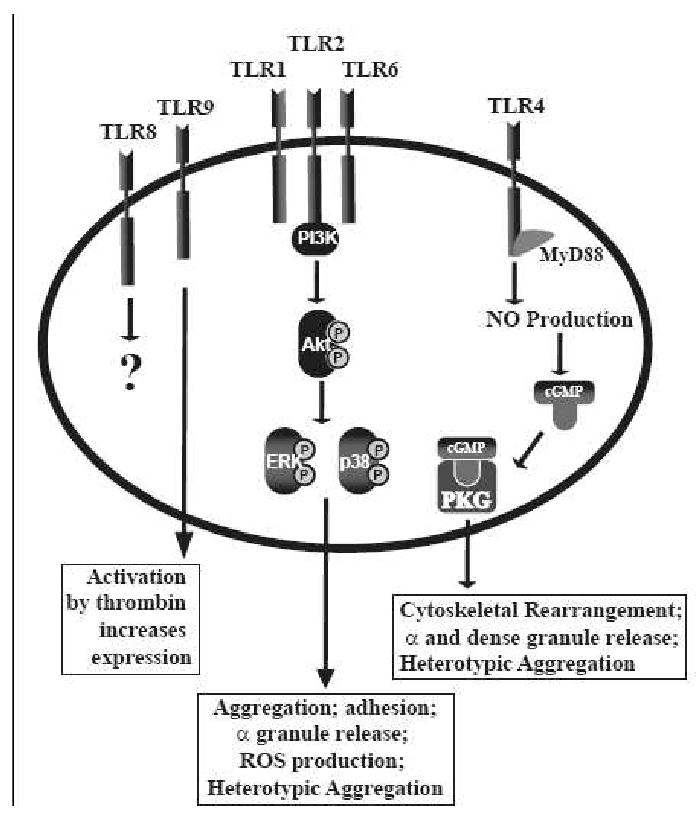
Summary of the TLRs shown to be expressed on platelets and the effects they have on platelet function.
The majority of studies looking into the functionality of TLRs in platelets have been focused on TLR4 and TLR2 as summarized in Figure 1. For TLR4, LPS stimulated platelets from mice deficient in TLR4 would not adhere to fibrinogen, suggestive that through TLR4, LPS can modulate platelet activation. [12] After a single injection of LPS, platelets did not accumulate in the lungs of TLR4 deficient mice compared to wildtype mice. [12] Similarly, LPS injected into a different TLR4 mouse model, showed that circulating platelet numbers were not affected [38-39], nor was there an increase in serum TNF-α levels compared to WT. [38] These studies suggest that through TLR4, platelets act as inflammatory sentinels, surrounding and isolating an infection, while modulating proinflammatory cytokine release. This idea is further supported by work showing platelet TLR4 induced platelet binding to adherent neutrophils. In this setting, however, LPS primed platelets and did not directly cause the formation of heterotypic aggregates. [40] In chicken thrombocytes, the avian equivalent to platelets, TLR4 stimulation with LPS increases IL6, COX-2, and PGE2 levels through NFκB and MAPK pathways. [41] Additionally, sCD40L [42-43] and PAF4 levels increase after platelet stimulation with LPS in a TLR4 dependent manner, however, RANTES, angiogenin, and PDGF-AB all decrease. [43] Children with enterohemorrhagic E. coli (EHEC) have platelets bound with EHEC-LPS. In vitro studies further confirmed that EHEC-LPS was binding to platelets through TLR4 and CD62 and were being activated as indicated by the increase in activate αIIbβ3 and fibrinogen binding. [44] All of these studies continue to support the idea of platelets having a role in inflammation. But TLR4 can also regulate the hemostatic function of platelets. LPS treatment in vitro increases platelet CD63 [43], one marker of activation, but not CD62P [12, 43], another activation marker. Through TLR4, LPS reduced the time to occlusion in an in vivo mouse thrombosis model.[45] Interestingly, in this study, adhesion of the platelets to the endothelium was shown to be dependent on the presence of neutrohils. [45] Contradictory to this study, LPS alone was shown to not activate platelets but enhanced agonist-induced aggregation through TLR4, TLR2, and MyD88. [46] Aggregation through TLR4 and 2 involved the production of NO, increase in cellular cGMP, and activation of PKG. [46] Additionaly, this study also showed LPS, alone, was able to increase ATP secretion from dense granules [46] and P-selectin (CD62P) from α granules [46], contrary to what has been previously demonstrated. [12, 43] Finally, a TLR4 polymorphism was found to be cardioprotective because individuals with this polymorphism had reduced platelet thromboxane biosynthesis which limited platelet function. [47] Therefore, these studies suggest that TLR4 and platelet function can affect cardiovascular disease as well.
