

Abstract
Water-insoluble regenerated silk materials are normally achieved by increasing β-sheet content (silk II). In the present study, water-insoluble silk films were prepared by controlling very slow drying of B. mori silk solutions, resulting in the formation of stable films with dominating silk I instead of silk II structure. Wide angle x-ray scattering (WAXS) indicated that the silk films stabilized by slow drying were mainly composed of silk I rather than silk II, while water- and methanol-annealed silk films had a higher silk II content. The silk films prepared through slow drying had a globule-like structure in the core with nano-filaments. The core region was composed of silk I and silk II, and these regions are surrounded by hydrophilic nano-filaments containing random, turns, and α-helix secondary structures. The insoluble silk films prepared by slow drying had unique thermal, mechanical and degradative properties. DSC results revealed that silk I crystals had stable thermal properties up to 250°C, without crystallization above the Tg, but degraded in lower temperature than silk II structure. Compared with water- and methanol-annealed films, the films prepared through slow drying achieved better mechanical ductility and more rapid enzymatic degradation, reflective of the differences in secondary structure achieved via differences in post processing of the cast silk films. Importantly, the silk I structure, a key intermediate secondary structure for the formation of mechanically robust natural silk fibers, was successfully generated in the present approach of very slow drying, mimicking the natural process. The results also point to a new mode to generate new types of silk biomaterials, where mechanical properties can be enhanced, and degradation rates increased, yet water insolubility is maintained along with low beta sheet content.
Keywords: silk I, silk II, water-insoluble, films
1. Introduction
Silk fibroin has been explored as a versatile protein biomaterial for the formation of films, fibers, microspheres, and porous scaffolds for various biomedical applications, due to biocompatibility, slow degradability and robust mechanical properties [1–7]. Regenerated silk-based materials are normally stabilized by the induction of β-sheet formation through the use of solvents [8, 9] or by physical stretching [10, 11]. The microstructure and porosity of these silk-based biomaterials, as well as features for cell interactions can be changed by chemical modifications, blending with biocompatible polymers or by exploiting various preparation processes. However further exploration is needed to address a broader range of biomedical needs, such as to reduce beta sheet content to promote more rapid degradability [12–14]. The beta sheets serve as physical crosslinks in silk-based biomaterials, providing mechanical stability, water insolubility and relating directly to rates of degradation via proteolytic action.
Water-insoluble silk films have been prepared with low β-sheet content [15]. These films were prepared in an all-aqueous process and degraded more rapidly when compared with the more traditional methanol-treated films. Thus, this approach satisfied some of the requirements for more rapidly degradabling biomaterials. These in vitro findings with proteolytic degradation, were later confirmed when these different processing methods were evaluated in vivo, where the aqueous processed silk sponges degraded within a few months in vivo, while solvent-processed sponges began to degrade over one year in vivo [16]. However, more studies are necessary to understand this water-annealing process and why these films degraded more rapidly as a result of water-annealing.
Thus, the goal of the present study was to gain more mechanistic insight into the role of water in silk material stabilization processes. In particular, the goal was to more closely mimic the natural process during silk processing, with silk fibroin concentrated very slowly at room temperature to allow sufficient time for the high molecular weight fibroin chains (almost 400 kDa in size) to self-assemble. The goal was to generate silk-based materials with a high silk I (hydrated type II beta turn crystal structures) [10,17] content and low β-sheet (beta sheet crystals) content, similar to the concentrated silk solution in the middle part of glands before spinning by silkworms and spiders.
2. Experimental Methods
2.1 Materials
Cocoons of Bombyx mori silkworm silk were kindly supplied by Tajima Shoji Co (Yokohama, Japan). Protease XIV (bacterial, from Streptomyces griseus, 5.9 units/mg solid) was purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were also from Sigma-Aldrich (St. Louis, MO)
2.2 Preparation of silk solutions
B. mori silk-fibroin solutions were prepared according to our previously published procedures [18]. Cocoons were boiled for 20 min in an aqueous solution of 0.02 M Na2CO3, and then rinsed thoroughly with distilled water to extract the sericin proteins. After drying, the extracted silk fibroin was dissolved in 9.3M LiBr solution at 60°C for 4 h, yielding a 20% (w/v) solution. This solution was dialyzed against distilled water using Slide-a-Lyzer dialysis cassettes (Pierce, MWCO 3,500) for 72 h to remove salt. The solution was optically clear after dialysis and was centrifuged to remove the small amount of silk aggregates that formed during the process. The final concentration of aqueous silk solution was ~7.5 wt%, determined by weighting the remaining solid after drying. The silk fibroin solution was then diluted to 4% and 2% with water, respectively.
