

Abstract
Cidofovir (HPMPC) is a broad-spectrum antiviral agent, currently used to treat AIDS-related human cytomegalovirus retinitis. Cidofovir has recognized therapeutic potential for orthopox virus infections, although its use is hampered by its inherent low oral bioavailability. Val-Ser-cyclic HPMPC (Val-Ser-cHPMPC) is a promising peptide prodrug which has previously been shown by us to improve the permeability and bioavailability of the parent compound in rodent models (Eriksson et al. Molecular Pharmaceutics, 2008 vol 5 598-609). Puromycin-sensitive aminopeptidase was partially purified from Caco-2 cell homogenates and identified as a prodrug activating enzyme for Val-Ser-cHPMPC. The prodrug activation process initially involves an enzymatic step where the l-Valine residue is removed by puromycin-sensitive aminopeptidase, a step that is bestatin-sensitive. Subsequent chemical hydrolysis results in the generation of cHPMPC. A recombinant puromycin-sensitive aminopeptidase was generated and its substrate specificity investigated. The kcat for Val-pNA was significantly lower than that for Ala-pNA, suggesting that some amino acids are preferred over others. Furthermore, the three-fold higher kcat for Val-Ser-cHPMPC as compared to Val-pNA suggests that the leaving group may play an important role in determining hydrolytic activity. In addition to its ability to hydrolyze a variety of substrates, these observations strongly suggest that puromycin-sensitive aminopeptidase is an important enzyme for activating Val-Ser-cHPMPC in vivo. Taken together, our data suggest that puromycin-sensitive aminopeptidase makes an attractive target for future prodrug design.
Keywords: prodrug, cidofovir, puromycin-sensitive, aminopeptidase, bioavailability, antiviral
1. Introduction
Prodrugs of therapeutically active agents have rightfully been receiving increased attention. The term prodrug describes chemicals with little or no pharmacological activity that undergo biotransformation to yield a therapeutically active metabolite (Albert, 1958). The chemical changes involved in creating prodrugs are usually designed to improve one or more physio-chemical properties that are lacking in the parent drug. Thus, numerous prodrugs of therapeutic agents have been developed to improve their original pharmaceutical, biopharmaceutical, and pharmacokinetic properties. Reliable and predictable in vivo activation is a critical aspect of the prodrug strategy; therefore, identification of the mechanisms of their in vivo activation is important from a prodrug design perspective. Recently our laboratory identified human valacyclovirase as one of the enzymes responsible for activation of the valyl ester prodrug forms of acyclovir (valacyclovir) and ganciclovir (valganciclovir) (Kim et al., 2003). The valyl ester prodrugs have previously been shown to significantly increase their parent drugs' oral absorption (Curran and Noble, 2001; Perry and Faulds, 1996; Pescovitz et al., 2000; Smiley et al., 1996). In the case of valacyclovir, for example, the oral bioavailability of acyclovir was increased 3- to 5-fold (Weller et al., 1993). It has been shown that the observed increase in bioavailability of acyclovir when administered as its prodrug valacyclovir is due to carrier-mediated intestinal absorption of valacyclovir via the human peptide transporter 1 (hPepT1) (Balimane et al., 1998; Ganapathy et al., 1998; Han et al., 1998). Analogous to the valacyclovir case mentioned above, we have now identified a puromycin-sensitive aminopeptidase as one of the activating enzymes of an amino acid-containing prodrug of cidofovir.
Cidofovir (Vistide®, HPMPC, 1, Fig. 1) is an antiviral agent that is clinically used for treatment of the AIDS-related herpes virus infection, cytomegalovirus retinitis. It is a broad-spectrum antiviral agent with therapeutic potential in the treatment of other herpes and DNA viruses, including polyoma-, papilloma-, adeno-, and poxvirus infections (De Clerco, 1997; De Clercq and Holy, 2005). Cidofovir is of particular interest due to its potential use as therapy in the event of an outbreak of smallpox (De Clercq, 2002). Currently, the drawback of using cidofovir in a large-scale emergency situation is its need for intravenous administration. The phosphonic acid group of cidofovir is ionized under physiological conditions and contributes substantially to its observed low oral bioavailability (< 5%) (Cundy et al., 1996; Wachsman et al., 1996). In the event of a smallpox outbreak, it would be essential to be able to conveniently administer effective dosages via the preferable oral route. Our research has therefore been focused on synthesizing orally available prodrugs of cidofovir that are efficiently activated in vivo.
Fig. 1.

Chemical structures of cidofovir (1) and Val-Ser-cHPMPC (2).
We have recently reported several examples of novel cyclic cidofovir (cHPMPC) prodrugs incorporating dipeptides and ethylene glycol-linked amino acids onto the cidofovir scaffold (Eriksson et al., 2006; Eriksson et al., 2007; Eriksson et al., 2008; McKenna et al., 2005; McKenna et al., 2006). One of our lead prodrugs, Val-Ser-cHPMPC (2, Fig. 1), shows significantly enhanced intestinal uptake (18.1% versus 2.2% for cHPMPC) in an in situ rat perfusion model (Eriksson et al., 2008; McKenna et al., 2005). Interestingly, the majority (≥ 90%) of Val-Ser-cHPMPC was found to be converted to cHPMPC during in situ rat perfusion experiments (Eriksson et al., 2008). In cell culture-based assays of antiviral activity, Val-Ser-cHPMPC (IC50 = 0.3 μM) performed nearly as well as HPMPC (IC50 = 0.26 μM), worse than cHPMPC (IC50 < 0.1 μM) and significantly better than ganciclovir (IC50 = 3 μM) against human cytomegalovirus (Eriksson et al., 2008). Val-Ser-cHPMPC did not perform nearly as well as either of the parent compounds against vaccinia and cowpox viruses, although this observation may be at least partly attributable to the lack of activating proteases in vitro, as well as the significantly shorter (3 vs. 10 days) incubation times required for assaying the poxviruses (Eriksson et al., 2008). Since Val-Ser-cHPMPC undergoes substantial in vivo activation in the rat intestine, we are interested in investigating plausible human activation pathway(s) for Val-Ser-cHPMPC. Herein we are reporting the purification and identification of a prodrug activating enzyme, puromycin-sensitive aminopeptidase (GenBank accession no. CAA68964), and show that it is responsible for the in vitro activation of Val-Ser-cHPMPC in Caco-2 cell homogenates. A recombinant puromycin-sensitive aminopeptidase (hereafter abbreviated APP-S1, for aminopeptidase, puromycin-sensitive) has been produced and the kinetic constants (Km and kcat) of the nucleotide prodrug hydrolysis have been determined. In addition, the APP-S prodrug activating pathway has been verified and will be discussed. Given the importance of antiviral agents in pharmacotherapy, the identification of enzymes responsible for activation of phosphonate-containing prodrugs can provide important new targets for the design of more effective therapeutic agents.