The functionality of the TLR2 has also been studied in platelets, but not as extensively as TLR4. In the setting of infection, TLR2 in concert with PAR-1 and endothelial-derived CX3CL1 was shown to react to Rickettsia africae to increase sCD40L levels from platelets, which could not be reduced with doxycycline, a widely used treatment for such infections. [48] Using TLR2-/- mice, the formation of platelet-neutrophil heterotypic aggregates was unaffected by the presence of P. gingivalis, a bacteria known to be recognized by TLR2. [49] Additionally, through TLR2 and PI3K/Akt, platelets formed platelet-neutrophil heterotypic aggregates that were dependent on the presence of P-selectin on the platelet cell surface. [49] These studies further confirm the role of platelets and platelet TLRs in infection.
Unlike TLR4, studies have shown decisively that stimulation of platelet TLR2 can directly activate platelets. Activation of this receptor with Pam3CSK4, a synthetic TLR2 ligand, results in platelet aggregation and adhesion [49-50] that is dependent on PI3K/Akt. [49] Akt signaling results in increased P-selectin levels, ROS production, and αIIbβ3 activation. [49] Interestingly, although the signaling cascades activated by TLR2 might be the same as thrombin, the activation of Akt, ERK1/2, and p38 occur at different times and with different levels of phosphorylation. [50] Not only does the signaling differ between agonists, the contents of the α-granule (specifically, FXIIIa, thrombospondin, fibrinogen β, gelsolin, PBP, and PF4) released by each agonist differ. [50] This work suggested that, depending on the type of agonist, platelet function could vary. Platelets stimulated with thrombin were able to form stable clots in vitro. However, stimulation with Pam3CSK4 resulted in greater formation of heterotypic aggregates compared to thrombin. [50]
Conclusions
In summary, megakaryocytes and platelets express various TLRs, however, the function of these receptors in megakaryocytes is not completely understood. The available data suggests that TLRs are the link between thrombopoiesis and infection, as seen with modulation of platelet production during endotoxemia. TLRs on platelets have been more extensively studied. Two TLRs, TLR2 and TLR4, both have been shown to augment platelet activation and alter its function from hemostatic regulator to immune sentinel. As further work is done in this field, we will further understand not only the basic function of both megakaryocytes and platelets, but also the involvement of these cells and TLRs in various disease pathologies.
Acknowledgments
This work was supported in part by NIH grant P50HL083801 (to J.E.F.) and T32 HL07224 (to L.M.B).