2.3 Film formation
To prepare silk films, 1.5 ml of silk solution was cast on polystyrene Petri dishes (diameter 30 mm). Without lids, silk solution generally dried within 12h, forming water-soluble films at room temperature. In this study, serials of lids with different holes were prepared and covered on dishes to control the drying rate. The area of each hole was 3.13 mm2 and the drying area was controlled through changing the number of holes. With the hood airflow maintained at 0.20 m/s, the drying time was related to silk concentration and the number of holes (hole surface area). The relationship between the number of holes, silk concentration and drying time is shown in Table 1. When silk concentration was above 4% and the number of holes was decreased to below 12 (the percentage of opened area <5.32%), the drying time would be above 48 h for water-insoluble silk films to form. The water-insoluble silk films formed from 7.6% silk solution with a dying time of 3 days was investigated as the primary focus in the study because the silk film had the best mechanical ductility under these conditions. As controls, water-soluble silk film was prepared directly through drying silk solutions on the dishes without lids. All the films were prepared at room temperature. Water-insoluble films were also prepared with the water-annealing and methanol-annealing methods, to serve as controls with the new slow drying method. In water-annealing process, the soluble silk film was placed in a water-filled desiccator with a 25 in. Hg vacuum for 6h to produce a water-insoluble film. In methanol-annealing process, the soluble silk film was immersed in 80% methanol solution for about 1h to increase β-sheet content. The water-insoluble silk film prepared by slow drying was termed SD-SF, while the water-soluble, water-annealed and methanol-annealed films were termed S-SF, WA-SF and MA-SF, respectively.
Table 1.
The relationship between the percentage of the opened area, silk concentration and the drying time and the stability of silk films through changing the drying rate (air flow 0.20 m/s)
| The percentage of the he opened (hole) area (%) | Silk Concentration |
|||||
|---|---|---|---|---|---|---|
| 2 wt% | 4 wt% | 7.6 wt% | ||||
| Drying time (day) | Stability | Drying time (day) | Stability | Drying time (day) | Stability | |
| 2.67 | 3 | Soluble | 4 | Insoluble | 5 | Insoluble |
| 4.00 | 2 | Soluble | 3 | Insoluble | 3 | Insoluble |
| 5.32 | 1.5 | Soluble | 2 | Soluble | 2 | Insoluble |
| 100 | 0.5 | Soluble | 0.5 | Soluble | 0.5 | Soluble |
2.4 Wide angle X-ray scattering (WAXS)
To investigate the crystalline structure of the silk films, WAXS experiments were performed in transmission mode at Brookhaven National Laboratory, National Synchrotron Light source (NSLS) at the X27C beam line. The wavelength, λ, was 0.1371 nm and the scattering vector, q (q = 4πsinθ/λ, for θ the half-scattering angle) was calibrated using aluminum oxide. The film samples were encapsulated using high temperature Kapton™ tape. Intensity was accumulated every 30 s, and then scattering patterns were recorded every 120 s in stepwise annealing experiments. The intensity was finally corrected for changes in the incident beam intensity, sample absorption, and Kapton™ background.
2.5 FTIR Analysis
The structure of the films was also analyzed by FTIR on a JASCO FTIR 6200 spectrometer (JASCO, Tokyo, Japan) equipped with a MIRacle™ attenuated total reflection (ATR) Ge crystal cell in reflection mode. For each measurement, 32 scans were coded with resolution 4 cm−1, with the wave number ranging from 400–4000 cm−1. Fourier self-deconvolution (FSD) of the infrared spectra covering the amide I region (1595–1705 cm−1) was performed by Opus 5.0 software to identify silk secondary structures [19]. Deconvolution was performed using Lorentzian line shape with a half-bandwidth of 25 cm−1 and a noise reduction factor of 0.3. FSD spectra were curve-fitted to measure the relative areas of the amide I region components [19].
2.6 Differential scanning calorimetry (DSC)
The thermal property of the silk films was measured in a TA Instrument Q100 DSC (TA Instruments, New Castle, DE) with a dry nitrogen gas flow of 50ml/min. Temperature-modulated differential scanning calorimetry (TMDSC) measurements were performed using a TA instrument Q100, equipped with a refrigerated cooling system. The samples were heated at 2°C/min from −30°C to 350°C with a modulation period of 60 s and temperature amplitudes of 0.318°C.
2.7 Thermogravimetric Measurements
The water content in different silk films was determined from thermogravimetric measurements (TGA), using a TA 500Q system. Samples were heated to 550°C with a step increase of 2°C/min under an inert nitrogen atmosphere.
2.8 Silk degradation
Water-insoluble films prepared through slow drying were incubated at 37°C in 40 ml PBS buffer containing 0.23U/ml protease XIV. Each solution contained an approximately equivalent mass (40±5 mg) of silk film. Solutions were replenished with enzyme and samples were collected daily. At designated time points, groups of samples were rinsed in distilled water and prepared for mass balance assessment. Samples without enzyme but in PBS served as controls.
2.9 SEM Imaging
The cross-sectional microstructure of different silk films was studied. Specimens were fractured in liquid nitrogen to avoid the deformation and sputter-coated with platinum, and then examined using a Zeiss Supra 55 VP SEM (Oberkochen, Germany).
2.10 Mechanical properties
The tensile properties of specimens (3.17 mm×25 mm×0.2 mm) was measured with a crosshead speed of 2 mm min−1 using an Instron 3366 testing frame (Instron, Norwood, MA) with a 10N capacity load cell. Samples were hydrated in 0.1 M phosphate buffered saline (PBS) for approximately 30 min before clamping and submerging in a temperature-controlled Biopuls bath (37±0.3°C) filled with PBS for at least 5 min prior to testing. The linear elastic modulus was calculated using a least-squares fitting between 0.1 N load and 2.5% strain past that point. Ultimate tensile strength was the highest stress value attained during the test and the elongation of failure was the last data point before a >10% decrease in the load.
