

Abstract
Endothelial nitric oxide synthase (eNOS) plays a crucial role in endothelial cell functions. SIRT1, a NAD+-dependent deacetylase, is shown to regulate endothelial function and hence any alteration in endothelial SIRT1 will affect normal vascular physiology. Cigarette smoke (CS)-mediated oxidative stress is implicated in endothelial dysfunction. However, the role of SIRT1 in regulation of eNOS by CS and oxidants are not known. We hypothesized that CS-mediated oxidative stress downregulates SIRT1 leading to acetylation of eNOS which results in reduced nitric oxide (NO)-mediated signaling and endothelial dysfunction. Human umbilical vascular endothelial cells (HUVECs) exposed to cigarette smoke extract (CSE) and H2O2 showed decreased SIRT1 levels, activity, but increased phosphorylation concomitant with increased eNOS acetylation. Pre-treatment of endothelial cells with resveratrol significantly attenuated the CSE- and oxidant-mediated SIRT1 levels and eNOS acetylation. These findings suggest that oxidant-mediated reduction of SIRT1 is associated with acetylation of eNOS which have implications in endothelial dysfunction.
Keywords: Cigarette smoke, oxidants, SIRT1, eNOS, endothelial dysfunction
Introduction
Reactive oxygen species (ROS) either generated endogenously or by exposure to cigarette smoke (CS) play a major role in the progression of several diseases including cardiovascular and pulmonary diseases [1,2]. CS is known to cause endothelial dysfunction which is characterized by reduced cell migration, angiogenesis vasodilation, proinflammatory and prothrombic properties associated with the development of cardiovascular diseases (CVD) [1,2] Endothelial nitric oxide synthase (eNOS), a vital signaling molecule, plays a crucial role in endothelial cell functions. eNOS regulates vascular tone, blood flow, platelet aggregation, cell adhesion and leukocyte-endothelial cell interactions which are characteristics of endothelial function [3]. It has been shown that accelerated ROS production leads to decreased eNOS levels, which play a central role in CS-mediated endothelial dysfunction [4]. We and others have recently shown that CS-mediated oxidative stress downregulate eNOS levels leading to decreased NO production and endothelium-dependent vasodilatation in endothelial cells [5-8]. However, it is not known whether eNOS is subjected to post-translational modifications, in particular acetylation, rendering it inactive in response to CS-mediated oxidative stress in endothelial cells.
SIRT1, which is homologous to yeast (S. cerevisiae) silent information regulator protein 2 (Sir 2), plays a critical role in aging, cell cycle regulation, apoptosis and inflammation [9, 10]. It is highly expressed in vascular endothelial cells and plays a key role in regulating the endothelial function [11-15]. Although SIRT1 has been shown to play a critical role in endothelial vascular biology, it is not known whether SIRT1 regulates endothelial function via acetylation/deacetylation of eNOS in response to CS and oxidants. We therefore hypothesized that CS-mediated oxidative stress downregulates SIRT1 by phosphorylation and degradation leading to eNOS acetylation in endothelial cells. To test this hypothesis, human umbilical vein endothelial cells (HUVECs) were exposed to cigarette smoke extract (CSE) and H2O2, and the role of SIRT1 in regulation of eNOS acetylation/deacetylation was determined. We also studied whether pharmacological activation and inhibition of SIRT1 can regulate eNOS acetylation in these cells.
Materials and Methods
Human umbilical vein endothelial cell culture
HUVECs culture was established as described previously [7, 8] by using human umbilical cords collected within 48 hr of delivery and were grown in EGM-2 (Lonza, Walkersville, MD, USA, previously known as Cambrex) media containing 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2. Cells were grown in 75 mm2 flask coated with 0.1% gelatin, and treatments were performed in 0.1% gelatin-coated 6-well plates [7, 8].
CSE preparation
Research grade cigarettes (1R3F) were obtained from the Kentucky Tobacco Research and Development Center at the University of Kentucky (Lexington, KY). 10% CSE was prepared by bubbling smoke from one cigarette into 10 ml of culture medium supplemented with 1% FBS at a rate of one cigarette per minute as described previously [7, 8].