2. Experimental Procedures
2.1. Chemicals and Reagents
Val-Ser-cHPMPC was synthesized as previously described (Eriksson et al., 2008). Cell culture reagents were obtained from Invitrogen/Gibco. EAH Sepharose 4B, Superdex-200, PD-10 and MonoQ columns were purchased from GE Healthcare. Bestatin, trifluoroacetic acid (TFA) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) were purchased from Sigma-Aldrich. Other chemicals were either ACS reagent grade, analytical or HPLC grade and purchased from ThermoFisher Scientific, Inc. unless otherwise noted.
2.2. Cell Culture
The human colon carcinoma cell line, Caco-2, was obtained from the American Type Culture Collection (ATCC HTB37, passage numbers 33-59). The cells were routinely maintained in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum, 25 mM D-glucose, 4 mM l-glutamine and 1 mM sodium pyruvate. The cells were grown in 150 mm tissue culture dishes, split every 6 days and seeded at 1.7 × 104 cells/ml. All cells were maintained in an atmosphere of 5% CO2 and 90% relative humidity at 37 °C.
2.3. Prodrug hydrolysis assay
The cell homogenates or subcellular fractions were preincubated with 10 mM sodium phosphate buffer (pH 7.4) for 3 minutes at 37 °C. Prodrug was added at a final concentration of 250 μM to initiate the enzymatic reaction. Samples were removed at predetermined time points (3-30 min) and quenched by the addition of 1-1.5 volumes of 10% ice-cold trifluoroacetic acid (TFA). The quenched samples were spun through 96-well 0.45 μm polyvinylidene difluoride (PVDF) membranes (Unifilter, Whatman GF/B) at 1,800 × g in a Jouan MR 22i tabletop centrifuge to remove the precipitates before HPLC analysis. The HPLC system (Waters) consisted of a reverse-phase column (XTerra RP18, 5 μm, 4.6 × 250 mm), a 515 pump, a 996 Photodiode Array UV detector and a 717 Plus Autosampler. The remaining prodrug and the production of parent drug as well as intermediate activation products were analyzed using a mobile phase consisting of 17 mM phosphate buffer containing 0.5 mM ion-pairing agent (tert-butyl ammonium dihydrogen phosphate) and 5% acetonitrile at the pH 7.3, with a flow rate of 1 ml/min and detection by absorbance at 274 nm. The specific activity of the homogenate was expressed as nanomoles/min·mg of protein based on the disappearance of the prodrug.
2.4. Purification of APP-S from Caco-2 cells
2.4.1. Caco-2 cell lysis and differential centrifugation
All centrifugations and column protein purifications were performed at 4 °C unless otherwise noted. Caco-2 cells from 20 confluent 150 mm-diameter dishes were washed once with phosphate-buffered saline (PBS) and harvested on ice by scraping with a rubber policeman using ice-cold PBS. The cells were pelleted 5 min at 1,000 × g and the supernatant discarded. Caco-2 cell homogenates were prepared in 10 mM phosphate buffer containing 0.25 M sucrose (pH 7.4) using a Dounce homogenizer. After achieving > 90% cell lysis by visual inspection (trypan blue staining), differential centrifugation was used to achieve various cell fractions. Briefly, the cell lysates were spun in an Allegra centrifuge (Beckman Coulture) for 10 min at 3,000 × g to give a pellet (P1) and a supernatant (S1). S1 was then centrifuged for 20 min at 10,000 × g → (P2) and (S2); S2 was centrifuged 30 min at 25,000 × g → (P3) and (S3); S3 was spun in a Beckman Coulture Optimax ultracentrifuge for 60 min at 100,000 × g → P4 and the final cytosolic fraction of the cell homogenate, which was stored at -80 °C until use. No protease or esterase inhibitors were added to the buffers throughout the purification scheme to avoid potential inhibition of the enzyme of interest.
2.4.2. Ammonium sulfate precipitation
Further concentration of the protein in the cytosolic fraction (final centrifugation supernatant; 174 mg total protein) was achieved with an ammonium sulfate precipitation at 4 °C (pH 7.4). The pellets containing the fractions responsible for the activation of Val-Ser-cHPMPC were found in the concentration range between 45-70% of (NH4)2SO4. The positive fractions were pooled and desalted using a PD-10 desalting column equilibrated with 50 mM Tris, pH 8.5 (buffer A) (55 mg total protein).
2.4.3. Bestatin affinity chromatography
The bestatin EAH Sepharose affinity column was synthesized according to the Acosta procedure (Acosta et al., 1998). Briefly, 6 ml of the EAH Sepharose 4B matrix were washed sequentially with a 10-fold volume excess each of water, 0.5 M NaCl, then water again (adjusted to pH 4.5 with HCl) and finally resuspended in 12 ml H2O (pH 4.5), to which was added an ∼45-fold molar excess of EDC (500 mg in 5 ml H2O). Bestatin (25 mg) was dissolved in 5 ml H2O and added dropwise over 3 hrs to the reaction suspension, and then incubated an additional 16 hrs at 25 °C with gentle agitation. The bestatin-modified Sepharose 4B beads were washed in the following order with 100 ml of 0.1 M sodium acetate containing 0.5 M NaCl (pH 4.0), 100 ml of 0.1 M Tris containing 0.5 M NaCl (pH 8.0), and 100 ml distilled H2O, before packing of the column. The Caco-2 cytosolic fraction (55 mg total protein) was run in the bestatin column equilibrated with 50 mM Tris, pH 8.5 and eluted with a stepwise gradient of 1 M NaCl (total 60 ml) in the same buffer at 1 ml/min. The active fractions, which eluted at a concentration of 0.25 M NaCl, were pooled and diluted with an equal volume of 20 mM Tris, pH 7 (buffer MQ) (5.1 mg total protein) and run in a MonoQ anion exchange column equilibrated in the same buffer. The MonoQ samples were eluted with 25 ml of MQ buffer followed by a gradient from 0 to 1 M NaCl in MQ (total 35 ml) at 0.5 ml/min. The active fractions were pooled together to generate 0.8 mg of protein.
Additional purifications from Caco-2 homogenates were conducted to achieve a total of 1.5 mg of protein. The combined protein samples were concentrated using a Centricon YM-3 centrifugal filter device (Amicon) and resolved in a Superdex-200 size exclusion column with 50 mM sodium phosphate containing 0.15 M NaCl, pH 6.8 at 0.4 ml/min.
2.4.4. SDS-PAGE analysis of purification
All purification steps were examined by 10% SDS-PAGE with Bio-Rad SDS-PAGE high molecular weight standards for the estimation of the molecular weight as described by Laemmli (Laemmli, 1970). The hydrolytic activity of each fraction was measured as described above (2.3. Prodrug hydrolysis assay), and the protein concentrations were based on the Lowry method (Lowry et al., 1951) using a Bio-Rad protein assay kit with bovine serum albumin as a standard.
2.5. Identification of the prodrug activating enzyme
The partially purified proteins from the active fractions of the Superdex-200 size exclusion chromatography were further resolved by SDS-PAGE and visualized by Coomassie blue staining. The protein band having an apparent molecular mass of ∼100 kDa was excised, digested with trypsin and analyzed in an ABI 4800 Proteome Analyzer (TOFTOF) mass spectrometer (Applied Biosystems) at the Michigan Proteome Consortium (www.proteomeconsortium.org), University of Michigan. The obtained amino acid sequence was used as a query for searching the non-redundant (nr) protein data base (NCBInr), and the protein of interest was identified with high confidence as NCBI accession number CAA68964 (Ion Score C.I. > 99%).