3. Results and Discussion
3.1 Structure characteristics
Changes in the structure of silk films prepared with the various treatments were determined by NMR, WAXS and FTIR-ATR. In previous studies, three silk fibroin conformations have been identified by X-ray diffraction, NMR, and infrared spectroscopy: random coil, α-form (silk I, type II β-turn) and β-form (silk II, anti parallel β-pleated sheet) [10, 20]. The corresponding d-spacings for silk I and II are as follows (in nanometers): 0.98 (II), 0.74 (I), 0.56 (I), 0.48 (II), 0.44 (I), 0.43 (II), 0.41 (I), 0.36 (I), 0.32 (I), 0.28 (I) [10]. In these d-spacings, the 0.74 nm peak occurs in a region of scattering space well removed from peaks found in the silk II structure [21]. Therefore, the peak at spacing near 0.72 nm is a strong evidence for silk I structure.
Fig. 1 shows the WAXS data of S-SF, SD-SF, WA-SF and MA-SF samples. The soluble silk film (S-SF) showed an amorphous state, characterized by the presence of a broad peak in the two-theta scattering angle range (for wavelength 0.1371 nm) from 10.5 to 25°. Different from S-SF, other water-insoluble films exhibited typical crystal structure. The SD-SF film showed typical X-ray diffractograms of the silk I structure, having diffraction peaks at 11.3, 14.4, 18.0, 19.8, 20.4, 22.2, 25.4 and 29.4°, corresponding to silk I crystalline spacing of 0.70, 0.55, 0.44, 0.40, 0.39, 0.36, 0.31 and 0.27 nm. No typical diffraction peaks of silk II were found in SD-SF film.
Figure 1.


Wide angle X-ray scattering (WAXS) data of silk films prepared through different processes: (a) Water-soluble silk film, S-SF; (b) Methanol-annealed silk film, MA-SF; (c) Water-annealed silk film, WA-SF; (d) Water-insoluble silk film through slow drying, SD-SF.
Water-annealed silk films (WA-SF) were characterized by diffraction peaks at two-theta values of 11.3, 18.0, 18.7, 22.2, 25.4 and 29.0°, corresponding to silk I and silk II crystalline d spacings of 0.70 (I), 0.44 (I), 0.42 (II), 0.36 (I), 0.31 (I) and 0.27 nm (I), respectively. The results indicate that silk I and silk II both formed after water-annealing. MA-SF also showed silk I and silk II crystal structures, having four diffraction peaks at 18.0, 18.6, 22.2 and 29.0°. Compared with WA-SF, the silk I peaks at 10.9 and 25.0° disappeared and the strength of silk II peaks at 18.7° increased, indicating more silk II formed in MA-SF. Interestingly, water-insoluble silk films gradually changed their crystal structure from silk I to silk II through slow drying, water annealing and methanol annealing processes. The results indicated that water had significant influence on the formation of silk I structure.
Structural changes in silk films after different processes were confirmed by FTIR-ATR (Fig. 2). The IR spectral region within 1700–1500 cm−1 is assigned to the peptide backbone of amide I (1700–1600 cm−1) and amide II (1600–1500 cm−1) absorption, which have been commonly used for the analysis of different secondary structures of silk fibroin. The peaks at 1610–1630 cm−1 (amide I) and 1510–1520 cm−1 (amide II) are characteristic of silk II secondary structure [19]. Although some researchers suggested that the absorptions at 1648–1554 cm−1 (amide I) and 1535–1542 cm−1 were indicative of random coil structure [22], more studies have revealed that these peaks were corresponded to silk I conformation [15, 23]. In the present study, the amide I band for S-SF showed one strong peak at 1644 cm−1, corresponding to random coil. Upon methanol treatment, the amide I band showed one strong peak at 1622 cm−1 with a shoulder at 1652 cm−1, while in water-annealed sample (WA-SF) one peak at 1652 cm−1 with a shoulder at 1622cm−1 appeared. When formed through slow drying, silk film (SD-SF) exhibited strong absorption at 1652 cm−1 with a minor shoulder at 1622 cm−1. The same trend in structural change was also found in the amide II region. From MA-SF, WA-SF to ST-SF, the peak at 1515 cm−1 (silk II) decreased, while the peak at 1539 cm−1 (silk I) increased. The FTIR results are consistent with WAXS results, confirming that the secondary structure of silk could be controlled through changing the processing utilized – slow drying, water annealing or methanol annealing.
Figure 2.



FTIR spectra of silk films prepared by different processes: (a) Water-soluble silk film, S-SF; (b) Methanol-annealed silk film, MA-SF; (c) Water-annealed silk film, WA-SF; (d) Water-insoluble silk film through slow drying, SD-SF.