Cell treatments
HUVECs were grown (90% confluent) in 6-well culture plates and starved for 6 hr in 0.1% FBS then treated with CSE (0.5% and 1.0%) or H2O2 (25, 50 and 100 μM) for 1 hr and 4 hr. For treatments with proteasome inhibitor (N-Acetyl-Leu-Leu-Nle-CHO, ALLN, 5 μM; Calbiochem, San Diego, CA), cells were pretreated for 1 hr followed by exposure to CSE or H2O2. To investigate the pharmacological regulation of SIRT1, HUVECs were pretreated for 1 hr with resveratrol, 10 μM (SIRT1 activator) and splitomicin, 10 μM (SIRT1 inhibitor) (Enzo lifesciences, PA, USA) alone or in combination with CSE (1.0%) or H2O2 (100 μM) for 4 hr. At the end of the incubations, cells were washed twice with ice-cold PBS and lysed using RIPA buffer containing protease inhibitor cocktail as described previously [8]. Finally, the whole cell lysates were sonicated for 10 seconds and centrifuged at 10,000 g for 15 min, and supernatant was separated and analyzed for protein content using BCA kit (Pierce).
Immunoblotting
Cellular protein (40 μg) was electrophoresed on 7.5% SDS-PAGE gel and transblotted on nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). Membranes were blocked with 5% (w/v) non-fat milk in PBS containing 0.1% (v/v) Tween 20 and incubated with SIRT1 (rabbit- anti-SIRT1, Cell Signaling) and phosphorylated SIRT1 (rabbit anti-phospho-SIRT1 (Ser27 and Ser47), Cell Signaling) antibodies. After washing, bound antibody was detected using anti-rabbit/anti-mouse antibody linked to horseradish peroxidase and bound complexes were detected using enhanced chemiluminescence (Perkin Elmer, Waltham, MA, USA).
SIRT1 activity assay
SIRT1 activity was assayed using a deacetylase colorimetric activity assay kit according to the manufacturer’s instructions (Biomol International, Plymouth Meeting, PA).
eNOS acetylation by immunoprecipitation
Whole cell lysate was used for eNOS immunoprecipitation. An antibody against eNOS (1:40 dilution, Cell Signaling) was added to 100 μg of cellular proteins in a final volume of 400 μl, and incubated for 1 hr. Protein-A/G agarose beads (20 μl) (Santa Cruz Biotechnology) were added to each sample and kept overnight at 4°C on a rocker. The beads were washed three times and then resuspended in 40 μl of RIPA buffer. The immunoprecipitated eNOS agarose bead suspension was resolved by 7.5% SDS-PAGE. To assess the acetylation of eNOS, the membranes were first probed against acetyl lysine antibody (Mouse monoclonal acetyl lysine, Cell Signaling) then reprobed for eNOS (rabbit anti-eNOS, Cell signaling).
Statistical analysis
The SigmaStat 3.0 statistical program was used to analyze the data. All pair-wise multiple comparisons were performed using the ANOVA; values of P < 0.05 were considered as significant.
Results
CSE and H2O2 decreased SIRT1 level in HUVECs
SIRT1 plays important role in vascular inflammation and endothelial dysfunction [15]. However, it is not known that whether SIRT1 level is altered in response to CS-mediated oxidative stress in endothelial cells. Therefore, we determined the effects of CSE on SIRT1 protein levels in HUVECs. Treatment of endothelial cells with CSE (0.5% and 1.0%) for 1 and 4 hr resulted significant dose- and time-dependent decrease in SIRT1 levels when compared to control treatments (P<0.001, Fig. 1A-C). Similarly, the SIRT1 level was reduced by dose- and time-dependent manner in HUVECs treated with H2O2 (25, 50 and 100 μM) for 1 and 4 hr (Fig. 1A-C).
Figure 1. Effect of CSE and H2O2 on SIRT1 levels and activity in HUVECs.

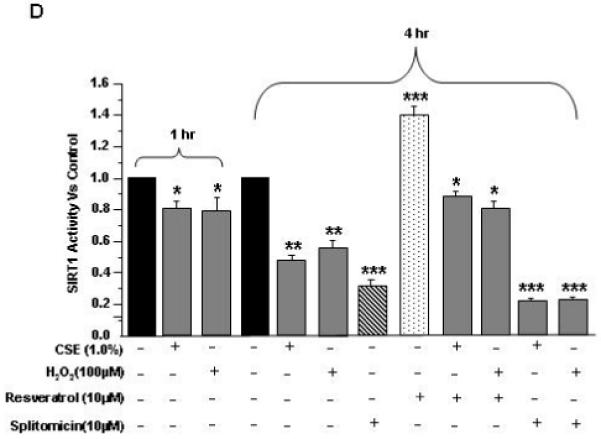
SIRT1 levels: A) HUVECs were treated with CSE (0.5% and 1.0%) and H2O2 (25, 50 and 100 μM) for indicated time points. B) and C) Histograms represent the relative intensity of SIRT1 levels (bands) compared to respective controls (n=3). SIRT1 activity: D) HUVECs were treated with CSE (1.0%) and H2O2 (100 μM) for 1 hr, and at 4 hr alone or in combination with pretreatment of resveratrol (10 μM) and splitomicin (10 μM), and SIRT1 deacetylase activity was determined. Data are Mean ± SE (n=3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. controls.