Abbreviations
- TLR
toll-like receptor
- NFκB
nuclear factor kappa B
- MAPK
mitogen activated protein kinase
- PAMP
pathogen associated molecular patterns
- LPS
lipopolysaccharide
- TIR
toll/interleukin-1 receptor
- MyD88
myeloid differentiation primary response gene 88
- IRAK1/4
interleukin-1 receptor associated kinase 1/4
- TRAF6
tumor necrosis factor associated factor 6
- LTA
lipoteichoic acid
- WT
wildtype
- ALS
anaphylaxis-like shock
- PAF
platelet activating factor
- TNF-α
tumor necrosis factor α
- IL6
interleukin 6
- COX-2
cyclooxygenase-2
- PGE2
prostaglandin E2
- sCD40L
soluble CD40 ligand
- RANTES
regulated upon activation, normal T cell expressed and secreted
- PDGF-AB
platelet-derived growth factor-AB
- EHEC
enterohemorrhagic E. coli
- NO
nitric oxide
- cGMP
cyclic guanosine monophosphate
- PKG
protein kinase G
- ATP
adenosine triphosphate
- PAR-1
protease activated receptor-1
- PI3K
phosphoinositide 3 kinase
- ROS
reactive oxygen species
- ERK1/2
extracellular signal related kinase 1/2
- FXIIIa
factor XIII activated
- PBP
platelet binding protein
- PF4
platelet factor-4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002 Apr 12;296(5566):298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997 Feb;9(1):4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997 Jul 24;388(6640):394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 4.Wetzler LM. The role of Toll-like receptor 2 in microbial disease and immunity. Vaccine. 2003 Jun 1;21 2:S55–60. doi: 10.1016/s0264-410x(03)00201-9. [DOI] [PubMed] [Google Scholar]
- 5.Beachey EH, Chiang TM, Ofek I, Kang AH. Interaction of lipoteichoic acid of group A streptococci with human platelets. Infect Immun. 1977 May;16(2):649–54. doi: 10.1128/iai.16.2.649-654.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg M, Offermanns S, Seifert R, Schultz G. Synthetic lipopeptide Pam3CysSer(Lys)4 is an effective activator of human platelets. Am J Physiol. 1994 Jun;266(6 Pt 1):C1684–91. doi: 10.1152/ajpcell.1994.266.6.C1684. [DOI] [PubMed] [Google Scholar]
- 7.Travlos GS. Normal structure, function, and histology of the bone marrow. Toxicol Pathol. 2006;34(5):548–65. doi: 10.1080/01926230600939856. [DOI] [PubMed] [Google Scholar]
- 8.Stohlawetz P, Folman CC, von dem Borne AE, Pernerstorfer T, Eichler HG, Panzer S, et al. Effects of endotoxemia on thrombopoiesis in men. Thromb Haemost. 1999 Apr;81(4):613–7. [PubMed] [Google Scholar]
- 9.Pick M, Perry C, Lapidot T, Guimaraes-Sternberg C, Naparstek E, Deutsch V, et al. Stress-induced cholinergic signaling promotes inflammation-associated thrombopoiesis. Blood. 2006 Apr 15;107(8):3397–406. doi: 10.1182/blood-2005-08-3240. [DOI] [PubMed] [Google Scholar]
- 10.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, et al. Expression of Toll-like receptors on human platelets. Thromb Res. 2004;113(6):379–85. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. 2005 Oct;94(4):831–8. [PubMed] [Google Scholar]
- 12.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood. 2005 Oct 1;106(7):2417–23. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 13.Maratheftis CI, Andreakos E, Moutsopoulos HM, Voulgarelis M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin Cancer Res. 2007 Feb 15;13(4):1154–60. doi: 10.1158/1078-0432.CCR-06-2108. [DOI] [PubMed] [Google Scholar]
- 14.Jayachandran M, Brunn GJ, Karnicki K, Miller RS, Owen WG, Miller VM. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: implications for thrombotic risk. J Appl Physiol. 2007 Jan;102(1):429–33. doi: 10.1152/japplphysiol.01576.2005. [DOI] [PubMed] [Google Scholar]
- 15.Alves-Filho JC. Toll-like receptors on platelets: the key for disseminated intravascular coagulation in sepsis? Thromb Res. 2005;115(6):537–8. doi: 10.1016/j.thromres.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 16.Funayama H, Huang L, Sato T, Ohtaki Y, Asada Y, Yokochi T, et al. Pharmacological characterization of anaphylaxis-like shock responses induced in mice by mannan and lipopolysaccharide. Int Immunopharmacol. 2009 Sep 13; doi: 10.1016/j.intimp.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Sheu JR, Hung WC, Kan YC, Lee YM, Yen MH. Mechanisms involved in the antiplatelet activity of Escherichia coli lipopolysaccharide in human platelets. Br J Haematol. 1998 Oct;103(1):29–38. doi: 10.1046/j.1365-2141.1998.00938.x. [DOI] [PubMed] [Google Scholar]