The secondary structure of silk films formed under different processes was further characterized by deconvolution of the IR spectra [19]. The relative ratios of different secondary conformations were calculated from amide I region (Table 2). S-SF had the highest content of random coil and turns, 29.5%, and 28.2%, respectively. When prepared through slow drying as well as after methanol or water treatments, random and turns transformed to silk I or silk II crystal structures, inducing the water-stability. Upon methanol treatment, the silk II content of insoluble films (MA-SF) increased from 20.0% to 40.8% while the silk I content (11.8%) was similar to that of soluble silk films (11.7%). Once prepared through slow drying, the silk I content increased from 11.8% to 18.5% while the silk II content increased slightly to 22.7%. The results indicate that the water-stability of MA-SF is due to the formation of silk II, while that of SD-SF comes from the formation of silk I. After water-annealing treatment, the increase of both silk I and silk II, from 11.8 and 20.0% to 16.0 and 30.3%, respectively, resulted in the water-stability. The development of crystalline structure in silk fibroin using osmotic stress has been studied [10, 17]. Silk I was a hydrated structure and the crystalline structure could be controlled by changing the ratio of lithium bromide, silk and water [17]. In the present study, the crystalline structure of silk was controlled by changing the content of water without lithium bromide. From methanol treatment, water treatment to slow drying processes, the water content in these processes gradually increased, inducing more silk I formation.
Table 2.
The relative ratio of secondary structures in water-soluble films and water-insoluble films prepared through different processes. (S-SF, soluble silk film; SD-SF, silk film prepared through slow drying; WA-SF, water-annealed silk film, and MA-SF, methanol-annealed silk film. N=3, average ± standard deviation)
| Assignment | S-SF (%) | SD-SF (%) | WA-SF (%) | MA-SF(%) |
|---|---|---|---|---|
| Side chains (1590–1605 cm−1) | 2.4±0.3 | 1.7±0.1 | 2.7±0.3 | 3.1±0.4 |
| Silk II, β-sheet (1610–1635 cm−1, 1695–1700 cm−1) | 20.0±0.3 | 22.7±0.2 | 30.3±0.7 | 40.8±2.1 |
| Random coil (1635–1645 cm−1) | 29.5±0.5 | 23.5±0.1 | 22.4±0.5 | 18.4±1.1 |
| Silk I, Type II β-turn (1647–1654 cm−1) | 11.8±0.6 | 18.5±0.3 | 16.0±0.4 | 11.7±1.3 |
| α-helix (1658–1664 cm−1) | 8.1±0.9 | 11.6±0.3 | 6.8±0.8 | 6.0±0.7 |
| Turns and bends (1666–1695 cm−1) | 28.2±1.1 | 22.0±0.6 | 21.8±1.3 | 20.0±1.5 |
In the natural silk spinning process, the silk I structure is considered a necessary intermediate for the pre-organization or pre-alignment of silk fibroin molecules [17]. The control of silk secondary structure has been studied with pH and ions [24, 25], however structures similar to natural concentrated silk solutions have not been fully recapitulated using regenerated silk fibroin. In the present study, silk I crystal structures were directly formed when silk fibroin was concentrated slowly to allow sufficient time for chain self-assembly. The WAXS data of SD-SF was almost the same as that of concentrated silk solution from the middle part of silkworm glands before spinning by silkworms [26]. More importantly, the silk I crystal structure was formed in aqueous neutral solution at room temperature. The result reveals that it is an inherent ability for silk fibroin to self-assemble to silk I structure in concentrated aqueous solution, although other factors such as ionic strength, temperature and pH can affect the self-assembly process [27–29].
3.2 Thermal stability
Fig 3A illustrates standard DSC curves of S-SF, SD-SF, WA-SF, and MA-SF films. S-SF films showed an endothermic peak at around 100°C, a nonisothermal crystallization peak at around 213°C and a degradation peak at 257°C. The endothermic peak at around 100°C is contributed by melt and evaporation of bound water and indicated that S-SF interacted with water. At the crystallization peak, unstable non-crystal structures transformed to β-sheet. After the appearance of the crystallization peak, the film started to degrade, with an endothermal peak at around 257°C. When annealed with methanol, the film (MA-SF) crystallized to form β-sheets. The endothermic peak at around 100°C disappeared, implying that the silk film became hydrophobic. The crystallization peak also disappeared because of the formation of β-sheet with methanol treatment prior to DSC scanning. The degradation peak increased to 260°C, meaning MA-SF had higher thermal stability of MA-SF than S-SF. Besides β-sheet crystals, silk I crystals formed in WA-SF and SD-SF. The endothermic peak at around 100°C was still maintained because the silk I crystal is a hydrated structure [17]. The crystallization peak disappeared, implying that silk I was a stable crystal, never transforming to β-sheet above the glass transition temperature, Tg. Both WA-SF and SD-SF showed two degradation peaks in the region from 240 to 260°C. The peaks of WA-SF appeared at 247 and 257°C while that of SD-SF appeared at 252 and 259°C. Interesting, the first degradation peak of WA-SF and SD-SF was lower than that of S-SF and MA-SF while the second degradation peak was similar with that of S-SF and MA-SF. Since β-sheet formed at the crystallization peak in S-SF and did not form in WA-SF and SD-SF before degradation, the degradation peak at 257–260°C should be related to stability induced by the β-sheets while the other degradation peak at 247–252°C belongs to the silk I structure.
Figure 3.