2.6. Generation of recombinant APP-S
2.6.1. Subcloning of APP-S cDNA
Human APP-S cDNA (IMAGE clone ID 6059589) in the mammalian expression vector pCMV-SPORT6 (Open Biosystems) was subcloned into the pET-28a vector (Novagen) for expression of the N-terminally His-tagged construct in E. coli. Briefly, the APP-S cDNA was excised from pCMV-SPORT6 and ligated into pET28a after digesting both with the restriction enzymes EcoRI and XhoI (New England Biolabs) and purifying by electrophoresis in a 1% agarose gel. The ∼2.8 kb band corresponding to APP-S and the ∼5.4 kb band corresponding to pET-28a were purified from the agarose using a QIAEX II gel extraction kit (QIAGEN) and ligated using T4 DNA ligase (New England Biolabs). To shift the inserted cDNA to the correct reading frame, one amino acid was inserted upstream of the APP-S cDNA using the primers 5′-GGCCTCGCCGCGAATGCCGGAG AAGAGG -3′ and 5′-CTCTTCTCCGGCATTCG CGGCGAGGCC -3′ (IDT) and QuikChange Site Directed Mutagenesis kit (Stratagene). The His-APP-S/pET-28a construct was then transformed into E. coli strain BL21-RIPL (Stratagene) followed by dideoxy sequencing (University of Michigan DNA Sequencing Core) to confirm the nucleotide sequence of the recombinant His-APP-S.
2.6.2. Recombinant APP-S expression and purification
His-APP-S protein expression was induced according to the method of Sengupta et al. (Sengupta et al., 2006) with modifications. Briefly, BL21-RIPL cells containing His-APP-S/pET-28a were grown to stationary phase in LB broth at 37 °C, and then expanded until cultures reached an optical density of 0.8-1.0 at 570 nm, at which point His-APP-S expression was induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) at 18 °C for 18-20 hr. Following centrifugation at 6,000 × g for 10 min at 4 °C, the cell pellet was resuspended in 50 mM sodium phosphate, 300 mM sodium chloride, and 20 mM imidazole, pH 8 (wash buffer) containing 1 mg/ml lysozyme (Sigma), followed by three cycles of freeze-thaw. After a 10 min incubation at 37 °C, the homogenate was pulsed for 30 seconds with a probe sonicator (Model KT40, Kontes) followed by centrifugation at 20,000 × g for 40 min. His-APP-S was purified from the supernatant using Ni-NTA agarose (QIAGEN), followed by washing with 100 bed volumes (∼ 250 ml) wash buffer. The recombinant APP-S was eluted with 100 U (∼15 μg) thrombin (GE Healthcare) in 1 ml PBS pH 7.4 for 18 hours at 25 °C with gentle agitation, which also served to remove the His tag. The thrombin and APP-S were separated from the Ni-NTA agarose by spinning at 1,500 × g for 5 min and the supernatant transferred to a clean tube, after which the Ni-NTA agarose was washed 3 × 1 ml with PBS, pH 7.4 and all four supernatants were combined. Thrombin was removed by incubating with 400 μl p-aminobenzamidine agarose (Sigma; binding capacity 4-8 mg thrombin) for 2 hours at 25 °C, followed by pelleting of the p-aminobenzamidine agarose at 1,500 × g. Protein concentration and relative purity were determined using the BCA assay (Pierce) and SDS-PAGE, respectively, after which the purified APP-S was aliquoted and stored at -80 °C.
2.7. Prodrug hydrolysis by recombinant APP-S
2.7.1. HPLC assay of metabolites
Recombinant APP-S (30 μg/ml) was preincubated with and without the inhibitor bestatin (Fluka, 20 μg/ml) in 10 mM HEPES, 100 mM NaCl (pH 7.4) for 5 minutes at 37 °C. Prodrug was added at final concentrations ranging from 0.125 to 1 mM to initiate the enzymatic reaction. Aliquots of 40 μl were removed at predetermined time points (0-15 min) and quenched by the addition of 80 μl of 10% ice-cold TFA. The samples were prepared and analyzed by HPLC essentially as described above for the prodrug hydrolysis assays using Caco-2 samples, except that the HPLC conditions consisted of an acetonitrile gradient (2-52%) mobile phase with a flow rate of 1 ml/min and detection at 274 nm. The specific activity was expressed as nanomoles/min·mg of protein based on the disappearance of the prodrug.
2.7.2. LC-MS identification of the metabolites
Further identification of the APP-S hydrolysis products was achieved on a Finnigan LCQ Deca XP Max mass spectrometer in positive mode with a Finnigan Surveyor PDA Plus detector and MS Pump Plus, all controlled using Xcalibur software. The samples (20 μl injections) were resolved in a Varian Microsorb-MV C-18 column (100-5, 250 × 4.6 mm) with UV detection at 274 nm. The eluent was diverted immediately before the mass spectrometer, such that only half of the flow was injected into the MS. The mobile phases consisted of 0.1 N ammonium acetate buffer, pH 5.5 containing either 0% acetonitrile (A) or 17.5% acetonitrile (B), run at 1 ml/min. Mobile phase gradients consisted of 100% A/ 0% B for 5 min, 50% A/ 50% B at 6 min (or 25% A/ 75% B at 7 min for prodrug alone samples), 20% A/ 80% B from 15 to 20 min.
2.7.3. APP-S hydrolysis of model substrates
The ability of APP-S to hydrolyze various amino acids was tested using the chromogenic substrates l-valine p-nitroanilide and l-alanine p-nitroanilide (Bachem). Recombinant APP-S was pre-incubated in PBS, pH 7.4 with and without 20 μg/ml bestatin in a final volume of 300 μl. The reaction was started by adding 0.05 – 1.6 mM substrate dissolved in dimethyl sulfoxide (DMSO) and carried out at 37 °C. The production of p-nitroaniline was measured spectrophotometrically at 405 nm every 30 sec for 15 min. The concentration of p-nitroaniline was determined using the Beer-Lambert equation (ε405 = 9500 L mol-1 cm-1). Km and Vmax were calculated for each substrate using GraphPad Prism 4.
3. Results
3.1. Purification of the prodrug activating enzyme
Since our initial perfusion studies showed that the activation step most likely occurred in or in the vicinity of the epithelial cells in the gastrointestinal tract of the rat (Eriksson et al., 2008), we decided to use Caco-2 cells as our protein source for identification of the human activating enzyme for Val-Ser-cHPMPC. The chemical as well as the enzymatic hydrolysis of Val-Ser-cHPMPC was investigated. The disappearance of Val-Ser-cHPMPC was observed and the half-life (t½) of the prodrug in the two systems (buffer and homogenate) were calculated from assuming first-order kinetics and the rate constant was derived from linear regression (r2 = 0.95) of first-order plots of prodrug concentration versus time. The half-life of Val-Ser-cHPMPC in the buffer system (10 mM PBS, pH 7.4) was determined to be 108.2 (±13.3) minutes compared to 13.6 (±3.6) minutes for the cytosolic fraction of the Caco-2 cell homogenate (2000 μg/ml protein concentration).
