

Abstract
Developmental biology, like many other areas of biology, has undergone a dramatic shift in the perspective from which developmental processes are viewed. Instead of focusing on the actions of a handful of genes or functional RNAs, we now consider the interactions of large functional gene networks and study how these complex systems orchestrate the unfolding of an organism, from gametes to adult. Developmental biologists are beginning to realize that understanding ontogeny on this scale requires the utilization of computational methods to capture, store and represent the knowledge we have about the underlying processes. Here we review the use of the Gene Ontology (GO) to study developmental biology. We describe the organization and structure of the GO and illustrate some of the ways we use it to capture the current understanding of many common developmental processes. We also discuss ways in which gene product annotations using the GO have been used to ask and answer developmental questions in a variety of model developmental systems. We provide suggestions as to how the GO might be used in more powerful ways to address questions about development. Our goal is to provide developmental biologists with enough background about the GO that they can begin to think about how they might use the ontology efficiently and in the most powerful ways possible.
Introduction
The ontogeny of an organism is a complex process requiring exquisite orchestration of gene expression and action. Over the last century, experimental embryologists have used embryo manipulation, genetics and molecular biology to understand the fundamental processes underlying specific steps along the path from fertilized egg to adult. These experiments have resulted in a large body of information that has been reported in the scientific literature. Often that information stands alone, describing a small set of genes, or a single isolated process, and how it affects a specific step in development. Recently, large-scale genomic and proteomic methods have moved the field from the paradigm of studying single genes in isolated experiments to studying sometimes thousands of genes, in a complex experiments. One challenge developmental biologists face in light of these new methodologies is how to integrate information obtained from large-scale experiments with information about specific developmental processes obtained using a variety of traditional experimental approaches. As experimental information becomes more complex and voluminous, it becomes impossible for a single person to comprehend and analyze. Instead, we must rely on computers for help in storing, retrieving, integrating, analyzing and interpreting data.
There are many ways to store information about any given thing. The simplest is to define lists of bins, or categories, used for description and grouping (Fig. 1). Keyword lists are a common example of categorization. Two limitations of this approach are that: (1) it is not possible to tell whether two things placed in two different bins have any relationship to one another, and (2) it does not necessarily allow for one thing (especially if it is a physical object) to be placed in more than one bin.
Figure 1.
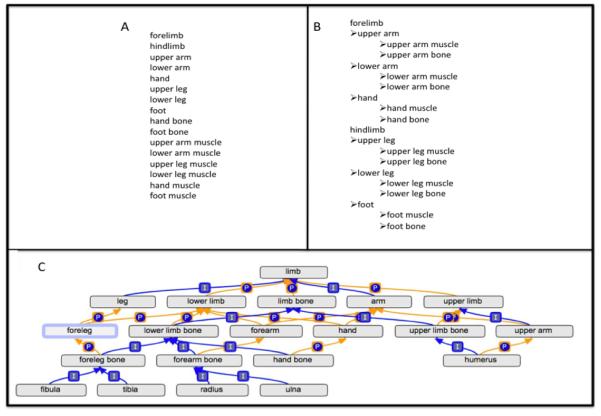
A) A simple list of keywords for anatomical structures in the limbs does not provide information about how the keywords relate to one another. B) The use of a hierarchy allows a simple view of how the structures of the limbs can relate to one another. In this case the hierarchy describes parts of the limbs, but does not relate the upper parts of the forelimb and hindlimb. C) The use of a directed acyclic graph permits terms to have more than one parent and provides a robust representation of the anatomy of limbs. In this tree-view of a simplified ontology, the upper leg bone is both a part_of the upper leg and is_a hindlimb bone. The boxed “I” denotes an is_a relationship and the circled “P” denotes a part_of relationship.
A better classification scheme would allow the individual categories to be related to one another in a meaningful way. Figure 1b illustrates the use of related categories for the classification of the limb anatomical structures listed in Figure 1a by representing them in a hierarchy. Using such a hierarchy, the femur, an upper leg bone, would be defined as being part of the upper leg, and the tibia and the fibula, both lower leg bones, as parts of the lower leg. The use of a hierarchy automatically goes beyond the use of a simple keyword list: not only does it provide information that an anatomical substructure is part of a superstructure, it also implies that anything associated with the substructure is related to the superstructure.
Ideally, when we store a piece of information about something like an anatomical structure, we would like to know how this bit of data relates to information we have stored about all other anatomical structures. One way to represent information about anatomical structures and the ways that they relate to each other is to use an ontology, a set of terms that represent objects or events and the relationships between those objects or events that describe how the terms tie in with one another in reality (Smith, 1998). By representing information using sound ontological principles, a computer can perform logical operations and infer information that goes beyond our original description (Uschold and Grüninger, 1996). Ontologies improve upon simple keyword lists and hierarchies because a single term may have relationships to many other terms instead of just one. In an ontology, terms and their relationships are often represented in a directed acyclic graph or DAG (Fig. 1c), a type of graph that allows us to draw the terms and their structure in a rich and logical way. The placement of terms in the graph relates them to, and defines them with respect to, all of the other terms in the graph. With well-defined relationships, we can write algebraic rules that a computer can use to draw conclusions about the information represented in the graph. For example, if we are interested in getting a list of all the bones that are part of a lower limb, we can use the information in the ontology to retrieve the radius, ulna, hand bones, tibia, fibula and foot bones. Recently, many ontologies of biological interest have been developed by a variety of groups to describe different aspects of biology and biomedicine (Fragoso et al., 2004; Baorto et al., 2009; Rothwell and Fritz, 1983; Whetzel et al., 2006; Smith et al., 2007; Temal et al., 2008). The Gene Ontology (GO), the focus of this review, is used to describe the biological roles of protein or functional RNA gene products.
General Aspects of The Gene Ontology
The GO consists of three ontologies: Molecular Function, Biological Process and Cellular Component (Ashburner et al., 2000) (Fig. 2). The Molecular Function ontology describes the biochemical reactions carried out by gene products at the molecular level, for example, binding or catalysis. The Biological Process ontology describes the biological objective of a gene product, such as germination. Biological processes are achieved by a series or a group of executed molecular functions. For example, the overall objective of the biological process histone phosphorylation (GO:0016572) can be accomplished by protein kinases carrying out their molecular function histone kinase activity (GO:0035173). The Cellular Component ontology describes where in a cell a particular molecular function may occur. In most cases, annotation of a gene product to a cellular component term means that the gene product has been found there and we infer that this may be where the gene product functions. The primary purpose of GO is to provide a standard way to describe the roles of gene products in any organism. The standardization gives us the ability to compare the roles gene products play across many species.
Figure 2.
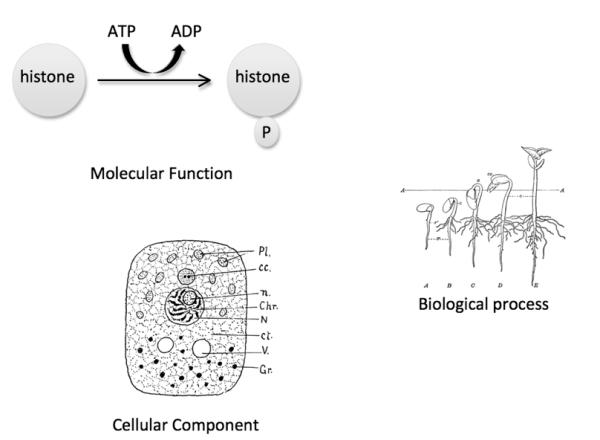
The Gene Ontology consists of three ontologies. Molecular Function describes the biochemical activity of a gene product, such as histone kinase activity. Biological Process describes an overall biological objective, such as seed germination (image obtained from http://etc.usf.edu/clipart/49400/49471/49471_seed_stages.htm). Cellular Component describes where in the cell a gene product is located, such as nucleus (image obtained from http://etc.usf.edu/clipart/47800/47857/47857_cell_struct.htm).
The GO is continually being modified and improved. The ontology changes on daily basis as ontology curators add finer detail to specific areas of the ontology and steadily work to ensure that the ontology is correct and consistent with current biological knowledge. Just as the ontology is constantly improving, annotators at contributing databases are continuously adding new information about gene products to the GO resource. As a result, the entire system, from the ontology to the gene product annotations, is dynamic.