DSC and TGA curves of silk films prepared through different processes: (A) Standard DSC scans; (B) Temperature-modulated DSC scans, TMDSC; and (C) TGA curves. The samples are as follows: (a) Water-soluble silk film, S-SF; (b) Water-insoluble silk film prepared through slow drying, SD-SF; (c) Water-annealed silk film, WA-SF; (d) Methanol-annealed silk film, MA-SF.
According to the FTIR and WAXS results, the crystal structure of SD-SF was mainly composed of silk I while WA-SF had a higher β-sheet content. The main degradation peak was the first peak at 252°C for SD-SF, but changed to the second peak at 257°C for WA-SF. The result confirms that the silk I crystal degraded at around 250°C, and silk II (β-sheet) degraded at around 260°C. Therefore, silk I crystal is a reasonably stable structure in terms of materials and function, but is more easily degraded than silk II.
Fig 3B shows the reversing DSC curves of S-SF, SD-SF, WA-SF and MA-SF films measured by TMDSC. In our previous studies we reported observation of a lower glass transition, Tg (1). Our previous studies revealed that Tg(1) provided information about the removal process of bound water, indicating the interaction between silk and bound water. [30]. S-SF had small Tg(1), meaning that litter water formed strong interactions with silk though water present in S-SF films as shown in standard DSC. Tg(1) formed in both WA-SF and SD-SF, and higher heat capacity at Tg(1) was achieved in the SD-SF films. Considering that SD-SF had a higher content of silk I, the stronger interactions between water and silk might be due to the formation of the silk I structure. Tg(2) provides information about the stability of the non-crystalline domains. The Tg(2) of WA-SF and SD-SF appeared at around 190°C, higher than that of S-SF and MA-SF at around 180°C. This result suggests that the relaxation time distribution for conformational rearrangements has been shifted, requiring higher temperature to active this motion.
The TGA results confirmed that stronger water-silk interactions formed following the increase in silk I content (Fig 3C). Compared with soluble silk films (S-SF), the water content increased from 7.2% to 7.5% and 8.5% for the WA-SF and SD-SF materials, respectively, and decreased to 6.3% for MA-SF.
3.3 Degradation
To functionally assess the differences in film processing, in-vitro degradation of the water-instable silk films prepared by controlling the drying rate was assessed. In previous research the degradation of methanol annealed and water annealed silk films had been studied [15]. The results indicated that there was no weight change in water-annealed or methanol annealed silk films after 2 weeks in PBS solution, serving as controls. After incubation in protease XIV solution (5.6 U/ml) at 37°C for 24h, weight losses from the water annealed and methanol annealed silk films were about 20% [15]. In the current study, SD-SF dissolved slowly with about 30% mass lost during 2 weeks in PBS solution (Fig 4a). Incubated in protease XIV solution (5.6 U/ml) at 37°C for 24h, SD-SF formed irregular debris that could not be washed and weighed. Therefore, SD-SF was incubated in protease XIV solution with a lower enzyme concentration (2.3 U/ml) to slow the degradation kinetics. At 24h of enzyme exposure, the weight loss from the SD-SF films was 39% in low protease concentration (Fig 4b), higher than that of WA-SF and MA-SF in high protease XIV concentration (5.6 U/ml). The results indicate that silk films containing higher silk I content degrade more rapidly in comparison to higher silk II content, and the degradation time can be controlled by changing the secondary structure of the silk films.
Figure 4.



Enzymatic degradation of water-insoluble silk films prepared through slow drying. (a) Silk film was cultivated in PBS solution without protease; (b) Silk film was cultivated in protease XIV solution. N=5, bars represent standard deviation.
3.4 Morphology
Water-insoluble silk films with different treatments were composed of nano-filaments, while no specific nanostructures were found in S-SF. After methanol or water treatments, nano-filaments formed (Fig 5b,c). When water-insoluble silk films were prepared through slow drying, more complicated nano-structures formed. The nano-filaments assembled to form globules with sizes about 200–1,000 nm in diameter (Fig 5d). The nano-filaments insides globules sprayed from the center and were surrounded by random nano-filaments (Fig 6a,b). The random nano-filaments were entangled with each other and linked the globules together, to form a stable network structure. The diameter of the nano-filaments inside the globules was 10–20 nm, different from outside globules where the feature sizes were about 20–80 nm in diameter. These results suggest that nano-filaments inside and outside the globules might have different secondary structures.
Figure 5.