CSE and H2O2 decreased SIRT1 deacetylase activity in HUVECs
We further investigated whether CSE-mediated oxidative stress which showed decreased SIRT1 levels also show decreased SIRT1 deacetylase activity. Indeed, SIRT1 deacetylase activity was reduced in a time-dependent manner at 1 and 4 hr by CSE (1.0%) and H2O2 (100 μM) treatments in endothelial cells (Fig. 1D). Pre-treatment of cells with resveratrol significantly increased the SIRT1 deacetylase activity alone or combination with CSE or H2O2 treatments whereas splitomicin alone or in combination with CSE or H2O2 treatments further reduced the SIRT1 deacetylase activity (Fig. 1D). These data showed that CSE-mediated reduction in SIRT1 level was associated with decrease in its deacetylase activity in HUVECs.
CSE and H2O2 increased SIRT1 phosphorylation in HUVECs
Recent studies suggest that alterations in mRNA level was not associated with reduced SIRT1 protein and activity levels [16,17], suggesting that post-translational modifications are involved in SIRT1 regulation. We therefore investigated the effect of CSE and H2O2 treatments on phosphorylation pattern of SIRT1 in HUVECs. Endothelial cells treated with increasing doses of CSE (0.5% and 1.0%) and H2O2 (25, 50 and 100 μM) for 1 and 4 hr showed significant phosphorylation of SIRT at Ser27 and Ser47 residues compared to control treatments (P<0.01 and P<0.001, Fig. 2A-C).
Figure 2. Effect of CSE and H2O2 on SIRT1 phosphorylation in HUVECs.
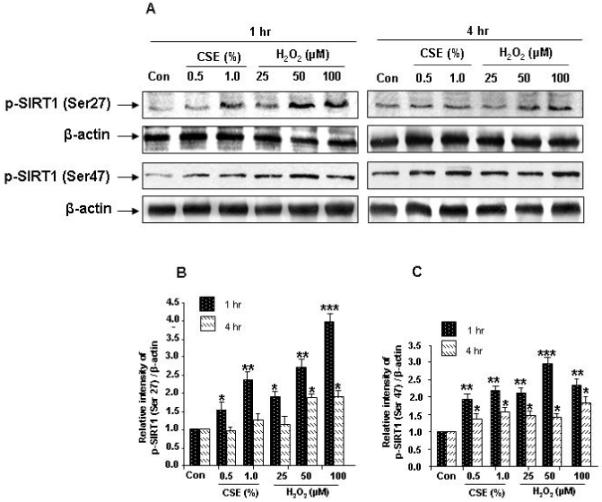
HUVECs were treated with CSE (0.5% and 1.0%) and H2O2 (25, 50 and 100 μM) for indicated time points. A) CSE and H2O2 increased the phosphorylation of SIRT1 (at Ser27 and Ser47). B) and C) Histograms represent relative intensity of phosphorylated SIRT1 levels compared to respective control treatments (n=3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. controls. p-SIRT1 (Ser27), phosphorylation of SIRT1 at Ser27; p- SIRT1 (Ser47), phosphorylation of SIRT1 at Ser 47.
CSE- and H2O2- mediated SIRT1 degradation in HUVECs is proteasome dependent
Since SIRT1 degradation is associated with its phosphorylation in response to CSE and oxidative stress. We therefore determined whether CSE- and H2O2- mediated reduction of SIRT1 levels was associated with its degradation in proteasomes. Pretreatment of HUVECs with proteasome inhibitor N-acetyl-Leu-Leu-Nle-CHO (ALLN; 5 μM) for 1 hr and followed by CSE (1.0%) or H2O2 (100 μM) treatment for 1 and 4 hr resulted in time-dependent attenuation of SIRT1 levels when compared to CSE and H2O2 treatments alone (Fig. 3A-C).