GO terms have 5 essential features (Fig. 3): 1) GO terms have unique names that unambiguously distinguish them from other terms. Whenever possible, terms are named as close to common usage in the community as possible. 2) GO terms have unique IDs of the format: GO:#######. These IDs are fixed and tied to the identity, definition, or meaning, of the term. Term names may change over time, but IDs remain constant. Therefore, the ID is the more stable identifier for a term in the ontology. If, upon further development of the GO, an existing term is deemed unsuitable for the ontology, the term is made obsolete and its ID is deprecated to a special obsolete category. 3) Terms have synonyms. Synonyms are very useful for searching purposes because they represent the many different ways that scientists refer to the same concept. For example, the terms ‘vitellogenesis’ and ‘yolk production’ mean the same thing, therefore a single GO term, vitellogenesis (GO:0007296) exists for this process with an exact synonym yolk production (GO:0007296). Synonym terms in GO can be exact, broad or narrow in relation to a primary term. 4) Terms have a textual definition. The textual definition is both necessary and sufficient to identify the term by its ID. Model Organism Database (MOD) biocurators and users of the ontology use textual definitions to understand the intended meaning of a term and thus the rationale for its association with a given gene product. 5) Terms have relationships to other terms such that each term is placed in the context of all of the other terms in the DAG.
Figure 3.

A partial OBO stanza displaying the term vitellogenesis. An OBO stanza is the textual description of an ontology term in the OBO format. Each term has an ID, a unique name, a textual definition that is supported by a reference, appropriate synonyms and relationships with other terms. Each term in the ontology is represented in an OBO stanza similar to this example. The definition of a term is tied to its ID. Although the string to describe a term may change, the ID of a term is stable and always represents the same biological object.
The GO uses six types of relationships is_a, part_of, regulates, positively_regulates, negatively_regulates and disjoint_from. With the exception of disjoint_from, in these binary relationships we refer to the less specific term as the parent and the more specific term as the child. The further a term is from the root of the graph, the more specific the term is. Therefore, terms that are furthest away from the root will convey more information than those that are closer. Automated methods are now being developed that take advantage of the information content of the ontology to help manage the placement of terms in the graph (Alterovitz et al., 2009). However, it is not particularly useful to discuss the ‘level’ of a term in the graph. Since the graph is a DAG, a single term may be at more than one level depending on the path that is taken to arrive at it from the root.
The different relationships describe the multiple ways in which 2 terms can be linked to each other. The is_a relationship in GO means that if A is_a B then every time we find an A in a natural biological setting, it is a kind of B and A is a child of B. For example, plasma membrane (GO:0005886) is_a membrane (GO:0016020) means that every plasma membrane we find is a kind of membrane. The part_of relationship in GO means that if A part_of B then every time we find an A in a natural biological setting it, along with other things, makes up a kind of B. Note that it does not mean that every time we find a B it must contain an A as a part. Consider the case of a process in the ontology that has a part_of child. This does not mean that every time the parent process occurs, the child part has to occur. Instead it means that every time the child part occurs, it has to be in the context of the parent process. For example, oogenesis (GO:0048477) has ovarian follicle cell development (GO:0030707) and ovarian nurse cell to oocyte transport (GO:0007300) as part_of children. Ovarian nurse cell to oocyte transport (GO:0007300) part_of oogenesis (GO:0048477) means that every time this transport occurs, it is part of the process of the formation and maturation of a female gamete. However, not all oocytes require transport from a nurse cell in order to undergo oogenesis (Biliński et al., 1998). In summary, then, if A part_of B, then every time A exists, it is a part of B, but not all kinds of B have to have a part A.
The regulates relationships are very important with respect to the representation of developmental processes in GO. The regulates relationship between processes means that a process A has a direct influence on another process B such that it controls some aspect of how process B unfolds. Process A can affect the rate at which B proceeds, the frequency at which B occurs, or how far along B is allowed to progress. For example, regulation of Notch signaling pathway (GO:0008593) regulates Notch signaling pathway (GO:0007219) means that every time a regulation of Notch signaling event occurs, it somehow modulates the rate, frequency or extent of Notch signaling. The negatively_regulates and positively_regulates relationships follow the regulates relationship in a straightforward manner by either increasing or decreasing the rate, frequency or extent of another process.
The disjoint_from relationship is a special relationship that is used to maintain the structural integrity of the DAG. If A is disjoint from B, then no A that exists can be a B. For example, cellular process (GO:0009987) disjoint_from multicellular organismal process (GO:0032501) means that if a process is a cellular process, it cannot also be a multicellular organismal process. It can, however, be a part_of a multicellular organismal process. For example, hepatocyte differentiation (GO:0070365) is_a cellular process (GO:0009987) and also is part_of liver development (GO:0001889) a multicellular organismal process (GO:0032501).
Rules governing the relationships between terms and how these relationships interact with each other are important because a computer can use them to draw conclusions about one term based on how it relates to other terms (Fig. 4). For example, the part_of relationship is transitive. If cell-cell signaling involved in cell fate specification (GO:0045168) is part of cell fate specification (GO:0001708) and cell fate specification (GO:0001708) is part of cell fate commitment (GO:0045165), then cell-cell signaling involved in cell fate specification (GO:0045168) is part of cell fate commitment (GO:0045165). Relationships like this can be linked together in a chain through multiple steps in the ontology. As a result, we can infer the relationship between two terms that might be quite distant from one another in the graph.
Figure 4.
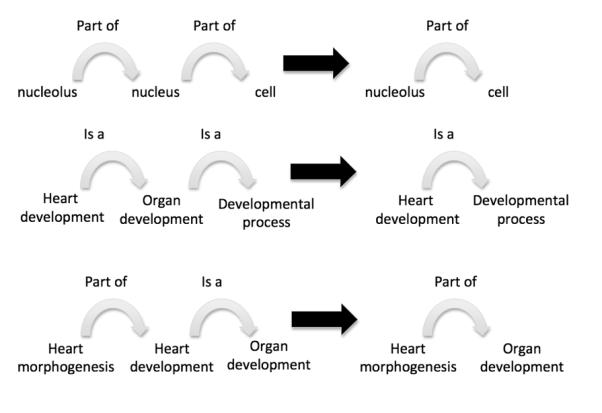
Rules governing relationships allow inferences to be made across the ontology. Here we show 3 rules that can be used for inference using the is_a and part_of relationships. The is_a and part_of relationships are transitive over themselves. The part_of relationship is transitive over the is_a relationship. The black arrow can be read as “therefore”.
Annotations Using the Gene Ontology
The primary purpose of developing GO is to describe the roles of gene products in an organism. MOD biocurators link gene products with GO terms through the process of annotation (Dwight et al., 2002; Hill et al., 2002, 2008; Berriman and Harris, 2004; Berardini et al., 2004; Haas et al., 2005; Aslett and Wood, 2006; Buza et al., 2007; Dimmer et al., 2007; Karp et al., 2007; Hong et al., 2008; Barrell et al., 2009; Tweedie et al., 2009). MOD curators are experts in the biology of their respective organisms, ensuring that the biology is accurately represented in the annotations. Model organism databases that are part of the Gene Ontology Consortium use a standardized method to annotate data from the primary literature (The Reference Genome Group of the Gene Ontology Consortium, 2009), translating experimental results to link GO terms with gene products. Annotations are maintained at both the participating MOD databases and at the central database maintained by the GO Consortium. GO annotations contain four essential elements: 1) the identity of the gene product being annotated, 2) the GO term chosen for the assertion, 3) the reference from which the annotation was made, and 4) the evidence that was used to indicate how an annotation to a given term is supported and how the term-gene product connection was inferred. Evidence for an annotation is captured through a set of evidence codes that broadly describe the types of experiments that can lead to a given conclusion. The use of evidence codes allows users of the annotation files to filter or sort annotations based on the types of evidence that support them.