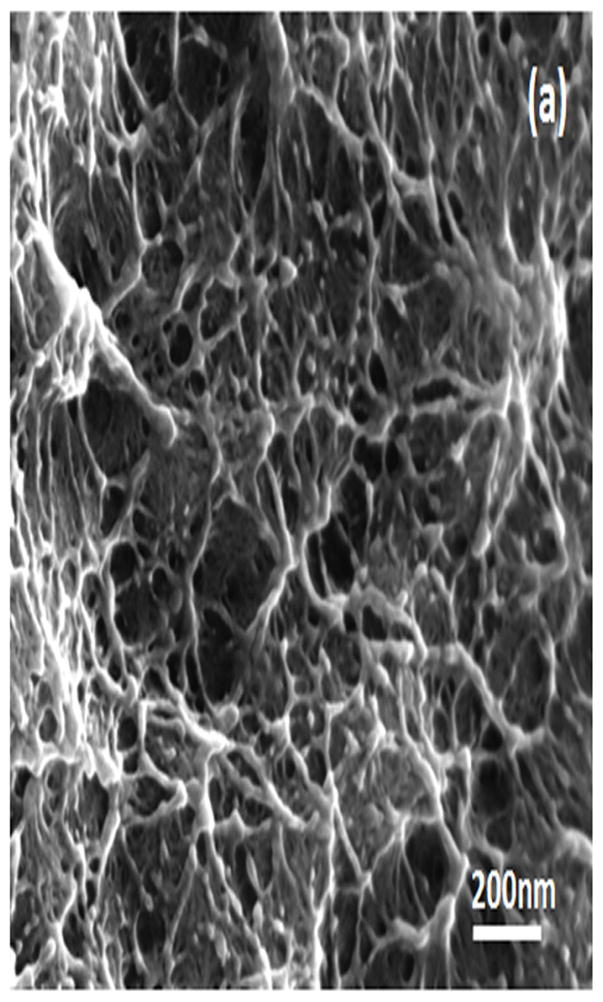
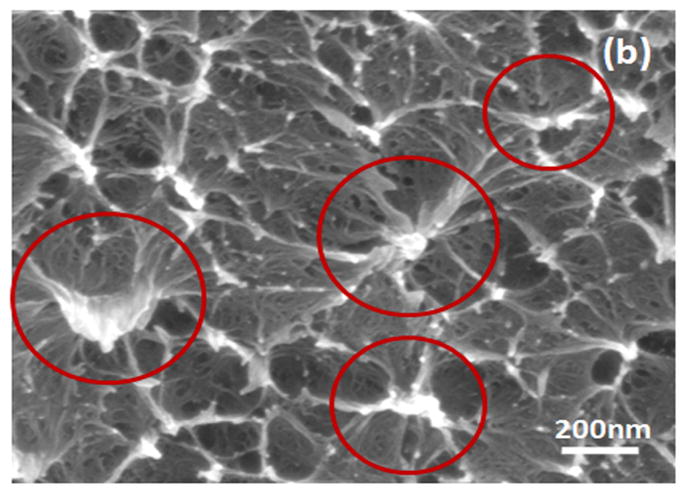
SEM images of silk films prepared through different processes: (a) soluble silk film, S-SF; (b) Methanol-annealed silk film, MA-SF; (c) Water-annealed silk film, WA-SF; (d) Water-insoluble silk film prepared through slow drying, SD-SF.
Figure 6.

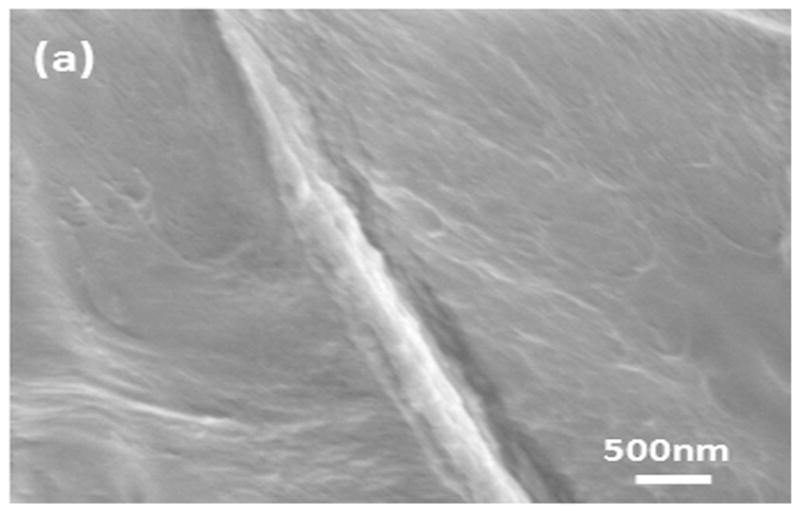
SEM images of the globules in silk film prepared through slow drying: (a) the nano-filament structure outside the globules; (b) the nano-filament structure inside the globules. The circles show the nanostructure inside the globules.
In order to investigate the differences in secondary structure in the nano-filaments, SD-SF films were incubated in protease XIV solution at 37°C to degrade the different nano-filaments separately. The nano-filaments outside the globules degraded first while the globules remained stable (Fig 7A(a–e)). Fig 7B shows the change in secondary structures during the degradation process. Following the degradation of the nano-filaments outside the globules, the non-crystalline structures (random coil, α-helix and turns) decreased while crystal structures (silk I and silk II) increased in the remaining samples. Although the exact content of secondary structures in that nano-filaments inside and outside the globules is not clear, the present reveals that nano-filaments outside globules are enriched in non-crystalline structures, while the features inside the globules are mainly composed of crystal structures. Water-insoluble silk films (SD-SF, WA-SF and MA-SF) were composed of nano-filaments, but only SD-SF further assembled into more complicated globule structures. A possible reason is that silk molecules have higher free mobility and space to assemble in SD-SF since SD-SF films formed in the aqueous state while MA-SF and WA-SF films were treated in the solid state. The higher mobility of silk molecules in the SD-SF forming process also means that controlling the secondary structure would be easier to regulate by controlling the drying rate, rather than with methanol annealing or water annealing processes. The present study also provides additional support for our previous mechanism of silk [10]. Through mimicking the natural process in silk processing glands we successfully regenerated the formation of silk I crystalline content. The silk self-assembled to form globules and the globules were surrounded by and connected through nano-filaments enriched in non-crystalline structures (rich hydrophilic blocks). These hydrophilic blocks formed strong interactions with water, preventing premature β-sheet formation. More importantly, since the globules are connected with nano-filaments, separation was avoided in shearing or stretching structural transitions, effectively clarifying why globules elongate and align rather than separate in our mechanism.