Figure 3. Effect of proteasome inhibitor on CSE- and H2O2-induced degradation of SIRT1 degradation in HUVECs.
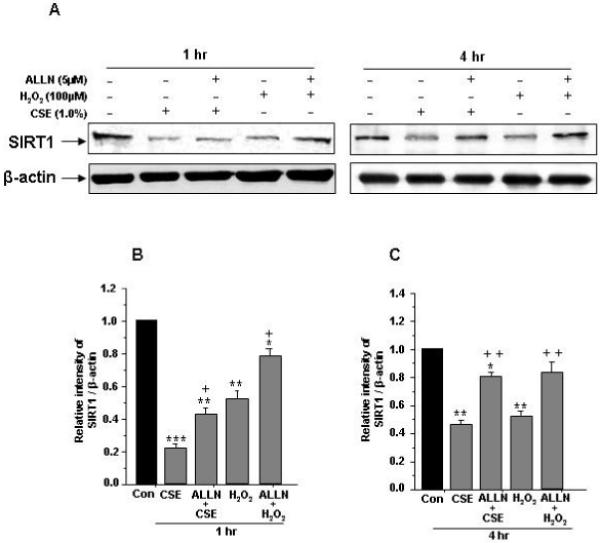
A) HUVECs cells were pre-treated with or without proteasomal inhibitor N-Acetyl-Leu- Leu-Nle-CHO (ALLN, 5 μM) for 1 hr and then treated with CSE (1.0%) or H2O2 (100 μM) for 1 and 4 hr. B) and C) Histograms represent Mean ± SE of relative intensity of SIRT1 levels compared to respective controls (n=3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. controls. +P < 0.05, ++P < 0.01 vs. CSE and H2O2 treatments alone.
CSE and H2O2 caused eNOS acetylation in HUVECs
eNOS plays an important role in endothelial function and is inactivated by oxidants [5-8]. We therefore determined whether CSE-and H2O2- mediated oxidative stress modifies eNOS, thereby rendering it inactive in HUVECs. We found that acetylation of eNOS (at lysine residues) was significantly increased in response to CSE (0.5% and 1.0%) and H2O2 treatments (50 and 100 μM) both at 1 and 4 hr (P<0.001, Fig. 4A-C). These data suggest that CS-mediated eNOS acetylation may lead to decreased NO bioavailability in endothelial cells.
Figure 4. Effect of CSE and H2O2 on eNOS acetylation and its regulation by SIRT1 in HUVECs.
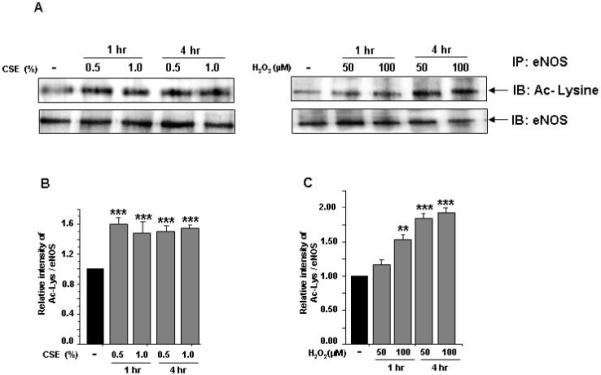
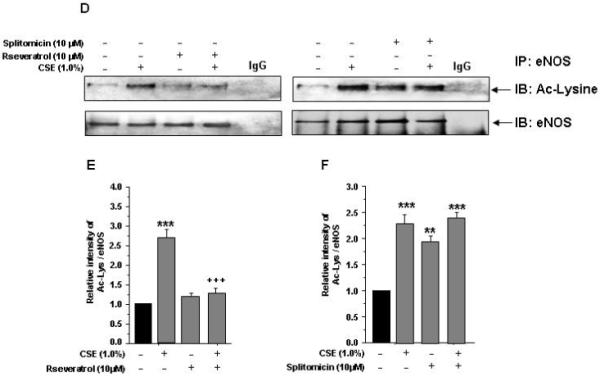
eNOS acetylation: A) HUVECs were treated with CSE (0.5% and 1.0%) and H2O2 (50 and 100 μM) for indicated time points. B) and C) Histograms represent the relative intensity of acetylated lysine levels of eNOS compared to respective controls (n=3). SIRT1 regulation of eNOS acetylation: D) HUVECs cells were pre-treated with or without SIRT1 activator (resveratrol, 10 μM) and inhibitor (splitomicin, 10 μM) for 1 hr and then treated with CSE (1.0%) for 4 hr. E) and F) Histograms represent the relative intensity of acetylated lysine levels of eNOS compared to respective controls (n=3). IgG was used as an isotype control. Data shown are Mean ± SE. **P < 0.01, ***P < 0.001 vs. control groups. +++P < 0.001 vs. CSE treatment alone.