Since manual annotation is both time-consuming and labor-intensive, most MODs have not comprehensively used their literature collections to annotate all of the published information about every gene product in their organism. To augment literature curation efforts, MODs also use data from large-scale methods or computational algorithms to predict assignment of GO terms to gene products (Biswas et al., 2002; Maeda et al., 2006; Tian et al., 2008; Okazaki et al., 2002). Most of these approaches rely on sequence similarity methods. For example, certain InterPro domains have been mapped to relevant GO terms because proteins containing these domains have been experimentally shown to perform a certain function or to be involved in a particular process. If a gene product has such an InterPro domain as part of its protein sequence, that gene product can then be assigned a GO term based on that functional domain (Mulder et al., 2003). These predicted annotations are almost always more general than curator-assigned annotations and use evidence codes, such as IEA (Inferred from Electronic Annotation), indicating that the annotation was made by a computational method. Although these annotations are reliable, it is good to keep in mind that they are indirectly inferred, generally not reviewed by curators, and the direct experimental evidence supporting them may be made from related proteins that might not have exactly the same biological role in different organisms. For a complete description of GO annotation evidence codes see: http://www.geneontology.org/GO.evidence.shtml
Developmental Biology in the Gene Ontology
A great deal of effort has been expended to ensure that there are appropriate terms describing developmental processes in the GO and, moreover, that these terms can be used consistently and correctly to annotate the many different organisms used to study this important area of biology. In GO, developmental process (GO:0032502) is defined as ‘A biological process whose specific outcome is the progression of an integrated living unit: an anatomical structure (which may be a subcellular structure, cell, tissue or organ), or organism over time from an initial condition to a later condition’. The words ‘developmental process’ and its definition are used rather than just the word ‘development’ to encompass the collection of developmental processes, rather than a single process that is broken down into parts. The distinction between a ‘collection’ and a ‘single process’ is important because it allows the GO term developmental process (GO:0032502) to cover the relevant processes that occur in all organisms, from unicellular fungi to plants and animals. The notion of a progression over time is an important concept in the definition of development. It distinguishes ‘transient processes’ such as the formation and retraction of a pseudopod in an amoeba, which most people would not consider a developmental process, from “an unfolding process’ such as the extension of an axon by a neuron, which most people would consider a developmental process.
Development can be thought of in two different ways: the anatomical sense and the procedural sense. In the anatomical sense, we think about the development of a structure like a limb, or an embryo. From a procedural perspective, we think about induction or morphogenesis. These two viewpoints are reflected in the definition of developmental process (GO:0032502) as well as in the definitions of this term’s children. One of the challenges for those of us who develop the GO is to represent these ideas universally and to structure them in a way that annotations of gene products from different model organisms can be used together to discover generalities about developmental processes. We must also structure and define terms so that annotators can use them in a manner consistent with the way the terms are used in the literature. As described below, the processes of cell differentiation, and developmental induction provide good examples of some of the challenges we face when we are forced to define terms precisely and fit them into a scheme like the Gene Ontology.
Cell Differentiation
Cell differentiation is one of the fundamental processes that is part of the life of any organism that develops, from single-celled yeast to multicellular organisms. It describes the process of cells becoming different from one another during development. In creating and maintaining the section of the GO describing cell differentiation, we asked ourselves: How do cells become different from one another? What exactly does the development of a cell encompass? When does development start and end? How do people use the term ‘cell differentiation’ in the scientific literature? By answering these questions, we can define the terms in GO that relate to cell differentiation in ways that fit into the logical context of the ontology. This way, they can be used in a straightforward way by annotators who are not necessarily experts in developmental biology.
We can start by asking what the term ‘cell development’ means. Previously, we stated that the term developmental process (GO:0032502) is defined as ‘A biological process whose specific outcome is the progression of an integrated living unit: an anatomical structure (which may be a subcellular structure, cell, tissue or organ), or organism over time from an initial condition to a later condition’. It logically follows that cell development (GO:0048468) would be defined as ‘The process whose specific outcome is the progression of the cell over time, from its formation to the mature structure’. The ‘progression of the cell’ part of the definition is easy to comprehend: it is intuitively obvious that this describes the changes that the cell goes through as it develops. The ‘initial and later conditions’ are more difficult to define. In particular, defining the initial condition of a cell is not entirely straightforward because during embryogenesis pre-existing cells already begin to set up the plans for the identity of the cells that will follow. Every cell undergoes two steps when changing its identity during development. The cell somehow decides what it is going to be and then it does so. Cells decide what they are going to be through the process of cell fate commitment (Gilbert, 2006).
Cell fate commitment can be broken down into two classically defined steps, cell fate specification and cell fate determination. Specification is the initial process where the information about the cell’s environment is set up. If a cell is left in this environment, it will differentiate correctly. Determination is the cell-autonomous process by which the cell is now committed to undergo a certain developmental program regardless of its environment. With respect to the structure of the ontology, we must ask, ‘When does the cell attain its identity?’ From a practical standpoint, if the developmental program of the cell can be changed, then it has not yet attained its identity. Therefore, cell development must occur after cell fate commitment, and the fate commitment is not part of the development of the cell.
What about cell differentiation? In the strictest sense, cell differentiation is separate from cell fate commitment (Gilbert, 2006). However, for practical reasons in the ontology and for annotation, it makes sense for cell differentiation (GO:0030154) to take on a more permissive definition that includes the process of cell fate commitment (GO:0045165). In some cases, cell fate commitment can begin in a cell that will give rise to a terminally differentiated cell. An example of this cell progression is the satellite cell -> myoblast -> skeletal muscle cell lineage (Zammit, 2008). In this lineage, satellite cells can be thought of as stem cells. They are self-renewing cells that can give rise to either more satellite cells or to myoblasts. Myoblasts can be considered skeletal muscle progenitors cells. They can divide, or they can give rise to cells that will develop into terminally differentiated skeletal muscle. The commitment of these cells to the skeletal muscle fate does not occur in the cell that terminally differentiates, but rather begins in the satellite cell precursors (Kanisicak et al., 2009). If a mutant is isolated in which mature skeletal muscle cells fail to form, the defect could be in the development of mature skeletal muscle cells, or it may be in the commitment of the satellite cell or myoblast pools, the precursor cell pools, to the muscle cell fate. However, it would not be unreasonable to describe such a mutant as having a phenotype where skeletal muscle cells failed to differentiate. Authors often refer to cells failing to differentiate, but it is difficult to distinguish whether this failure is at the level of commitment of those cells to their fate, or their subsequent development (Wanderling et al., 2007; Vaahtomeri et al., 2008; Zannino and Appel, 2009).
The representation of differentiation in GO not only allows annotators to use terminology that is most globally consistent with the way the word ‘differentiation’ is used in the literature, but also permits annotators to use more specific terms such as cell fate specification (GO:0001708) where experimental data supports that conclusion. Figure 5 shows the arrangement of cell differentiation (GO:0030154) and its children in the ontology. Cell differentiation (GO:0030154) has two part_of children, cell fate commitment (GO:0045165) and cell development (GO:0048468). Cell development (GO:0048468) has four part_of children, developmental cell growth (GO:0048588), cell maturation (GO:0048469), developmental programmed cell death (GO:0010623), and cell morphogenesis involved in cell differentiation (GO:0000904). Developmental cell growth (GO:0048588) is the non-polarized growth of a cell that is part of its development and does not result in a cell shape change. An example of this is a plant cell that grows symmetrically to increase the size of a structure, but does not change its shape as it grows (Kondorosi et al., 2000). Cell maturation (GO:0048469) is the process whereby a cell becomes biochemically fully functional. It does not include growth or shape change, but just a change in the biochemical machinery such that the cell can function in its mature state. An example of cell maturation is the biochemical maturation of intestinal epithelial cells that occurs as they move from the intestinal crypt to the villus (Chang et al., 2008). Developmental programmed cell death (GO:0010623) is programmed cell death that is a natural consequence of a developmental process. For example, the cells in the ‘webs’ between the digits in a vertebrate limb undergo developmental programmed cell death as their last step in development (Chen and Zhao, 1998). Cell morphogenesis involved in differentiation (GO:0000904) describes the shape changes that a cell goes through as it progresses towards a mature stable state. For example, guard cells in corn plants elongate differentially to create a stomatal pore (Galatis, 1980).
Figure 5.

This graphical representation shows cell differentiation and its part_of children. To make the graph easier to view, we have removed the is_a parents from the terms.