Interestingly, a new prodrug activation peak appeared in the Caco-2 cell homogenate HPLC-chromatogram that was solely observed in the Caco-2 cell system. The activation peak was analyzed by mass spectrometry and determined to correspond to Ser-cHPMPC (data not shown), and later confirmed with purified recombinant APP-S. The formation of this activation peak can be inhibited by the addition of bestatin, an aminopeptidase inhibitor, whereupon the half-life of the prodrug in the presence of cell homogenate increased to 100.1 (±1.6) minutes. Preliminary data showed that other protease inhibitors did not affect the half-life of Val-Ser-cHPMPC in the presence of Caco-2 cell homogenates to nearly the same extent as bestatin: 1 mM AEBSF (t½ = 56 min), 10 μg/ml aprotinin (t½ = 14 min), 10 μg/ml leupeptin (t½ = 25 min), 10 μg/ml pepstatin A (t½ = 18 min) or 10 μg/ml E-64 (t½ = 25 min), consistent with previously published data (Sengupta et al., 2006). We utilized these results and the knowledge that bestatin is a reversible inhibitor to make our purification procedure from the cytosolic fraction of the Caco-2 cell homogenate more efficient. By incorporating an FPLC step containing a bestatin-modified column (an in-house synthesized affinity column) in between the ammonium precipitation and the MonoQ anion exchange column, the partial purification procedure for APP-S was greatly enhanced.
The specific activity (SA) from the active fractions obtained following the MonoQ column was determined to be 142 nmol/min·mg protein, corresponding to a 22-fold purification from the initial cytosolic cell homogenate (Table 1). Interestingly, when Val-Ser-cHPMPC was incubated for a prolonged time period (2 hr) at 37 °C in the presence of the active fractions from the MonoQ column, the activation peak corresponding to Ser-cHPMPC disappeared.
Table I.
Partial purification scheme of APP-S from Caco-2 cells. Caco-2 cell homogenate was sequentially purified by ammonium sulfate precipitation, bestatin affinity chromatography, and MonoQ anion exchange chromatography. Following each purification step, the active fraction was incubated with Val-Ser-cHPMPC at 37 °C (10 mM sodium phosphate buffer, pH 7.4) with aliquots taken at predetermined time points (3-30 min). Disappearance of the prodrug peak was monitored by HPLC (detection at 274 nm) and used to calculate specific activity (nmol/min·mg protein) and half-life (min).
| Purification step | Total protein | Specific activity | Half-life of Val-Ser-cHPMPC | Purification |
|---|---|---|---|---|
| mg | nmol/min·mg protein | min | -fold | |
| Cytosolic fraction | 174 | 6.8 | 13.6 | 1.0 |
| (NH4)2SO4 precipitation | 55 | 11.2 | 7.2 | 1.7 |
| Bestatin column | 5.1 | ND a | ND | ND |
| MonoQ column | 0.8 | 142.5 | 4.6 | 22 |
ND = not determined
3.2. Identification of the prodrug activating enzyme
The active fractions following MonoQ and size-exclusion chromatography (lane 4-6, Fig. 2) mentioned above were collected and analyzed by 10% SDS-PAGE and Coomassie staining. By visually comparing the SDS-PAGE gels through the purification process, deducting bands present in non-active fractions from bands present in the active fractions, the band at ∼100 kDa was identified exclusively to be present in the active fractions. An SDS-PAGE gel was submitted to the Michigan Proteome Consortium for MS/MS analysis, and the identity of the band was determined with high confidence to be APP-S (NCBI accession number CAA68964).
Fig. 2.
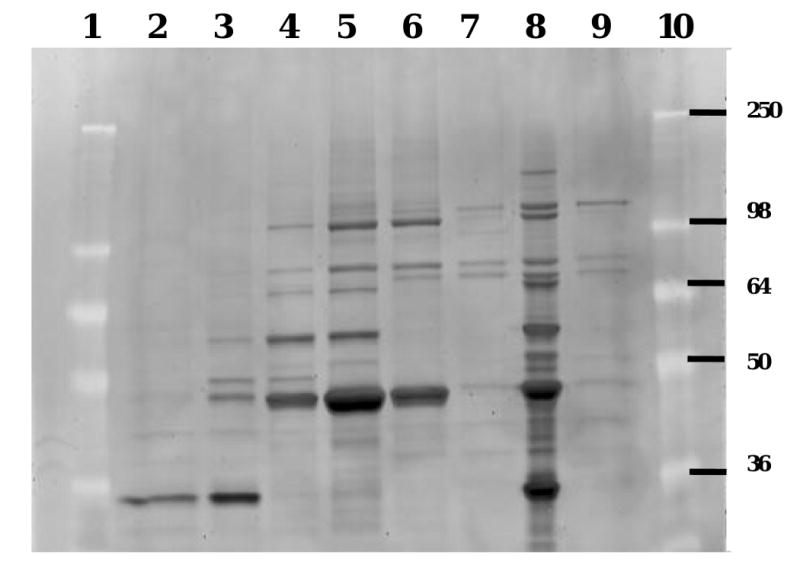
Superdex-200 purification of puromycin-sensitive aminopeptidase from Caco-2 cell homogenates. Active and non-active fractions from the Superdex-200 purification were analyzed by 10% SDS-PAGE, here stained with SYPRO Red. Lanes 4-6 contain active fractions hydrolyzing Val-Ser-cHPMPC, while lanes 2, 3, 7 and 9 are non-active fractions. Lane 8 corresponds to the pooled active MonoQ fractions that were initially applied to the Superdex-200 column. Lanes 1 and 10 are size markers with molecular mass expressed in kDa. The band visible at ∼ 100 kDa in lane 4-6 and 8 was exclusively present in the active fractions and its identity was determined by tandem mass spectrometry and database searches to be puromycin-sensitive aminopeptidase (APP-S).
3.3 Recombinant APP-S hydrolysis investigations
To confirm that APP-S is an activating enzyme of Val-Ser-cHPMPC, the APP-S cDNA (Open Biosystems) was subcloned into the bacterial expression vector pET-28a (Novagen). Recombinant His-tagged APP-S was purified to > 98% purity (Fig. 3) using Ni-NTA agarose (QIAGEN) and thrombin cleavage to remove the His-tag. The purified recombinant APP-S migrated as two bands in SDS-PAGE when eluted from Ni-NTA by thrombin digestion (Fig. 3), but as a single band when eluted with imidazole (data not shown). Both thrombin-eluted bands were identified as APP-S by peptide mass analysis at the University of Michigan Proteome Core, and the presence of a potential thrombin-cleavable sequence, in addition to the expected pET-28a vector thrombin cleavage site, was subsequently identified at amino acid 15 in APP-S's N-terminus, consistent with the ∼1.5 kDa MW difference observed in these gels. The activity of purified recombinant APP-S was first analyzed using the model substrates l-alanine p-nitroanilide (Ala-pNA) and l-valine p-nitroanilide (Val-pNA) in the presence and absence of the inhibitor bestatin. Val-pNA was shown to have a lower Km than Ala-pNA (0.28 ± 0.19 mM vs 0.51 ± 0.14 mM, respectively) and a greater than 18-fold lower Vmax (289 ± 85.1 nmol/min·mg protein vs 5,365 ± 610.0 nmol/min·mg protein, respectively) as shown in Table 2. APP-S hydrolysis of the model substrates was completely inhibited by the addition of bestatin.