Pharmacological activation of SIRT1 regulates CSE-induced eNOS acetylation in HUVECs
We further studied whether CS-mediated eNOS acetylation was associated with decreased SIRT1 levels and pharmacological activation of SIRT1 can modulate CSE-induced eNOS acetylation. Pretreatment of HUVECs with resveratrol (10 μM) for 4 hr alone or in combination with CSE (1.0%) resulted in reduction in eNOS acetylation when compared to CSE alone (P<0.001, Fig. 4 D-F). Furthermore, pretreatment of endothelial cells with splitomicin alone or in combination with CSE increased eNOS acetylation. We also showed phosphorylation of eNOS at ser1177 in response to CSE [8], which was reversed by resveratrol (Arunachalam et al. unpublished data). These data support the notion that activation of SIRT1 attenuates eNOS post-translational modifications (phosphoacetylation of eNOS) which may regulate endothelial function during oxidative stress.
Discussion
Oxidative stress-mediated chronic inflammation in smokers renders damage to endothelial cells thus increases response to vascular injury [6]. Previously, we have shown that CS-induced oxidative stress impaired the angiogenic function of endothelial cells by downregulating phosphorylation of Akt and eNOS in endothelial cells [7, 8]. Recently, it has been shown that activation of SIRT1 protects endothelial cells against CS-induced oxidative stress [18]. However, the downstream target of SIRT1 in protection against oxidative stress-mediated endothelial dysfunction is not fully known. We hypothesized that CS and oxidative stress downregulate SIRT1 and increase eNOS acetylation leading to endothelial dysfunction. Indeed, CSE and H2O2 treatments caused dose- and time-dependent reduction of SIRT1 levels and its deacetylase activity in HUVECs. Our data are in agreement with the recent reports showing that CS-mediated oxidative stress decreased SIRT1 levels and its deacetylase activity in vitro in macrophages and epithelial cells as well as in lungs of patients with chronic obstructive pulmonary disease (COPD) [19-22].
Oxidative stress cause extensive post-translational modifications to signaling proteins leading to loss of their functions [23]. Recent studies have identified several phosphorylation sites on SIRT1 [16, 17] and the phosphorylation on these residues alters the SIRT1 protein levels [22, 24]. We proposed that CS-mediated reduction of SIRT1 level is associated with its phosphorylation in endothelial cells. CSE treatments dose-dependently increased phosphorylation of SIRT1 on Ser27 and Ser47 residues. Ubiquitin-dependent proteasomal degradation plays a critical role in degradation of oxidatively modified proteins [25]. Hence phosphorylated SIRT1 may undergo proteasomal degradation. Indeed, we showed that proteasomal inhibition by a potent proteasome inhibitor attenuated the SIRT1 levels suggesting a possible role of ubiquitin-dependent degradation of SIRT1. This observation is supported by a recent study demonstrating that ionizing radiation induces proteasomal degradation of SIRT1 [26].