The GO intentionally does not include lineage relationships in the development portion of the graph. Although lineage relationships are often well understood, it becomes problematic to describe them in terms of the part_of relationship that is used in GO. For example, a cell from a mesenchyme condensation develops into an osteoprogenitor cell that develops into an osteoblast. We could theoretically trace the lineage of the mesenchymal cell all the way back to the fertilized egg. If we represented these lineages in the ontology as parts of the differentiation of the last cell, then we would conclude that the differentiation of the fertilized egg is a part_of the development of the osteoblast. Although this could be considered to be technically true, it would be a disservice for users of the GO to group gene products that are involved in the differentiation of the first few cells of the embryo under osteoblast differentiation. One could also make the argument that the differentiation of the osteoblast cell is actually the last step in the development of the osteoprogenitor cell and is therefore a part_of osteoprogenitor cell development. This line of reasoning serves no practical purpose when we consider retrieving gene products that are involved in osteoprogenitor cell development. Instead developmental lineage relationships are much better described as a develops_from relationship between two discrete structures, as they are represented in the cell type ontology (Bard et al., 2005).
Developmental Induction
Of course, developmental biologists don’t only care about the progression of a structure from its beginning to end, but also about how these progressions relate to one another and are coordinated. We are interested in teasing out how these processes are regulated and controlled, all the way from the types of interactions that cells have with each other and their environment resulting in the unfolding of a developmental process, to the control of gene regulatory networks underlying almost all of development (Santner et al., 2009; Davidson and Levine, 2008; Simpson et al., 2009). For this reason, the description of regulatory networks in developmental biology is important in the developmental hierarchy of GO.
Sometimes, the regulatory relationship between a process and the process it regulates is very clear. For example, the androgen signaling pathway prevents mammary glands from fully developing in male mammals (Veltmaat et al., 2003). It is straightforward that the signaling pathway negatively regulates the development of the mammary gland, but we would not want to think of it as part of the development of the gland itself. However, the representation of other regulatory processes such as developmental induction is not as clear-cut.
One of the earliest descriptions of tissue interactions that are involved in development was the identification of inductive interactions that occur in amphibian embryos (Spemann and Mangold, 1924). Since this initial description it has been discovered that inductive interactions occur over and over again in developmental systems. In particular, the interaction between an instructive tissue and a receptive tissue leads to the development of many organs (Pispa and Thesleff, 2003; Jacobson and Sater, 1988). However, in an argument analogous to the cell specification argument made above, the inductive events are not considered part_of the development of those organs since they occur before the precursors to those organs are present or determined to become those organs. For this reason, we have defined the specification of a field of cells as the initial step in organ formation. For example, in the development of the mammalian lung, the first recognizable structure that will develop into the lung is the field of cells in the foregut that will undergo budding. However, it would be a mistake not to somehow relate the inductive events of the mesenchyme signaling to the foregut endoderm that result in the specification of the lung field in the foregut, to the process of lung development. To do this, we can take advantage of the positively_regulates relationship. By making induction of an organ (GO:0001759) positively regulate specification of organ identity (GO:0010092), we can now describe how the inductive process relates to the formation of the field of cells that will develop into an organ. This relationship is one of positive regulation, because it results in the initiation of the process, increasing both the process’ rate and extent. Once a field of cells that will form an organ is specified, it will continue to develop into the organ under normal circumstances.
Coordinating the Gene Ontology with Other Ontologies
One of the ways that terms are made more specific is to describe developmental processes based on the anatomical structures (such as the heart) or the cell types that are involved (such as cardiomyoblasts). Because of this, curators strive to coordinate GO with other ontologies developed and used by MODs and other biological databases. Ontologies that describe cell type and anatomies are particularly useful (Bard et al., 1998, 2005; Sprague et al., 2001; Grumbling and Strelets, 2006; Hayamizu et al., 2005; Lee and Sternberg, 2003; Gaudet et al., 2008). GO curators can use the knowledge of experts in other fields to provide a consistent representation of biology across disciplines. For example, GO curators check the structure of GO terms relating to the anatomical representation of the central nervous system against the developmental description of the central nervous system (Fig. 6). This type of cross-checking provides great power in ensuring the accuracy of, and consistency between, both ontologies (Hill et al., 2002). Furthermore, since many anatomical dictionaries contain develops_from relationships, aligning the ontologies allows for the execution of interesting developmental biology queries that cannot be achieved by querying the GO alone since, as we have described above, GO does not contain developmental lineage information. Using lineage information from an anatomical dictionary, a user could search for a structure and all of the structures that it develops from in order to get an insight into the gene products that might give rise to a mutant phenotype. For example, if a mutant zebrafish showed a developmental defect in chondrocyte development, we might want to use the lineage information from the zebrafish anatomy ontology to search on both chondroblast differentiation (GO:0060591) and chondrocyte differentiation (GO:0002062), since chondroblasts are the precursors to chondrocytes.
Figure 6.

A tree-view of a portion of central nervous system development in GO and the portion of the Mouse Anatomical Dictionary for the brain. Ontology editors for both resources coordinate their efforts so that the description of anatomical structure development in GO is consistent with the structure of the anatomical dictionary. For example, diencephalon development and telencephalon development are represented as part_of forebrain development in GO and diencephalon and telencephalon are represented as part_of the forebrain in the anatomical dictionary.
Uses of Gene Ontology Annotations
Currently the Gene Ontology and the annotations of gene products made using terms from the ontology are used for three primary purposes: (1) browsing the ontology for biological information about a specific GO process, function or component, (2) retrieving information about single genes of interest, and (3) obtaining functional information about large numbers of genes that have been identified in large-scale proteomics or microarray studies.
There are a variety of online resources for browsing the ontology and its annotations, either by GO term or by gene product. In addition to the AmiGO browser (Carbon et al., 2009), available from the Gene Ontology home page at http://amigo.geneontology.org/cgi-bin/amigo/go.cgi, most of the databases that contribute annotations to the GO resource also host GO browsers and display GO annotations at their own sites. An extensive list of tools for analyzing gene products annotated using GO can be found at http://www.geneontology.org/GO.tools.shtml?all.
AmiGO can be used to search the GO database for a GO term (Fig. 7). For example, if ‘limb development’ is entered into the query box and the ‘GO terms’ radio button is selected, the search returns a page that highlights the two terms that match the query string limb development (GO:0060173) and limbic system development (GO:0021761). From this page, users can link to information about each term, genes that are annotated to the terms, a textual tree view of the term, and its definition. Several options are available from the drop down menu, including access to graphical views of the term in the context of the ontology and many other options.
Figure 7.
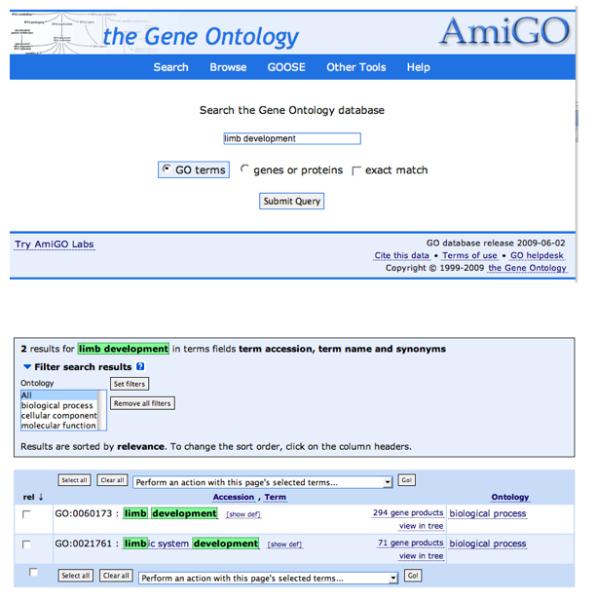
A screen capture showing the search interface for the AmiGO tool that is provided by The Gene Ontology Consortium. The tool can be used to search for either GO terms or genes or proteins in the GO database. The results of the search are shown in the bottom panel.
AmiGO can also be used to retrieve information about gene products (Fig. 8). Gene symbols or names can be entered into the search field and a list of genes that have annotations in the GO database is returned when the query is performed. Users can then link to the GO terms associated with those gene products, the sequences associated with those gene products, or a BLAST search using the sequence of the gene product. Using the advanced query form, queries for more than one gene at a time can be performed.