- 18.Davis RB, Meeker WR, Mc QD. Immediate effects of intravenous endotoxin on serotonin concentrations and blood platelets. Circ Res. 1960 Jan;8:234–9. doi: 10.1161/01.res.8.1.234. [DOI] [PubMed] [Google Scholar]
- 19.Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. 2002 Dec;70(12):6524–33. doi: 10.1128/IAI.70.12.6524-6533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klinger MH, Jelkmann W. Role of blood platelets in infection and inflammation. J Interferon Cytokine Res. 2002 Sep;22(9):913–22. doi: 10.1089/10799900260286623. [DOI] [PubMed] [Google Scholar]
- 21.Liu CZ, Huang HY, Tsai PJ, Shih MH. Blockade of glycoprotein IIb/IIIa by crotavirin, a member of disintegrins, prevents platelet from activation and aggregation by Staphylococcus aureus bacteria. Thromb Res. 2005;116(2):145–55. doi: 10.1016/j.thromres.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 22.Sjobring U, Ringdahl U, Ruggeri ZM. Induction of platelet thrombi by bacteria and antibodies. Blood. 2002 Dec 15;100(13):4470–7. doi: 10.1182/blood-2002-01-0069. [DOI] [PubMed] [Google Scholar]
- 23.Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, et al. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol. 2005 Aug;57(3):804–18. doi: 10.1111/j.1365-2958.2005.04731.x. [DOI] [PubMed] [Google Scholar]
- 24.Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, et al. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology. 2003 Jun;124(7):1846–54. doi: 10.1016/s0016-5085(03)00397-4. [DOI] [PubMed] [Google Scholar]
- 25.Kerrigan SW, Douglas I, Wray A, Heath J, Byrne MF, Fitzgerald D, et al. A role for glycoprotein Ib in Streptococcus sanguis-induced platelet aggregation. Blood. 2002 Jul 15;100(2):509–16. doi: 10.1182/blood.v100.2.509. [DOI] [PubMed] [Google Scholar]
- 26.Chia JS, Lin YL, Lien HT, Chen JY. Platelet aggregation induced by serotype polysaccharides from Streptococcus mutans. Infect Immun. 2004 May;72(5):2605–17. doi: 10.1128/IAI.72.5.2605-2617.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeller JA, Lenz A, Eschenfelder CC, Zunker P, Deuschl G. Platelet-leukocyte interaction and platelet activation in acute stroke with and without preceding infection. Arterioscler Thromb Vasc Biol. 2005 Jul;25(7):1519–23. doi: 10.1161/01.ATV.0000167524.69092.16. [DOI] [PubMed] [Google Scholar]
- 28.Matera C, Falzarano C, Berrino L, Rossi F. Effects of tetanus toxin, Salmonella typhimurium porin, and bacterial lipopolysaccharide on platelet aggregation. J Med. 1992;23(5):327–38. [PubMed] [Google Scholar]
- 29.Kalvegren H, Majeed M, Bengtsson T. Chlamydia pneumoniae binds to platelets and triggers P-selectin expression and aggregation: a causal role in cardiovascular disease? Arterioscler Thromb Vasc Biol. 2003 Sep 1;23(9):1677–83. doi: 10.1161/01.ATV.0000084810.52464.D5. [DOI] [PubMed] [Google Scholar]
- 30.Kalvegren H, Andersson J, Grenegard M, Bengtsson T. Platelet activation triggered by Chlamydia pneumoniae is antagonized by 12-lipoxygenase inhibitors but not cyclooxygenase inhibitors. Eur J Pharmacol. 2007 Jul 2;566(13):20–7. doi: 10.1016/j.ejphar.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol Immunol. 2000 Dec;15(6):393–6. doi: 10.1034/j.1399-302x.2000.150610.x. [DOI] [PubMed] [Google Scholar]
- 32.Pham K, Feik D, Hammond BF, Rams TE, Whitaker EJ. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets. 2002 Feb;13(1):21–30. doi: 10.1080/09537100120104863. [DOI] [PubMed] [Google Scholar]
- 33.Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R, et al. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood. 2001 Jun 15;97(12):3790–7. doi: 10.1182/blood.v97.12.3790. [DOI] [PubMed] [Google Scholar]