eNOS activation leads to NO release which exerts vasoprotective and cardioprotective effects in endothelial cells by regulating blood flow, inhibition of platelet aggregation and inflammatory cell adhesion [27]. Moreover, ROS-mediated downregulation of eNOS plays a critical role in CS-mediated endothelial dysfunction [4, 5, 7, 28]. SIRT1 is recently identified as a critical regulator of endothelial function [29]. In the light of our data showing the reduction of SIRT1 levels by CS, we hypothesized that CS would cause increased eNOS acetylation in endothelial cells (acetylation leads to inactivation of eNOS activity). Indeed, we show increased eNOS acetylation in endothelial cells in response to CSE treatments. Furthermore, SIRT1 activation by pre-treatment with a non-specific SIRT1 activator resveratrol attenuated CS-induced eNOS acetylation whereas SIRT1 inhibition by splitomicin further increased eNOS acetylation in HUVECs. We further showed phosphorylation of eNOS at Ser1177 in response to CSE [8], which was reversed by resveratrol suggesting that CSE induces phosphoacetylation of eNOS. This is supported by other observations that SIRT1 activation promotes endothelial-dependent vasodilatation by targeting eNOS for deacetylation to enhance NO production [30] whereas SIRT1 knock-down showed decreased NO production and impaired endothelial-dependent vasodilatation [31]. Our data support the concept that activation of SIRT1 pharmacologically or by dietary polyphenol resveratrol may protect against deleterious effects of CS/oxidants on endothelial dysfunction [18, 31]. Hence it is hypothesized that the impairment of eNOS due to acetylation under oxidative stress would lead to endothelial dysfunction, and resveratrol or a specific SIRT1 activator would deacetylate eNOS and maintain the NO bioavailability/endothelial function. This may be one of the possible mechanisms of resveratrol in maintaining or improving endothelial function. However, further studies are required to investigate whether eNOS acetylation alters its activity and NO bioavailability in response to CS in endothelial cells leading to endothelial dysfunction and alterations in proliferation, migration and angiogenesis. It is also worthwhile to determine whether the acetylation of eNOS has any impact on caveolin-1 acetylation as these molecules play a synergistic role in NO bioavailability and hence endothelial function/angiogenesis [32]. Preliminary data from our lab also indicated acetylation of caveolin-1 in response to CS exposure in endothelial cells.
In summary, we show that CS and oxidative stress downregulate SIRT1 levels and increase its phosphorylation on Ser27 and Ser47 residues concomitant with increased eNOS acetylation in endothelial cells. Pre-treatment of endothelial cells with resveratrol attenuated the CSE-mediated SIRT1 levels and eNOS acetylation. Identification of eNOS as a substrate of SIRT1 under the conditions of oxidative stress is important in understanding the beneficial effect of SIRT1 on endothelial vascular biology in terms of preventing stress-induced endothelial dysfunction, an early step in the pathogenesis of cardiopulmonary diseases.
Acknowledgements
This study was supported by the NIH R01-HL085613, 1R01HL097751-01 and NIEHS Environmental Health Science Center grant ES-01247.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15:1983–1992. doi: 10.1097/01.ASN.0000132474.50966.DA. [DOI] [PubMed] [Google Scholar]
- [2].Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- [3].Le Brocq M, Leslie SJ, Milliken P, Megson IL. Endothelial dysfunction: from molecular mechanisms to measurement, clinical implications, and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1631–1674. doi: 10.1089/ars.2007.2013. [DOI] [PubMed] [Google Scholar]
- [4].Raij L, DeMaster EG, Jaimes EA. Cigarette smoke-induced endothelium dysfunction: role of superoxide anion. J Hypertens. 2001;19:891–897. doi: 10.1097/00004872-200105000-00009. [DOI] [PubMed] [Google Scholar]
- [5].Su Y, Cao W, Han Z, Block ER. Cigarette smoke extract inhibits angiogenesis of pulmonary artery endothelial cells: the role of calpain. Am J Physiol Lung Cell Mol Physiol. 2004;287:L794–800. doi: 10.1152/ajplung.00079.2004. [DOI] [PubMed] [Google Scholar]
- [6].Dinh-Xuan AT, Higenbottam TW, Clelland CA, Pepke-Zaba J, Cremona G, Butt AY, Large SR, Wells FC, Wallwork J. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung disease. N Engl J Med. 1991;324:1539–1547. doi: 10.1056/NEJM199105303242203. [DOI] [PubMed] [Google Scholar]