Figure 8.
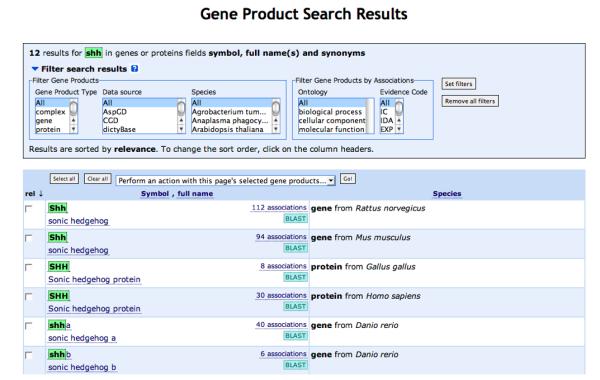
A screen capture showing the results of a gene search using the AmiGO tool. The results can be filtered in a number of ways or can be used to link to annotations, gene information or BLAST.
By far, the most common use of GO in the published literature is for the analysis of large datasets resulting from proteomic or microarray studies. There are far too many of these studies for us to comprehensively review them here. The GO consortium maintains an extensive bibliography of published literature using GO: http://www.geneontology.org/cgi-bin/biblio.cgi
We will, however, review a few select studies that illustrate the use of GO to study development in Mus musculus, Arabidopsis thaliana, Danio rerio, and Caenorhabditis elegans.
Using GO to study development in mice
For a typical large-scale expression experiment that is used to study development in mice, RNA is isolated from a tissue at different stages of development, or RNA is isolated from wild-type and mutant embryos that have a developmental defect. Then, the sets of transcripts are analyzed by microarray or other large-scale analyses to identify transcripts that have significantly different expression levels. This type of experiment often leads to the identification of hundreds of differences in the expression of genes. Statistical analysis tools using GO annotations in the context of the GO hierarchy are used to cluster those genes into functional categories (Jensen et al., 2004; Xu et al., 2007; Hecht et al., 2007; Zhu et al., 2007; Matsuki et al., 2005; van Lunteren et al., 2006; Clemente et al., 2006; Agbemafle et al., 2005; Ivins et al., 2005; Choi et al., 2007; Baguma-Nibasheka et al., 2007; Vaes et al., 2006; Zhang et al., 2004; Li et al., 2006). An example of this type of experiment is the one carried out by James et al to study gene expression during chondrocyte differentiation (James et al., 2005). Micromass cultures were used to differentiate mesenchyme cells into chondrocytes. A number of well-known differentiation markers were used to monitor development. RNA was isolated from the cultures at 3-day intervals for 15 days and the RNA was subjected to microarray analysis. Once genes were identified with significant differences in expression levels, they were clustered using the FatiGO program (Al-Shahrour et al., 2004). Consistent with the differentiation program, a large fraction of genes were shown to function in catalysis, signal transduction, molecular transport, transcription and structural activity. The authors state that this trend in gene expression provides additional support for the validity of their micromass culture system.
The use and validation of systems like the micromass culture are critical in the study of development in mice, because they allow for the analysis of a homogeneous cell type over time. Further clustering of the expression data also confirmed the validity of their approach, for example, genes involved in muscle differentiation were down-regulated. Down regulation of muscle genes would be expected as these cells are instructed to differentiate into a non-muscle cell type. The authors also discuss the importance of the initial analysis of expression patterns in the interpretation of the data. Any study using GO to infer information from microarray data is dependent both on the analysis of the expression data itself and on the amount of knowledge that is embedded in the GO system through the structure of the ontology and the depth/comprehensiveness of the annotations.
Recently, the use of GO has gone beyond analysis of mRNA expression profiles in large-scale array experiments and it has been used to study more targeted questions. For example, mutational analysis has shown that many of the homeobox-containing genes in mice are required for developmental patterning processes (Favier and Dollé, 1997). However, little was known about exactly how the homeobox-containing genes executed their ability to control cell fates and set up regional identity in embryos. Salsi et al recently used the Hoxd13 gene product in a chromatin precipitation experiment to identify target genes of this transcription factor (Salsi et al., 2008). The authors used GO to functionally categorize the genes that were identified. Not surprisingly, many of the genes were involved in development and cell proliferation. Studies like this move the field a step further by linking the targets of a step in a gene regulatory network to the biology represented in the GO. By continuing studies like this, we will eventually be able to understand the gene regulatory networks and the effects that they may be having on development at a cellular level.
Using GO to study development in plants
Arabidopsis thaliana has been used as a model organism to study many plant processes, including development. Again, as in other experimental systems, a popular use of GO has been in the functional categorization of groups of up- or down-regulated genes or in categorizing the transcriptome of a particular cell or group of cells and comparing those results with the categorization of the entire Arabidopsis genome (Day et al., 2008; Wang et al., 2008; Borges et al., 2008; Menges et al., 2008). In one particular study, Cai and Lashbrook (2008) investigated the abscission zone (AZ) transcriptome, looking for regulators of the process of the controlled detachment of an organ (Cai and Lashbrook, 2008). They looked specifically at the genes expressed in laser capture microdissected abscission zones from the stamens of Arabidopsis flowers. By using the laser-capture technique, they were able to take their analysis to a level of detail that was much finer than through the use of a gross dissection. In comparing the enriched GO categories of the upregulated genes in the test set against the distribution of the same categories in the entire Arabidopsis genome, they noted that enrichment of the data set in the GO cellular compartment categories of cell wall (GO:0005618) and extracellular matrix (GO:0031012) and in the GO molecular function category transcription factor activity (GO:0003700). One such transcription factor, AtZFP2 was investigated further. Expression analyses show that the gene product is localized not only in stamen abscission zones but in petal and sepal AZs as well. Additionally, mutant plants that overexpress AtZFP2 display delayed dehiscence of floral organs suggesting involvement of this transcription factor in the regulation of abscission. In this case, the use of GO analysis expedited the identification of a gene product for further study.
Another set of experiments looked at the genes expressed in fluorescence-activated cell sorted Arabidopsis sperm cells (Borges et al., 2008). Aside from being able to pinpoint gene products that are expressed specifically in the sperm cells, the authors wanted to identify what kinds of genes were overrepresented in the sperm transcriptome. Classification of sperm gene products based on GO annotations showed enrichment in the categories of DNA replication (GO:0006260), DNA repair (GO:0006281), cell cycle (GO:0007049), and ubiquitin-dependent protein catabolic process (GO:0006511). The first three categories make sense because Arabidopsis sperm cells spend most of their development in the S phase of the cell cycle, during which DNA synthesis occurs (Friedman, 1999). Plant sperm have also been shown to have upregulated expression of polyubiquitin (Singh et al., 2002), which is relevant to the last category. These studies and many others show the utility of GO annotations in enhancing data analysis.
Using GO to study development in fish
Studies using knockout mice have clearly demonstrated that the MAPK signaling cascade has a prominent role in the early development of vertebrates. These studies have suggested that despite sharing many common activators, Mapk1 and Mapk3 may play distinct roles during embryogenesis (Saba-El-Leil et al., 2003; Pagès et al., 1999). External fertilization, rapid embryonic development, transparency during embryogenesis, and the ability to knock down the expression of specific genes with antisense morpholino oligonucleotides make the zebrafish (D. rerio) ideal for such molecular studies of early vertebrate development. Krens et al. (2008) used morpholinos to specifically knock down mapk3 and mapk1 in zebrafish embryos. Microarray-based gene expression profiling of these knockdowns at various stages of embryonic development followed by cross referencing of the up- and down- regulated genes in each case with the GO was then used to determine the biological roles played by the differentially expressed genes. Consistent with the distinct phenotypes generated in knockout mice, the mapk1 and mapk3 knockdown zebrafish embryos revealed that these two signaling kinases have overlapping but distinct downstream effects. Knockdown of mapk3 was found to affect genes involved in dorsal-ventral patterning and embryonic cell migration, while the mapk1 knockdown affected genes involved in cell migration as well as mesoderm differentiation and patterning (Krens et al., 2008).