Fig. 3.
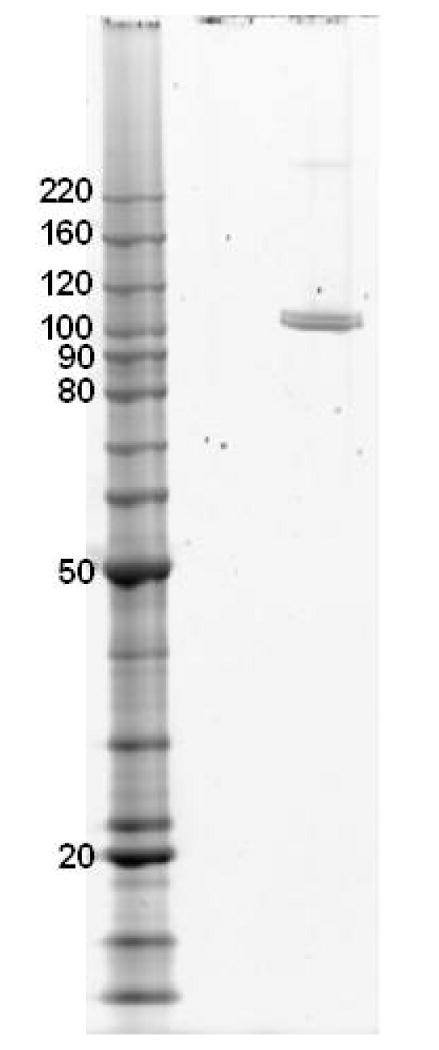
Purified recombinant APP-S. Recombinant human APP-S was expressed in BL21-RIPL cells and purified using a Ni-NTA affinity column. APP-S was eluted via cleavage by thrombin between the His-tag and the recombinant APP-S. Following quantification of total protein using the BCA assay (Pierce), 80 ng of APP-S was run in a 4-12% Bis-Tris gel (Invitrogen) and stained with Krypton Protein Stain (Pierce Biotechnology, Inc.). Lane 1 contains BenchMark Protein Ladder (Invitrogen) and lane 2 contains the purified recombinant APP-S, which was purified to > 97% purity as determined by ImageQuant Analysis (Molecular Dynamics).
Table II.
Kinetics of Val-Ser-cHPMPC hydrolysis by recombinant APP-S. Recombinant APP-S was incubated with Ala-pNA, Val-pNA or Val-Ser-cHPMPC at 37 °C in the presence or absence of the inhibitor bestatin. Hydrolysis of the p-nitroanilide compounds was monitored spectrophotometrically at 405 nm every 30 sec for 15 min. Hydrolysis of Val-Ser-cHPMPC was determined by monitoring the disappearance of the prodrug peak by HPLC (detection at 274 nm) at t = 0, 1, 2, 3, 5, 10, and 15 min. Km and Vmax were calculated using GraphPad Prism 4.0.
| Substrate | Km (mM) | Vmax (nmol/min·mg protein) | kcat (min-1)a | kcat/ Km (min-1·M-1) |
|---|---|---|---|---|
| Ala-pNA | 0.51 ± 0.14 | 5365 ± 610 | 1071 | 2.1 × 106 |
| Val-pNA | 0.28 ± 0.19 | 289 ± 85 | 58 | 0.21 × 106 |
| Val-Ser-cHPMPC | 0.85 ± 0.33 | 1873 ± 400 | 187 | 0.22 × 106 |
kcat values are calculated from Vmax values with the assumption that all enzyme molecules are catalytically active.
To determine the Km and Vmax of APP-S for Val-Ser-cHPMPC, APP-S was incubated with a range of substrate concentrations with aliquots withdrawn at predetermined time points. Using HPLC to determine the concentration of Val-Ser-cHPMPC at various time points, V0 was calculated using the disappearance of prodrug. The data from four independent experiments were plotted (Fig. 4) and analyzed by non-linear regression (GraphPad Prism 4, GraphPad Software, Inc) to determine Vmax and Km. APP-S was shown to hydrolyze Val-Ser-cHPMPC with a Vmax of 1873 ± 400 nmol/min·mg protein and a Km of 0.85 ± 0.33 mM. As expected, this hydrolysis was almost completely inhibited by addition of bestatin. APP-S was not able to appreciably hydrolyze d-Val-d-Ser-cHPMPC beyond that which was detected in buffer alone; similar to what was observed with Caco-2 homogenates (data not shown).
Fig. 4.
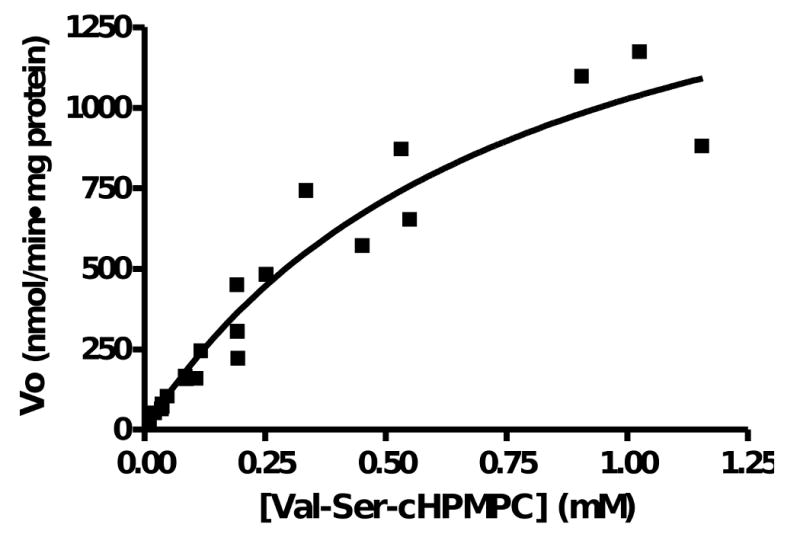
Michaelis-Menten plot of Val-Ser-cHPMPC hydrolysis by APP-S. Recombinant APP-S (30 μg/ml) was incubated in 10 mM HEPES, 100 mM NaCl, pH 7.4 at 37 °C with 0.125 to 1 mM Val-Ser-cHPMPC. Aliquots of 40 μl were removed at predetermined time points (0-15 min) and quenched by the addition of 80 μl of 10% ice-cold TFA. The samples were analyzed by HPLC with a 2-52% acetonitrile gradient mobile phase at 1 ml/min and detection at 274 nm. The concentration detected at 0 min was used as the initial concentration in the plot above to control for variability between sample sets. The V0 was calculated and the plot was generated using GraphPad Prism 4.0.