- [7].Edirisinghe I, Yang SR, Yao H, Rajendrasozhan S, Caito S, Adenuga D, Wong C, Rahman A, Phipps RP, Jin ZG, Rahman I. VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. FASEB J. 2008;22:2297–2310. doi: 10.1096/fj.07-099481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Edirisinghe I, Arunachalam G, Wong C, Yao H, Rahman A, Phipps RP, Jin ZG, Rahman I. Cigarette Smoke-Induced Oxidative/Nitrosative Stress Impairs VEGF- And Fluid Shear Stress-Mediated Signaling In Endothelial Cells. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2874. In press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [9].Dryden SC, Nahhas FA, Nowak JE, Goustin AS, Tainsky MA. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol. 2003;23:3173–3185. doi: 10.1128/MCB.23.9.3173-3185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- [11].Ota H, Akishita M, Eto M, Iijima K, Kaneki M, Ouchi Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J Mol Cell Cardiol. 2007;43:571–579. doi: 10.1016/j.yjmcc.2007.08.008. [DOI] [PubMed] [Google Scholar]
- [12].Ota H, Eto M, Kano MR, Ogawa S, Iijima K, Akishita M, Ouchi Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1634–1639. doi: 10.1161/ATVBAHA.108.164368. [DOI] [PubMed] [Google Scholar]
- [13].Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, Vasa-Nicotera M, Ippoliti A, Novelli G, Melino G, Lauro R, Federici M. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation. 2009;120:1524–1532. doi: 10.1161/CIRCULATIONAHA.109.864629. [DOI] [PubMed] [Google Scholar]
- [14].Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, Pearson KJ, de Cabo R, Ungvari Z. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev. 2009;130:518–527. doi: 10.1016/j.mad.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, Minor W, Scrable H. Phosphorylation regulates SIRT1 function. PLoS One. 2008;3:e4020. doi: 10.1371/journal.pone.0004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zschoernig B, Mahlknecht U. Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem Biophys Res Commun. 2009;381:372–377. doi: 10.1016/j.bbrc.2009.02.085. [DOI] [PubMed] [Google Scholar]
- [18].Csiszar A, Labinskyy N, Podlutsky A, Kaminski PM, Wolin MS, Zhang C, Mukhopadhyay P, Pacher P, Hu F, de Cabo R, Ballabh P, Ungvari Z. Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. Am J Physiol Heart Circ Physiol. 2008;294:H2721–2735. doi: 10.1152/ajpheart.00235.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol. 2007;292:L567–576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
- [20].Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nakamaru Y, Vuppusetty C, Wada H, Milne JC, Ito M, Rossios C, Elliot M, Hogg J, Kharitonov S, Goto H, Bemis JE, Elliott P, Barnes PJ, Ito K. A protein deacetylase SIRT1 is a negative regulator of metalloproteinase-9. Faseb J. 2009;23:2810–2819. doi: 10.1096/fj.08-125468. [DOI] [PubMed] [Google Scholar]
- [22].Caito SW, Cook S, Yang S-R, Rajendrasozhan S, Rahman I. Sirtuin, an oxidant sensitive deacetylase, is posttranslationally modified and degraded by the proteasome in response to cigarette smoke in lung epithelial cells. FASEB J. 2008;22 Abstract 747.1. [Google Scholar]
- [23].Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem. 2008;283:21837–21841. doi: 10.1074/jbc.R700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ford J, Ahmed S, Allison S, Jiang M, Milner J. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle. 2008;7:3091–3097. doi: 10.4161/cc.7.19.6799. [DOI] [PubMed] [Google Scholar]
- [25].Farout L, Friguet B. Proteasome function in aging and oxidative stress: implications in protein maintenance failure. Antioxid Redox Signal. 2006;8:205–216. doi: 10.1089/ars.2006.8.205. [DOI] [PubMed] [Google Scholar]
- [26].Hong EH, Lee SJ, Kim JS, Lee KH, Um HD, Kim JH, Kim SJ, Kim JI, Hwang SG. Ionizing radiation induces cellular senescence of articular chondrocytes via negative regulation of SIRT1 by p38 kinase. J Biol Chem. 285:1283–1295. doi: 10.1074/jbc.M109.058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Braam B, Verhaar MC. Understanding eNOS for pharmacological modulation of endothelial function: a translational view. Curr Pharm Des. 2007;13:1727–1740. doi: 10.2174/138161207780831275. [DOI] [PubMed] [Google Scholar]
- [28].Barbera JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med. 2001;164:709–713. doi: 10.1164/ajrccm.164.4.2101023. [DOI] [PubMed] [Google Scholar]
- [29].Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ungvari Z, Labinskyy N, Mukhopadhyay P, Pinto JT, Bagi Z, Ballabh P, Zhang C, Pacher P, Csiszar A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H1876–1881. doi: 10.1152/ajpheart.00375.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu Y, Buikema H, van Gilst WH, Henning RH. Caveolae and endothelial dysfunction: filling the caves in cardiovascular disease. Eur J Pharmacol. 2008;585:256–260. doi: 10.1016/j.ejphar.2008.02.086. [DOI] [PubMed] [Google Scholar]