Figure 7.

Morphology and secondary structure changes of silk films prepared through slow drying following degradation: (A) SEM images of fracture surface of silk film explosion to protease XIV solution for 6h (a–c) and 12h (d–e): (a) Globule structure at low magnification. The white arrow indicates the direction from the surface to the inside; (b) Nano-structures near the surface area of silk films at higher magnification; (c) Nano-structures inside of the silk films at higher magnification; (d) Globule structure at low magnification, incubation time 12 h; and (e) Nano-structure at higher magnification, incubation time 12h. (B) Secondary structure changes with degradation time. The content of secondary structures was calculated from the deconvoluted amide I band using the method of Fourier self-deconvolution. N=3, bars represent standard deviation.
3.5 Mechanical properties
The mechanical properties of the water-insoluble silk films (SD-SF, WA-SF and MA-SF) were measured in the wet state (Table 3). The SD-SF films were more ductile than water annealed or methanol annealed films, even though the tensile modulus and strength of SD-SF films were lower. Following the decrease in β-sheet content, ductility increased while strength decreased. Therefore, mechanical properties of silk films can be controlled by different processing approaches.
Table 3.
Mechanical properties of water-insoluble silk films prepared through different processes (N=3, average ± standard deviation)
| Silk film | Tensile modulus [MPa] | Tensile strength [MPa] | Elongation at break [%] |
|---|---|---|---|
| Slow drying (SD-SF) | 92±6 | 2.2±0.2 | 260±24 |
| Water annealing (WA-SF) | 102±17 | 3.1±0.5 | 200±20 |
| Methanol annealing (MA-SF) | 207±39 | 4.4±0.6 | 125±40 |
4. Conclusions
By controlling the drying rate, water-insoluble silk films can be prepared directly from aqueous silk solutions. Similar with the soluble silk films (20.0%), water-insoluble silk films had reduced β-sheet content (22.7%), compared with methanol and water annealed silk films (30.3% and 40.8%, respectively. Since with very slow drying silk fibroin has sufficient time to self-assemble into stable structures, microstructures composed of nano-filaments formed and resulted in improved water and thermal stability. WAXS results indicated that the films formed similar crystal structures to natural concentrated silk solutions found in the middle part of the glands of the silkworm. The reduced β-sheet and high silk I structure also resulted in silk films that degraded more rapidly than those generated by water annealing or methanol annealing, which would lend to an extended range of biomedical utility where more rapid degradation in vivo can be useful for some applications.
Acknowledgments
The authors thank the NSF, the AFOSR and the NIH for support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vepari C, Kaplan DL. Silk as a biomaterial. Prog Polym Sci. 2007;32:991–1007. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang XQ, Wenk E, Matsumoto A, Meinel L, Li CM, Kaplan DL. Silk microspheres for encapsulation and controlled release. J Control Release. 2007;117:360–370. doi: 10.1016/j.jconrel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 3.Karageorgiou V, et al. Porous silk fibroin 3-D scaffolds for delivery of bone morphologenetic protein-2 in vitro and in vivo. J Biomed Mater Res A. 2006;78A:324–334. doi: 10.1002/jbm.a.30728. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Jin HJ, Kaplan DL, Rutledge GC. Mechanical properties of electrospun silk fibers. Macromolecules. 2004;37:6856–6864. [Google Scholar]
- 5.Jin HJ, Park J, Valluzi R, Cebe P, Kaplan DL. Biomaterial films of Bombyx Mori silk fibroin with poly (ethylene oxide) Biomacromolecules. 2004;5:711–717. doi: 10.1021/bm0343287. [DOI] [PubMed] [Google Scholar]
- 6.Jiang CY, et al. Mechanical properties of robust ultrathin silk fibroin films. Adv Funct Mater. 2007;17:2229–2237. [Google Scholar]
- 7.Liu HF, Fan HB, Wang Y, Toh SL, Goh JCH. The interaction between a combined knitted silk scaffold and microporous silk sponge with human mesenchymal stem cells for ligament tissue engineering. Biomaterials. 2008;29:662–674. doi: 10.1016/j.biomaterials.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 8.Cao ZB, Chen X, Yao J, Huang L, Shao ZZ. The preparation of regenerated silk fibroin microspheres. Soft Matter. 2007;3:910–915. doi: 10.1039/b703139d. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Cao CB, Ma XL, Tang Y, Zhu HS. Preparation of non-woven mats from all-aqueous silk fibroin solution with electrospinning method. Polymer. 2006;47:6322–6327. [Google Scholar]