- 34.Curtis MA, Macey M, Slaney JM, Howells GL. Platelet activation by Protease I of Porphyromonas gingivalis W83. FEMS Microbiol Lett. 1993 Jun 15;110(2):167–73. doi: 10.1111/j.1574-6968.1993.tb06315.x. [DOI] [PubMed] [Google Scholar]
- 35.Naito M, Sakai E, Shi Y, Ideguchi H, Shoji M, Ohara N, et al. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not Rgp proteinase. Mol Microbiol. 2006 Jan;59(1):152–67. doi: 10.1111/j.1365-2958.2005.04942.x. [DOI] [PubMed] [Google Scholar]
- 36.Montrucchio G, Bosco O, Del Sorbo L, Fascio Pecetto P, Lupia E, Goffi A, et al. Mechanisms of the priming effect of low doses of lipopoly-saccharides on leukocyte-dependent platelet aggregation in whole blood. Thromb Haemost. 2003 Nov;90(5):872–81. doi: 10.1160/TH03-02-0085. [DOI] [PubMed] [Google Scholar]
- 37.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005 Apr;83(2):196–8. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 38.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood. 2006 Jan 15;107(2):637–41. doi: 10.1182/blood-2005-06-2202. [DOI] [PubMed] [Google Scholar]
- 39.Shibazaki M, Nakamura M, Endo Y. Biphasic, organ-specific, and strain-specific accumulation of platelets induced in mice by a lipopolysaccharide from Escherichia coli and its possible involvement in shock. Infect Immun. 1996 Dec;64(12):5290–4. doi: 10.1128/iai.64.12.5290-5294.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007 Apr;13(4):463–9. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 41.Scott T, Owens MD. Thrombocytes respond to lipopolysaccharide through Toll-like receptor-4, and MAP kinase and NF-kappaB pathways leading to expression of interleukin-6 and cyclooxygenase-2 with production of prostaglandin E2. Mol Immunol. 2008 Feb;45(4):1001–8. doi: 10.1016/j.molimm.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 42.Cognasse F, Lafarge S, Chavarin P, Acquart S, Garraud O. Lipopolysaccharide induces sCD40L release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med. 2007 Feb;33(2):382–4. doi: 10.1007/s00134-006-0488-8. [DOI] [PubMed] [Google Scholar]
- 43.Cognasse F, Hamzeh-Cognasse H, Lafarge S, Delezay O, Pozzetto B, McNicol A, et al. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br J Haematol. 2008 Apr;141(1):84–91. doi: 10.1111/j.1365-2141.2008.06999.x. [DOI] [PubMed] [Google Scholar]
- 44.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006 Jul 1;108(1):167–76. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rumbaut RE, Bellera RV, Randhawa JK, Shrimpton CN, Dasgupta SK, Dong JF, et al. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a TLR4-dependent, neutrophil-independent mechanism. Am J Physiol Heart Circ Physiol. 2006 Apr;290(4):H1671–9. doi: 10.1152/ajpheart.00305.2005. [DOI] [PubMed] [Google Scholar]
- 46.Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, et al. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. 2009 Jun 15;182(12):7997–8004. doi: 10.4049/jimmunol.0802884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patrignani P, Di Febbo C, Tacconelli S, Moretta V, Baccante G, Sciulli MG, et al. Reduced thromboxane biosynthesis in carriers of toll-like receptor 4 polymorphisms in vivo. Blood. 2006 May 1;107(9):3572–4. doi: 10.1182/blood-2005-12-4811. [DOI] [PubMed] [Google Scholar]
- 48.Damas JK, Jensenius M, Ueland T, Otterdal K, Yndestad A, Froland SS, et al. Increased levels of soluble CD40L in African tick bite fever: possible involvement of TLRs in the pathogenic interaction between Rickettsia africae, endothelial cells, and platelets. J Immunol. 2006 Aug 15;177(4):2699–706. doi: 10.4049/jimmunol.177.4.2699. [DOI] [PubMed] [Google Scholar]
- 49.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009 Feb 13;104(3):346–54. doi: 10.1161/CIRCRESAHA.108.185785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, et al. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb Haemost. 2009 Jul;102(1):97–110. doi: 10.1160/TH08-08-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]