The kcat for Val-pNA was approximately 18-fold lower (p < 0.06) than the kcat for Ala-pNA and approximately three-fold lower than the kcat for Val-Ser-cHPMPC. The kcat/Km values for Val-Ser-cHPMPC cleavage by APP-S (0.22 × 106 M-1·min-1) are comparable to those obtained for the para-nitroanilide derivatives of l-alanine and l-valine as well as by others (Sengupta et al., 2006).
3.4. Activation pathways for Val-Ser-cHPMPC and its metabolites
Using LC-MS analysis it was observed that in the presence of the recombinant APP-S, the peptide bond in Val-Ser-cHPMPC was cleaved to remove the N-terminal amino acid (l-valine, Fig. 5) to generate the intermediate, Ser-cHPMPC, (3). When Val-Ser-cHPMPC was incubated for a prolonged time period (2h at 37 °C in the presence of the MonoQ active fractions), the activation peak corresponding to 3 disappeared while the peak corresponding to 4 steadily increased. When Val-Ser-cHPMPC was incubated in buffer alone or with APP-S in the presence of the inhibitor bestatin the peak corresponding to compound 3 was not present. Besides the above-mentioned activating products, a minor species (≤ 15%) with a mass of 480 (positive ion mode) was also observed (5). This mass corresponds to the intact dipeptide conjugate attached to the parent compound and not the cyclic version of cidofovir.
Fig. 5.

Chemical structures of the observed activating metabolites obtained during the hydrolysis of Val-Ser-cHPMPC by recombinant APP-S. Samples from APP-S hydrolysis of Val-Ser-cHPMPC were analyzed by LC/MS. It was found that the peak corresponding to 3 was present in the samples hydrolyzed by APP-S, but not present in the negative control samples (prodrug in buffer alone or with APP-S and bestatin). Enzymatic (APP-S), as well as chemical hydrolysis, is involved in the overall prodrug activation process.
Discussion
By stepwise purification from the human intestinal cell line Caco-2 and MS/MS analysis we have demonstrated that APP-S is involved in activation of the antiviral prodrug Val-Ser-cHPMPC. While our data do not exclude the possibility that other proteases may be involved, the observation that bestatin, a known inhibitor of APP-S (Constam et al., 1995; Sengupta et al., 2006), is able to inhibit enzymatic hydrolysis of Val-Ser-cHPMPC in Caco-2 cell homogenates further suggests that APP-S is important in the in vivo activation of the prodrug. Furthermore, the relative inability of the inhibitors aprotinin, leupeptin, and pepstatin A to reduce Val-Ser-cHPMPC hydrolysis in Caco-2 cell homogenate (< 10% inhibition of specific activity, data not shown) is consistent with previous reports that these are not potent inhibitors of APP-S (Constam et al., 1995; Sengupta et al., 2006). Moreover, it has been reported that APP-S prefers basic and hydrophobic amino acids and has relatively low affinity for acidic residues (Constam et al., 1995; Wagner et al., 1981), consistent with our observation that the valine residue is efficiently cleaved from Val-Ser-cHPMPC in Caco-2 cell homogenates. Finally, APP-S has been shown to hydrolyze a variety of amino acid substrates, with the exception of: (i) those that have acidic side chains, (ii) d-amino acid isomers, or (iii) N-terminally-blocked amino acids (26). Consistent with these observations, the recombinant APP-S was unable to hydrolyze the d-amino acid version of 2, d-Val-d-Ser-cHMPC (data not shown). Furthermore, there was no significant hydrolysis of d-Val-d-Ser-cHPMPC in Caco-2 homogenate as compared to buffer alone (data not shown).
The recombinant APP-S was able to efficiently hydrolyze Val-Ser-cHPMPC in vitro, reinforcing our hypothesis concerning APP-S's role in Val-Ser-cHPMPC activation in vivo. Similar to that observed in Caco-2 cell homogenates, bestatin was able to inhibit APP-S hydrolysis of Val-Ser-cHPMPC. The approximately 18-fold lower kcat for Val-pNA as compared to Ala-pNA further confirmed that some amino acids are preferred over others. Furthermore, the three-fold higher kcat for Val-Ser-cHPMPC as compared to Val-pNA suggests that the leaving group may play an important role in determining hydrolytic activity. Interestingly, the Km value for the hydrolysis of Val-Ser-cHPMPC by APP-S is in the sub-millimolar range, suggesting that under in vivo conditions the conversion of Val-Ser-cHPMPC is likely to occur well below saturating substrate concentrations. The kcat/Km values for Val-Ser-cHPMPC cleavage by APP-S (0.22 × 106 min-1·M-1) are comparable to those obtained for the para-nitroanilide derivatives of l-alanine and l-valine as well as by others (Sengupta et al., 2006), suggesting that the prodrug hydrolysis is likely to occur with a reasonable efficiency even in the presence of other substrates.
LC-MS analysis revealed that the peptide bond in Val-Ser-cHPMPC was cleaved to remove the N-terminal valine to generate the Ser-cHPMPC intermediate (3). This intermediate disappeared with prolonged incubation (2 hr at 37 °C) in the presence of the MonoQ column active fractions while the peak associated with cHPMPC (cyclic cidofovir, 4) correspondingly increased. One likely mechanism for this relative instability of Ser-cHPMPC is nucleophilic attack on the phosphodiester linkage by the primary amine of serine that is produced after the removal of valine by APP-S. Indeed, Lazarus et al. have previously proposed and demonstrated such a mechanism to explain the intramolecular hydrolysis of amine-containing phosphoryl esters (Lazarus and Benkovic, 1979; Lazarus et al., 1980). Based on these findings, and the fact that the major species found in rat plasma after a modified in situ single pass perfusion is indeed cHPMPC (Eriksson et al., 2008), we suspected that the activation of Val-Ser-cHPMPC occurs through both enzymatic and chemical pathways. It is worth noting that cyclic cidofovir itself undergoes a biotransformation to generate cidofovir when exposed to endogenous cyclic cytidine 3′,5′-monophosphate (cCMP) phosphodiesterase (Bischofberger et al., 1994; Mendel et al., 1997), and can therefore be regarded as a prodrug of cidofovir. In addition, cyclic cidofovir has been reported to be less nephrotoxic than cidofovir, while also exhibiting potent antiviral activity (Bischofberger et al., 1994). Nevertheless, cyclic cidofovir itself shows low oral bioavailability (Cundy et al., 1996), indicating the need to mask the residual phosphonate negative charge present at physiological pH, which the reported Val-Ser-cHPMPC prodrug has been designed to do.