- 10.Jin HJ, Kaplan DL. Mechanism of silk processing in insects and spiders. Nature. 2003;424:1057–1061. doi: 10.1038/nature01809. [DOI] [PubMed] [Google Scholar]
- 11.Freddi G, Tsukada M, Beretta S. Structure and physical properties of silk fibroin/polyacrylamide blend films. J Appl Polym Sci. 1999;71:1563–71. [Google Scholar]
- 12.Jin HJ, Fridrikh SV, Rutledge GC, Kaplan DL. Electrospining Bombyx mori silk with poly (ethylene oxide) Biomacromolecules. 2002;3:1233–1239. doi: 10.1021/bm025581u. [DOI] [PubMed] [Google Scholar]
- 13.Lv Q, Hu K, Feng QL, Cui FZ. Fibroin/collagen hybrid hydrogels with crosslinking method: Preparation, properties and cytocompatibility. J Biomed Mater Res A. 2008;84A:198–207. doi: 10.1002/jbm.a.31366. [DOI] [PubMed] [Google Scholar]
- 14.Lu Q, Zhang SJ, Hu K, Feng QL, Cao CB, Cui FZ. Cytocompatibility and blood compatibility of multifunctional fibroin/collagen/heparin scaffolds. Biomaterials. 2007;28:2306–2313. doi: 10.1016/j.biomaterials.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 15.Jin HJ, Park J, Karageorgiou V, Kim UJ, Valluzzi R, Cebe P, Kaplan DL. Water-insoluble silk films with reduced β-sheet content. Adv Funct Mater. 2005;15:1241–1247. [Google Scholar]
- 16.Wang YZ, et al. In vivo degradation of three-dimensional silk fibroin scaffolds. Biomaterials. 2008;29:3415–3428. doi: 10.1016/j.biomaterials.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sohn Sk, Strey HH, Gido SP. Phase behavior and hydration of silk fibroin. Biomacromolecules. 2004;5:751–757. doi: 10.1021/bm0343693. [DOI] [PubMed] [Google Scholar]
- 18.Kim UJ, Park J, Kim HJ, Wada M, Kaplan DL. Three-dimensional aqueous-derived biomaterial scaffolds from silk fibroin. Biomaterials. 2005;26:2775–2785. doi: 10.1016/j.biomaterials.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 19.Hu X, Kaplan DL, Cebe P. Determining Beta-sheet crystallinity in fibrous proteins by thermal analysis and infrared spectroscopy. Macromolecules. 2006;39:6161–6170. [Google Scholar]
- 20.Demura M, Asakura T, Kuroo T. Immobilization of biocatalysts with Bombyx mori silk fibroin by several kinds of physical treatment and its application to glucose sensors. Biosensors. 1989;4:361–372. doi: 10.1002/bit.260330513. [DOI] [PubMed] [Google Scholar]
- 21.Anderson JP. Morphology and crystal structure of a recombinant silk-like molecule, SLP4. Biopoly. 1998;45:307–321. [Google Scholar]
- 22.Um IC, Kweon HY, park YH, Hudson S. Structure characteristics and properties of the regenerated silk fibroin prepared from formic acid. Int J Biol Macromol. 2001;29:91–97. doi: 10.1016/s0141-8130(01)00159-3. [DOI] [PubMed] [Google Scholar]
- 23.Wilson D, Valluzzi R, Kaplan D. Conformational transition in model silk peptides. Biophys J. 2000;78:2690–2701. doi: 10.1016/S0006-3495(00)76813-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Li, Chen X, Shao ZZ, Huang Y, Knight DP. Effect of metallic ions on silk formation in the mulberry silkworm, bombyx mori. J Phys Chem B. 2005;109:16937–16945. doi: 10.1021/jp050883m. [DOI] [PubMed] [Google Scholar]
- 25.Kim UJ, Park JY, Li CM, Jin HJ, Valluzzi R, Kaplan DL. Structure and properties of silk hydrogels. Biomacromolecules. 2004;5:786–92. doi: 10.1021/bm0345460. [DOI] [PubMed] [Google Scholar]
- 26.Kratky O, Schauenstein E, Sekora A. An unstable lattice in silk fibroin. Nature. 1950;165:319–320. [Google Scholar]
- 27.Dicko C, Kenney JM, Knight D, Vollrath F. Transition to a beta-sheet-rich structure in spidroin in vitro: the effects of pH and cations. Biochemistry. 2004;43:14080–14087. doi: 10.1021/bi0483413. [DOI] [PubMed] [Google Scholar]
- 28.Pyda M, Hu X, Cebe P. Heat capacity of silk fibroin based on the vibrational motion of poly (amido acid)s in the presence and absence of water. Macromolecules. 2008;41:4786–4793. [Google Scholar]
- 29.Terry AE, Knight DP, Porter D, Vollrath F. pH induced changes in the rheology of silk fibroin solution from the middle division of bombyx mori silkworm. Biomacromolecules. 2004;5:768–772. doi: 10.1021/bm034381v. [DOI] [PubMed] [Google Scholar]
- 30.Hu X, Kaplan DL, Cebe P. Dynamic protein-water relationships during β-sheet formation. Macromolecules. 2008;41:3939–3948. [Google Scholar]