Using GO to study development in worms
One of the most fundamental questions in developmental biology is how totipotent germ cells are specified and maintained during organismal development. In the self-fertilizing hermaphroditic nematode C. elegans, a key piece of this biological puzzle, namely knowledge of differential gene expression between germline and somatic cells, has been effectively studied by combining temporal and genetic mutant analyses with DNA microarrays (Reinke et al., 2004). Using RNA prepared from animals at various stages of postembryonic development as well as from those containing mutations that result in: 1) fewer than normal germ cells, 2) feminized animals producing only oocytes, or 3) masculinized animals producing only sperm, researchers have identified genes generally expressed during germ cell development as well as those enriched in either developing oocytes or sperm. To determine the functions of the variably expressed genes, and thus gain insight into the molecular signature of these cell types, researchers have used GO to determine what activities are enriched in germ cells versus somatic cells, and in oocytes versus spermatocytes. Results of this analysis indicate that germline-enriched and oocyte-enriched gene sets contain roughly the same distribution of functions, for example ~30% of the genes in each set encode nucleic acid-binding proteins. Spermatocytes, however, contain more signaling molecules such as kinases and phosphatases, but fewer DNA and RNA metabolism factors, when compared to other germline-enriched genes. In contrast, studies that assessed the functional differences between hermaphrodite and male larvae somatic tissues found little enrichment in functional categories for differentially expressed gene products (Thoemke et al., 2005).
Two additional studies have used the GO to investigate genome organization and regulation of gene expression during germ cell and somatic cell development. In their study on the organization and expression of genes contained within C. elegans operons, Reinke and Cutter, 2009 determined that regardless of molecular function, as determined by Gene Ontology annotation, genes contained within C. elegans operons (~15% of protein-coding genes) are expressed in the germline, but not during spermatogenesis (Reinke and Cutter, 2009). Thus, germline expression has a greater influence on operon composition than gene function, suggesting that expression in the germline somehow influences operon organization. In another study, researchers were interested in determining the genomic distribution of the histone variant HTZ-1 during embryogenesis (Whittle et al., 2008). By mapping sites of HTZ-1 occupation, they determined that during embryogenesis, HTZ-1 occupies the promoters of only 23% of C. elegans genes and that these genes are enriched for GO annotations related to metazoan development and positive regulation of growth, consistent with their expression during embryogenesis.
Future Directions/Beyond Large Scale Analyses
In this review, we have described how the GO is developed with particular respect to representing developmental processes. We have also discussed a number of studies where GO annotations have been used to answer developmental questions, particularly for large-scale genomic analysis. Where do we go from here? We must continue to reach out to the users of the GO so that the power and utility of the system is used to its maximum advantage. It is only through user education that the resource can be used to its fullest extent. For example, when performing any GO analysis, the data sources for both the annotations and the ontology should be given, and annotations should be filtered and processed in ways that are appropriate for the study (Rhee et al., 2008). Otherwise, GO analyses presented in papers cannot be replicated. Secondly, we must continue to develop both the ontology and gene product annotations.
Despite our work to iron out the logic in representing developmental processes in GO, there are many specific aspects of development that need attention. In particular, the development of many structures is still represented at a generic level. For example liver development (GO:0001889) currently has only five children, hepatocyte differentiation (GO:0070365), its regulatory children and liver trabecula formation (GO:0060344). Clearly this area of the ontology needs to be expanded. The mouse anatomical dictionary has 27 children for the term liver. It would be straightforward to create an anatomical representation of liver development based on anatomy. But pure anatomical descriptions will not suffice if one of our goals is to understand how common processes are used in different contexts to achieve ontogeny. For this type of work, we must dovetail anatomical descriptions with process-based descriptions. In particular we must be able to describe developmental processes at the cellular level. We have begun this effort by inviting experts in a targeted field to meet with us and discuss their specialty. So far, we have used this approach to expand the areas of the GO pertaining to central nervous system, lung, and muscle development (Feltrin et al., 2009). We have also been systematically targeting specific developmental processes by using reviews and the current literature. A large amount of effort has recently been spent on expanding the GO with terms describing the development of branching organs like the prostate, mammary gland, and salivary glands (Hill et al., 2009). Targeting similar processes allows us to better capture those processes in a consistent manner. It also allows us to better represent the similarities and differences in what morphologically appear to be similar processes. This style of ontology development should allow us to annotate specific gene products to these processes and understand how orthologs and paralogs do or do not contribute to similar developmental events. Of course, any ontology development must also be accompanied by annotation if we are to understand the roles of gene products in these processes. In the end, if the ontology is used for gene product analysis, the information in the ontology is only as useful as the depth of annotations made to the ontology. Curators at MODs are constantly working to refine gene product annotations and to accurately reflect the large amount of information that already exists and is constantly being generated in the scientific literature. But the research communities of some organisms used for many developmental studies do not have resources that will allow for the detailed annotation of their gene products in a dedicated database. For resources like GO to be maximally useful for the study of these organisms, the ontology must accurately reflect the developmental processes in these organisms, researchers must either be able to contribute detailed annotations for their gene products to a central repository, or infer annotations for their gene products to specific terms in the ontology.
As we develop the ontology with more and more detail, we can retrieve information from the ontology itself. One avenue we are particularly interested in pursuing is the description of the cell signaling processes that are involved in developmental processes. At some point, we would like to be able to ask questions like ‘show me all of the developmental processes in which the smoothened signaling pathway plays a role’. We could then further refine the query to ask whether these processes have similarities, for example, ‘Are all of these processes part of mesenchymal-to-epithelial transitions or branching morphogenesis?’. We could also overlay the gene annotations onto these types of queries to explore the evolutionary relationships between genes and the developmental processes in which they play roles (Berardini et al., 2008).
As the GO continues to develop and becomes connected to other ontologies, we will be able to use GO to generate hypotheses that can then be tested in the laboratory. For example, if we have experimental evidence that a specific pathway is involved in some aspect of the development of an anatomical structure, we can use GO annotations in conjunction with the graph structure to identify all of the genes that are known or predicted to play roles in that pathway. By combining that knowledge and the integrated knowledge of anatomy, we can query gene expression data to determine which of those genes are expressed in the right place to be acting in the development of the anatomical structure. We can also do analogous types of experiments combining phenotype information with process or function from GO. If mutation of a locus displays a defect in a specific anatomical structure, GO can be used to infer what cellular or molecular processes might underlie the phenotypic defect. This will become especially important when the phenotypic defect models a genetic disease or an agriculturally important trait.
Of course, GO does not evolve independently of experimental science. Large-scale studies and microarray analyses continue to improve. In the examples described above, GO–based analyses were used to answer specific questions about development by either using single cell types, or by mutating known genes. As micro-dissection techniques and array analysis techniques continue to improve, transcript profiles will be able to be studied at a cellular level, and over very tight time courses. This will allow us to analyze the results at a much greater resolution. Instead of clustering in areas like metabolism or cell signaling, we will be able to pinpoint the specific processes, or maybe even the specific steps in those processes, that are occurring or being affected.
Acknowledgements
The authors wish to thank Monica McAndrews-Hill, Constance Smith and Judith Blake for critically reading the manuscript. DPH, KMVA and TZB are supported by The GO Consortium grant HG002273 from the National Human Genome Research Institute (NHGRI) at the NIH to M. Ashburner, J. Blake, JM. Cherry and S. Lewis. TZB is also supported by grant DBI-0417062 from the NSF. KMVA is supported by grant #P41-HG02223 from the National Human Genome Research Institute (NHGRI) at the United States National Institutes of Health. DGH is supported by grant #P41 HG002659 from the National Human Genome Research Institute (NHGRI) at the United States National Institutes of Health.