Puromycin-sensitive aminopeptidase has been extensively studied and has been implicated in a number of physiological processes, including normal cellular turnover (Bhutani et al., 2007; Botbol and Scornik, 1983; Goldberg and Rock, 1992), cell cycle regulation (Constam et al., 1995), processing of antigenic peptides for display on MHC class I (Stoltze et al., 2000; Towne et al., 2008), and degradation of neuropeptides and brain function (Osada et al., 1999; Schulz et al., 2001). However, to our knowledge, this is the first reported finding that APP-S is able to hydrolyze an antiviral prodrug. The broad tissue distribution of APP-S and other neutral aminopeptidases, as well as their homology and expression in a variety of species (Constam et al., 1995; McLellan et al., 1988; Schulz et al., 2001; Tobler et al., 1997) can be advantageous to ensure complete and rapid prodrug activation, as was previously noted for Val-Ser-cHPMPC in situ (Eriksson et al., 2008). Additionally, APP-S has been shown to have a broad substrate specificity with preference for hydrophobic and basic amino acids, (Hui et al., 1983; Johnson and Hersh, 1990; Schnebli et al., 1979; Sengupta et al., 2006; Wagner et al., 1981) making it an attractive target for future prodrug design.
Acknowledgments
This research was supported by grant number GM007767 from NIGMS and NIH grant U01 AI061457. Proteomics data were provided by the Michigan Proteome Consortium (www.proteomeconsortium.org) which is supported in part by funds from The Michigan Life Sciences Corridor.
The abbreviations used are
- APP-S
puromycin-sensitive aminopeptidase
- HPMPC
cidofovir, (Methyl (S)-2-((S)-2-Amino-3-methyl-butyrylamino)-3-[(S)-5-(4-amino-2-oxo-2H-pyrimidin-1-ylmethyl)-2-oxido-1,4,2-dioxaphosphinan-2-yloxy]propanoate)
- cHPMPC
cyclic cidofovir
- Val-Ser-cHPMPC
l-valine-l-serine cyclic cidofovir
- Val-pNA
l-valine para-nitroanilide
- Ala-pNA
l-alanine para-nitroanilide
- HPMPC
cidofovir
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide
- TFA
trifluoroacetic acid
- IPTG
isopropyl β-D-thiogalactopyranoside
- DMSO
dimethyl sulfoxide
- PVDF
polyvinylidene difluoride
- PBS
phosphate-buffered saline
Footnotes
While PSA is the most commonly used abbreviation for puromycin-sensitive aminopeptidase, the authors chose instead to use APP-S in order to avoid confusion with the most prevalent definition of PSA in the scientific literature, prostate-specific antigen.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta D, Goni F, Carmona C. Characterization and partial purification of a leucine aminopeptidase from Fasciola hepatica. Journal of Parasitology. 1998;84(1):1–7. [PubMed] [Google Scholar]
- Albert A. Chemical aspects of selective toxicity. Nature. 1958;182(4633):421–2. doi: 10.1038/182421a0. [DOI] [PubMed] [Google Scholar]
- Balimane PV, Tamai I, Guo A, Nakanishi T, Kitada H, Leibach FH, Tsuji A, Sinko PJ. Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir. Biochemical and Biophysical Research Communications. 1998;250(2):246–251. doi: 10.1006/bbrc.1998.9298. [DOI] [PubMed] [Google Scholar]
- Bhutani N, Venkatraman P, Goldberg AL. Puromycin-sensitive aminopeptidase is the major peptidase responsible for digesting polyglutamine sequences released by proteasomes during protein degradation. EMBO Journal. 2007;26(5):1385–1396. doi: 10.1038/sj.emboj.7601592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger N, Hitchcock MJM, Chen MS, Barkhimer DB, Cundy KC, Kent KM, Lacy SA, Lee WA, Li ZH, et al. 1-[((S)-2-Hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl]cytosine, an intracellular prodrug for (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine with improved therapeutic index in vivo. Antimicrobial Agents and Chemotherapy. 1994;38(10):2387–91. doi: 10.1128/aac.38.10.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botbol V, Scornik OA. Peptide intermediates in the degradation of cellular proteins. Bestatin permits their accumulation in mouse liver in vivo. The Journal of Biological Chemistry. 1983;258(3):1942–9. [PubMed] [Google Scholar]
- Constam DB, Tobler AR, Rensing-Ehl A, Kemler I, Hersh LB, Fontana A. Puromycin-sensitive aminopeptidase. Sequence analysis, expression, and functional characterization. The Journal of Biological Chemistry. 1995;270(45):26931–9. doi: 10.1074/jbc.270.45.26931. [DOI] [PubMed] [Google Scholar]
- Cundy KC, Bidgood AM, Lynch G, Shaw JP, Griffin L, Lee WA. Pharmacokinetics, bioavailability, metabolism, and tissue distribution of cidofovir (HPMPC) and cyclic HPMPC in rats. Drug Metabolism and Disposition: the biological fate of chemicals. 1996;24(7):745–52. [PubMed] [Google Scholar]
- Cundy KC, Li ZH, Hitchcock MJ, Lee WA. Pharmacokinetics of cidofovir in monkeys. Evidence for a prolonged elimination phase representing phosphorylated drug. Drug Metabolism and Disposition: the biological fate of chemicals. 1996;24(7):738–744. [PubMed] [Google Scholar]
- Curran M, Noble S. Valganciclovir. Drugs. 2001;61(8):1145–50. doi: 10.2165/00003495-200161080-00013. discussion 1151-2. [DOI] [PubMed] [Google Scholar]
- De Clerco E. In search of a selective antiviral chemotherapy. Clinical Microbiology Reviews. 1997;10(4):674–693. doi: 10.1128/cmr.10.4.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Research. 2002;55(1):1–13. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E, Holy A. Case history: Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nature Reviews. Drug Discovery. 2005;4(11):928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- Eriksson U, Peterson LW, Kim Js, Mitchell S, Kijek P, Hilfinger JM, Drach JC, Kashemirov BA, McKenna CE. Ehylene glycol-linked amino acid conjuagted of cyclic cidofovir: synthesis and biological activity. Antiviral Research. 2006;70:A58. [Google Scholar]
- Eriksson U, Hilfinger JM, Kim JS, Mitchell S, Kijek P, Borysko KZ, Breitenbach JM, Drach JC, Kashemirov BA, McKenna CE. Synthesis and biological activation of an ethylene glycol-linked amino acid conjugate of cyclic cidofovir. Bioorganic & Medicinal Chemistry Letters. 2007;17(3):583–586. doi: 10.1016/j.bmcl.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson U, Peterson LW, Kashemirov BA, Hilfinger JM, Drach JC, Borysko KZ, Breitenbach JM, Kim JS, Mitchell S, Kijek P, McKenna CE. Serine Peptide Phosphoester Prodrugs of Cyclic Cidofovir: Synthesis, Transport, and Antiviral Activity. Molecular Pharmaceutics. 2008;5(4):598–609. doi: 10.1021/mp8000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochemical and Biophysical Research Communications. 1998;246(2):470–5. doi: 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Rock KL. Proteolysis, proteasomes and antigen presentation. Nature. 1992;357(6377):375–9. doi: 10.1038/357375a0. [DOI] [PubMed] [Google Scholar]