References
- Agbemafle BM, Oesterreicher TJ, Shaw CA, Henning SJ. Immediate early genes of glucocorticoid action on the developing intestine. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2005;288:G897–906. doi: 10.1152/ajpgi.00454.2004. [DOI] [PubMed] [Google Scholar]
- Al-Shahrour F, Díaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics (Oxford, England) 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- Alterovitz G, Xiang M, Hill DP, Lomax J, Liu J, Cherkassky M, Mungall C, Harris MA, Dolan ME, Blake JA, Ramoni MF. Ontology Engineering. Nature Biotech. 2009 doi: 10.1038/nbt0210-128. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslett M, Wood V. Gene Ontology annotation status of the fission yeast genome: preliminary coverage approaches 100% Yeast (Chichester, England) 2006;23:913–919. doi: 10.1002/yea.1420. [DOI] [PubMed] [Google Scholar]
- Baguma-Nibasheka M, Angka HE, Inanlou MR, Kablar B. Microarray analysis of Myf5-/-:MyoD-/- hypoplastic mouse lungs reveals a profile of genes involved in pneumocyte differentiation. Histology and Histopathology. 2007;22:483–495. doi: 10.14670/HH-22.483. [DOI] [PubMed] [Google Scholar]
- Baorto D, Li L, Cimino JJ. Practical experience with the maintenance and auditing of a large medical ontology. Journal of Biomedical Informatics. 2009 doi: 10.1016/j.jbi.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JL, Kaufman MH, Dubreuil C, Brune RM, Burger A, Baldock RA, Davidson DR. An internet-accessible database of mouse developmental anatomy based on a systematic nomenclature. Mechanisms of Development. 1998;74:111–120. doi: 10.1016/s0925-4773(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Bard JL, Rhee SY, Ashburner M. An ontology for cell types. Genome Biology. 2005;6:R21. doi: 10.1186/gb-2005-6-2-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrell D, Dimmer E, Huntley RP, Binns D, O’Donovan C, Apweiler R. The GOA database in 2009--an integrated Gene Ontology Annotation resource. Nucleic Acids Research. 2009;37:D396–403. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini TZ, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller LA, Yoon J, Doyle A, Lander G, Moseyko N, Yoo D, Xu I, et al. Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiology. 2004;135:745–755. doi: 10.1104/pp.104.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini TZ, Hill DP, Rhee SY, Blake JA. Homeodomain proteins in mice and plants: What we know and what we don’t. Developmental Biology. 2008;319:564. [Google Scholar]
- Berriman M, Harris M. Annotation of parasite genomes. Methods in Molecular Biology (Clifton, N.J.) 2004;270:17–44. doi: 10.1385/1-59259-793-9:017. [DOI] [PubMed] [Google Scholar]
- Biliński SM, Büning J, Simiczyjew B. The ovaries of Mecoptera: basic similarities and one exception to the rule. Folia Histochemica Et Cytobiologica / Polish Academy of Sciences, Polish Histochemical and Cytochemical Society. 1998;36:189–195. [PubMed] [Google Scholar]
- Biswas M, O’Rourke JF, Camon E, Fraser G, Kanapin A, Karavidopoulou Y, Kersey P, Kriventseva E, Mittard V, Mulder N, Phan I, Servant F, Apweiler R. Applications of InterPro in protein annotation and genome analysis. Briefings in Bioinformatics. 2002;3:285–295. doi: 10.1093/bib/3.3.285. [DOI] [PubMed] [Google Scholar]
- Borges F, Gomes G, Gardner R, Moreno N, McCormick S, Feijó JA, Becker JD. Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiology. 2008;148:1168–1181. doi: 10.1104/pp.108.125229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buza TJ, McCarthy FM, Burgess SC. Experimental-confirmation and functional-annotation of predicted proteins in the chicken genome. BMC Genomics. 2007;8:425. doi: 10.1186/1471-2164-8-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Lashbrook CC. Stamen abscission zone transcriptome profiling reveals new candidates for abscission control: enhanced retention of floral organs in transgenic plants overexpressing Arabidopsis ZINC FINGER PROTEIN2. Plant Physiology. 2008;146:1305–1321. doi: 10.1104/pp.107.110908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics (Oxford, England) 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Chance MR, Nicholas C, Ahmed N, Guilmeau S, Flandez M, Wang D, Byun D, Nasser S, Albanese JM, Corner GA, Heerdt BG, Wilson AJ, et al. Proteomic changes during intestinal cell maturation in vivo. Journal of Proteomics. 2008;71:530–546. doi: 10.1016/j.jprot.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao X. Shaping limbs by apoptosis. The Journal of Experimental Zoology. 1998;282:691–702. [PubMed] [Google Scholar]
- Choi Y, Qin Y, Berger MF, Ballow DJ, Bulyk ML, Rajkovic A. Microarray analyses of newborn mouse ovaries lacking Nobox. Biology of Reproduction. 2007;77:312–319. doi: 10.1095/biolreprod.107.060459. [DOI] [PubMed] [Google Scholar]
- Clemente EJ, Furlong RA, Loveland KL, Affara NA. Gene expression study in the juvenile mouse testis: identification of stage-specific molecular pathways during spermatogenesis. Mammalian Genome: Official Journal of the International Mammalian Genome Society. 2006;17:956–975. doi: 10.1007/s00335-006-0029-3. [DOI] [PubMed] [Google Scholar]
- Davidson EH, Levine MS. Properties of developmental gene regulatory networks. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20063–20066. doi: 10.1073/pnas.0806007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day RC, Herridge RP, Ambrose BA, Macknight RC. Transcriptome analysis of proliferating Arabidopsis endosperm reveals biological implications for the control of syncytial division, cytokinin signaling, and gene expression regulation. Plant Physiology. 2008;148:1964–1984. doi: 10.1104/pp.108.128108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer E, Berardini TZ, Barrell D, Camon E. Methods for gene ontology annotation. Methods in Molecular Biology (Clifton, N.J.) 2007;406:495–520. doi: 10.1007/978-1-59745-535-0_24. [DOI] [PubMed] [Google Scholar]
- Dwight SS, Harris MA, Dolinski K, Ball CA, Binkley G, Christie KR, Fisk DG, Issel-Tarver L, Schroeder M, Sherlock G, Sethuraman A, Weng S, Botstein D, et al. Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO) Nucleic Acids Research. 2002;30:69–72. doi: 10.1093/nar/30.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier B, Dollé P. Developmental functions of mammalian Hox genes. Molecular Human Reproduction. 1997;3:115–131. doi: 10.1093/molehr/3.2.115. [DOI] [PubMed] [Google Scholar]
- Feltrin E, Campanaro S, Diehl AD, Ehler E, Faulkner G, Fordham J, Gardin C, Harris M, Hill D, Knoell R, Laveder P, Mittempergher L, Nori A, et al. Muscle Research and Gene Ontology: New standards for improved data integration. BMC Medical Genomics. 2009;2:6. doi: 10.1186/1755-8794-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso G, de Coronado S, Haber M, Hartel F, Wright L. Overview and Utilization of the NCI Thesaurus. Comparative and Functional Genomics. 2004;5:648–654. doi: 10.1002/cfg.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman W. Expression of the cell cycle in sperm of Arabidopsis: implications for understanding patterns of gametogenesis and fertilization in plants and other eukaryotes. Development. 1999;126:1065–1075. doi: 10.1242/dev.126.5.1065. [DOI] [PubMed] [Google Scholar]
- Galatis B. Microtubules and guard-cell morphogenesis in Zea mays L. Journal of Cell Science. 1980;45:211–244. doi: 10.1242/jcs.45.1.211. [DOI] [PubMed] [Google Scholar]
- Gaudet P, Williams JG, Fey P, Chisholm RL. An anatomy ontology to represent biological knowledge in Dictyostelium discoideum. BMC Genomics. 2008;9:130. doi: 10.1186/1471-2164-9-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SF. Developmental Biology. Sinauer Associates Inc.; 2006. [Google Scholar]