- Han H, de Vrueh RL, Rhie JK, Covitz KM, Smith PL, Lee CP, Oh DM, Sadee W, Amidon GL. 5′-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharmaceutical Research. 1998;15(8):1154–9. doi: 10.1023/a:1011919319810. [DOI] [PubMed] [Google Scholar]
- Hui KS, Wang YJ, Lajtha A. Purification and characterization of an enkephalin aminopeptidase from rat brain membranes. Biochemistry. 1983;22(5):1062–7. doi: 10.1021/bi00274a010. [DOI] [PubMed] [Google Scholar]
- Johnson GD, Hersh LB. Studies on the subsite specificity of the rat brain puromycin-sensitive aminopeptidase. Archives of Biochemistry and Biophysics. 1990;276(2):305–9. doi: 10.1016/0003-9861(90)90724-d. [DOI] [PubMed] [Google Scholar]
- Kim I, Chu XY, Kim S, Provoda CJ, Lee KD, Amidon GL. Identification of a human valacyclovirase: biphenyl hydrolase-like protein as valacyclovir hydrolase. The Journal of Biological Chemistry. 2003;278(28):25348–25356. doi: 10.1074/jbc.M302055200. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lazarus RA, Benkovic SJ. Mechanism of hydrolysis of phosphorylethanolamine triesters. Multiple catalytic effects of an intramolecular amino group. Journal of the American Chemical Society. 1979;101(15):4300–12. [Google Scholar]
- Lazarus RA, Benkovic PA, Benkovic SJ. Mechanism of hydrolysis of phosphorylethanolamine diesters. Intramolecular nucleophilic amine participation. Journal of the Chemical Society. 1980;(2):373–9. [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of Biological Chemistry. 1951;193(1):265–75. [PubMed] [Google Scholar]
- McKenna CE, Kashemirov BA, Eriksson U, Amidon GL, Kish PE, Mitchell S, Kim JS, Hilfinger JM. Cidofovir peptide conjugates as prodrugs. Journal of Organometallic Chemistry. 2005;690(10):2673–2678. [Google Scholar]
- McKenna CE, Kashemirov BA, Peterson LW, Eriksson U, Saejung K, Kim JS, Mitchell S, Kijek P, Hilfinger JM, Drach JC. Cidofovir and Foscarnet peptide prodrugs. Antiviral Research. 2006;70:A37. [Google Scholar]
- McLellan S, Dyer SH, Rodriguez G, Hersh LB. Studies on the tissue distribution of the puromycin-sensitive enkephalin-degrading aminopeptidases. Journal of Neurochemistry. 1988;51(5):1552–9. doi: 10.1111/j.1471-4159.1988.tb01124.x. [DOI] [PubMed] [Google Scholar]
- Mendel DB, Cihlar T, Moon K, Chen MS. Conversion of 1-[((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl]cytosine to cidofovir by an intracellular cyclic CMP phosphodiesterase. Antimicrobial Agents and Chemotherapy. 1997;41(3):641–6. doi: 10.1128/aac.41.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada T, Ikegami S, Takiguchi-Hayashi K, Yamazaki Y, Katoh-Fukui Y, Higashinakagawa T, Sakaki Y, Takeuchi T. Increased anxiety and impaired pain response in puromycin-sensitive aminopeptidase gene-deficient mice obtained by a mouse gene-trap method. Journal of Neuroscience. 1999;19(14):6068–78. doi: 10.1523/JNEUROSCI.19-14-06068.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry CM, Faulds D. Valaciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections. Drugs. 1996;52(5):754–72. doi: 10.2165/00003495-199652050-00009. [DOI] [PubMed] [Google Scholar]
- Pescovitz MD, Rabkin J, Merion RM, Paya CV, Pirsch J, Freeman RB, O'Grady J, Robinson C, To Z, Wren K, Banken L, Buhles W, Brown F. Valganciclovir results in improved oral absorption of ganciclovir in liver transplant recipients. Antimicrobial Agents and Chemotherapy. 2000;44(10):2811–5. doi: 10.1128/aac.44.10.2811-2815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnebli HP, Phillipps MA, Barclay RK. Isolation and characterization of an enkephalin-degrading aminopeptidase from rat brain. Biochimica et Biophysica Acta. 1979;569(1):89–98. doi: 10.1016/0005-2744(79)90084-6. [DOI] [PubMed] [Google Scholar]
- Schulz C, Perezgasga L, Fuller MT. Genetic analysis of dPsa, the Drosophila orthologue of puromycin-sensitive aminopeptidase, suggests redundancy of aminopeptidases. Development Genes and Evolution. 2001;211(12):581–8. doi: 10.1007/s00427-001-0194-z. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Horowitz PM, Karsten SL, Jackson GR, Geschwind DH, Fu Y, Berry RW, Binder LI. Degradation of Tau Protein by Puromycin-Sensitive Aminopeptidase in Vitro. Biochemistry. 2006;45(50):15111–15119. doi: 10.1021/bi061830d. [DOI] [PubMed] [Google Scholar]
- Smiley ML, Murray A, de Miranda P. Valacyclovir HCl (Valtrex): an acyclovir prodrug with improved pharmacokinetics and better efficacy for treatment of zoster. Advances in Experimental Medicine and Biology. 1996;394:33–9. doi: 10.1007/978-1-4757-9209-6_4. [DOI] [PubMed] [Google Scholar]
- Stoltze L, Schirle M, Schwarz G, Schroter C, Thompson MW, Hersh LB, Kalbacher H, Stevanovic S, Rammensee HG, Schild H. Two new proteases in the MHC class I processing pathway. Nature Immunology. 2000;1(5):413–8. doi: 10.1038/80852. [DOI] [PubMed] [Google Scholar]
- Tobler AR, Constam DB, Schmitt-Graff A, Malipiero U, Schlapbach R, Fontana A. Cloning of the human puromycin-sensitive aminopeptidase and evidence for expression in neurons. Journal of Neurochemistry. 1997;68(3):889–97. doi: 10.1046/j.1471-4159.1997.68030889.x. [DOI] [PubMed] [Google Scholar]
- Towne CF, York IA, Neijssen J, Karow ML, Murphy AJ, Valenzuela DM, Yancopoulos GD, Neefjes JJ, Rock KL. Puromycin-Sensitive Aminopeptidase Limits MHC Class I Presentation in Dendritic Cells but Does Not Affect CD8 T Cell Responses during Viral Infections. J Immunol. 2008;180(3):1704–1712. doi: 10.4049/jimmunol.180.3.1704. [DOI] [PubMed] [Google Scholar]
- Wachsman M, Petty BG, Cundy KC, Jaffe HS, Fisher PE, Pastelak A, Lietman PS. Pharmacokinetics, safety and bioavailability of HPMPC (cidofovir) in human immunodeficiency virus-infected subjects. Antiviral Research. 1996;29(23):153–161. doi: 10.1016/0166-3542(95)00829-2. [DOI] [PubMed] [Google Scholar]
- Wagner GW, Tavianini MA, Herrmann KM, Dixon JE. Purification and characterization of an enkephalin aminopeptidase from rat brain. Biochemistry. 1981;20(13):3884–90. doi: 10.1021/bi00516a034. [DOI] [PubMed] [Google Scholar]
- Weller S, Blum MR, Doucette M, Burnette T, Cederberg DM, de Miranda P, Smiley ML. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clinical Pharmacology and Therapeutics. 1993;54(6):595–605. doi: 10.1038/clpt.1993.196. [DOI] [PubMed] [Google Scholar]