- Grumbling G, Strelets V. FlyBase: anatomical data, images and queries. Nucleic Acids Research. 2006;34:D484–488. doi: 10.1093/nar/gkj068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Wortman JR, Ronning CM, Hannick LI, Smith RK, Maiti R, Chan AP, Yu C, Farzad M, Wu D, White O, Town CD. Complete reannotation of the Arabidopsis genome: methods, tools, protocols and the final release. BMC Biology. 2005;3:7. doi: 10.1186/1741-7007-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayamizu TF, Mangan M, Corradi JP, Kadin JA, Ringwald M. The Adult Mouse Anatomical Dictionary: a tool for annotating and integrating data. Genome Biology. 2005;6:R29. doi: 10.1186/gb-2005-6-3-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht J, Seitz V, Urban M, Wagner F, Robinson PN, Stiege A, Dieterich C, Kornak U, Wilkening U, Brieske N, Zwingman C, Kidess A, Stricker S, et al. Detection of novel skeletogenesis target genes by comprehensive analysis of a Runx2(-/-) mouse model. Gene Expression Patterns: GEP. 2007;7:102–112. doi: 10.1016/j.modgep.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Hill DP, Blake JA, Richardson JE, Ringwald M. Extension and integration of the gene ontology (GO): combining GO vocabularies with external vocabularies. Genome Research. 2002;12:1982–1991. doi: 10.1101/gr.580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DP, Smith B, McAndrews-Hill MS, Blake JA. Gene Ontology annotations: what they mean and where they come from. BMC Bioinformatics. 2008;9(Suppl 5):S2. doi: 10.1186/1471-2105-9-S5-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DP, Sitnikov D, Blake JA. Using gene ontology to study branching morphogenesis in mice. Developmental Biology. 2009;331:454. [Google Scholar]
- Hong EL, Balakrishnan R, Dong Q, Christie KR, Park J, Binkley G, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Krieger CJ, et al. Gene Ontology annotations at SGD: new data sources and annotation methods. Nucleic Acids Research. 2008;36:D577–581. doi: 10.1093/nar/gkm909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivins S, van Beuren K Lammerts, Roberts C, James C, Lindsay E, Baldini A, Ataliotis P, Scambler PJ. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Developmental Biology. 2005;285:554–569. doi: 10.1016/j.ydbio.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Jacobson AG, Sater AK. Features of embryonic induction. Development (Cambridge, England) 1988;104:341–359. doi: 10.1242/dev.104.3.341. [DOI] [PubMed] [Google Scholar]
- James CG, Appleton CTG, Ulici V, Underhill TM, Beier F. Microarray analyses of gene expression during chondrocyte differentiation identifies novel regulators of hypertrophy. Molecular Biology of the Cell. 2005;16:5316–5333. doi: 10.1091/mbc.E05-01-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen P, Magdaleno S, Lehman KM, Rice DS, Lavallie ER, Collins-Racie L, McCoy JM, Curran T. A neurogenomics approach to gene expression analysis in the developing brain. Brain Research. Molecular Brain Research. 2004;132:116–127. doi: 10.1016/j.molbrainres.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Kanisicak O, Mendez JJ, Yamamoto S, Yamamoto M, Goldhamer DJ. Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD. Developmental Biology. 2009 doi: 10.1016/j.ydbio.2009.05.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp PD, Keseler IM, Shearer A, Latendresse M, Krummenacker M, Paley SM, Paulsen I, Collado-Vides J, Gama-Castro S, Peralta-Gil M, Santos-Zavaleta A, Peñaloza-Spínola MI, Bonavides-Martinez C, et al. Multidimensional annotation of the Escherichia coli K-12 genome. Nucleic Acids Research. 2007;35:7577–7590. doi: 10.1093/nar/gkm740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondorosi E, Roudier F, Gendreau E. Plant cell-size control: growing by ploidy? Current Opinion in Plant Biology. 2000;3:488–492. doi: 10.1016/s1369-5266(00)00118-7. [DOI] [PubMed] [Google Scholar]
- Krens SFG, Corredor-Adámez M, He S, Snaar-Jagalska BE, Spaink HP. ERK1 and ERK2 MAPK are key regulators of distinct gene sets in zebrafish embryogenesis. BMC Genomics. 2008;9:196. doi: 10.1186/1471-2164-9-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RYN, Sternberg PW. Building a cell and anatomy ontology of caenorhabditis elegans. Comparative and Functional Genomics. 2003;4:121–126. doi: 10.1002/cfg.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Cui X, Kim N. Transcription profile during maternal to zygotic transition in the mouse embryo. Reproduction, Fertility, and Development. 2006;18:635–645. doi: 10.1071/rd06015. [DOI] [PubMed] [Google Scholar]
- van Lunteren E, Moyer M, Leahy P. Gene expression profiling of diaphragm muscle in alpha2-laminin (merosin)-deficient dy/dy dystrophic mice. Physiological Genomics. 2006;25:85–95. doi: 10.1152/physiolgenomics.00226.2005. [DOI] [PubMed] [Google Scholar]
- Maeda N, Kasukawa T, Oyama R, Gough J, Frith M, Engström PG, Lenhard B, Aturaliya RN, Batalov S, Beisel KW, Bult CJ, Fletcher CF, Forrest ARR, et al. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genetics. 2006;2:e62. doi: 10.1371/journal.pgen.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Hori G, Furuichi T. Gene expression profiling during the embryonic development of mouse brain using an oligonucleotide-based microarray system. Brain Research. Molecular Brain Research. 2005;136:231–254. doi: 10.1016/j.molbrainres.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Menges M, Dóczi R, Okrész L, Morandini P, Mizzi L, Soloviev M, Murray JAH, Bögre L. Comprehensive gene expression atlas for the Arabidopsis MAP kinase signalling pathways. The New Phytologist. 2008;179:643–662. doi: 10.1111/j.1469-8137.2008.02552.x. [DOI] [PubMed] [Google Scholar]
- Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Barrell D, Bateman A, Binns D, Biswas M, Bradley P, Bork P, Bucher P, Copley RR, Courcelle E, et al. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Research. 2003;31:315–318. doi: 10.1093/nar/gkg046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Pagès G, Guérin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science (New York, N.Y.) 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Pispa J, Thesleff I. Mechanisms of ectodermal organogenesis. Developmental Biology. 2003;262:195–205. doi: 10.1016/s0012-1606(03)00325-7. [DOI] [PubMed] [Google Scholar]
- Reinke V, Cutter AD. Germline expression influences operon organization in the Caenorhabditis elegans genome. Genetics. 2009;181:1219–1228. doi: 10.1534/genetics.108.099283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development (Cambridge, England) 2004;131:311–323. doi: 10.1242/dev.00914. [DOI] [PubMed] [Google Scholar]
- Rhee SY, Wood V, Dolinski K, Draghici S. Use and misuse of the gene ontology annotations. Nature Reviews. Genetics. 2008;9:509–515. doi: 10.1038/nrg2363. [DOI] [PubMed] [Google Scholar]
- Rothwell D, Fritz A. SNOMED microcomputer software system. Pathologist. 1983;37:15–18. [PubMed] [Google Scholar]
- Saba-El-Leil MK, Vella FDJ, Vernay B, Voisin L, Chen L, Labrecque N, Ang S, Meloche S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Reports. 2003;4:964–968. doi: 10.1038/sj.embor.embor939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsi V, Vigano MA, Cocchiarella F, Mantovani R, Zappavigna V. Hoxd13 binds in vivo and regulates the expression of genes acting in key pathways for early limb and skeletal patterning. Developmental Biology. 2008;317:497–507. doi: 10.1016/j.ydbio.2008.02.048. [DOI] [PubMed] [Google Scholar]
- Santner A, Calderon-Villalobos LIA, Estelle M. Plant hormones are versatile chemical regulators of plant growth. Nature Chemical Biology. 2009;5:301–307. doi: 10.1038/nchembio.165. [DOI] [PubMed] [Google Scholar]
- Simpson F, Kerr MC, Wicking C. Trafficking, development and hedgehog. Mechanisms of Development. 2009;126:279–288. doi: 10.1016/j.mod.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Singh M, Xu H, Bhalla P, Zhang Z, Swoboda I, Russell S. Developmental expression of polyubiquitin genes and distribution of ubiquitinated proteins in generative and sperm cells. Sexual Plant Reproduction. 2002;14:325–329. [Google Scholar]
- Smith B. The Basic Tools of Formal Ontology. In: Guarino N, editor. Formal Ontology in Information Systems. IOS Press; 1998. pp. 19–28. [Google Scholar]
- Smith B, Ashburner M, Rosse C, Bard J, Bug W, Ceusters W, Goldberg LJ, Eilbeck K, Ireland A, Mungall CJ, Leontis N, Rocca-Serra P, Ruttenberg A, et al. The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nat Biotech. 2007;25:1251–1255. doi: 10.1038/nbt1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spemann H, Mangold H. Ueber die Induktion von Embryonalanlagen durch Implantation artfremder Organisatoren. 1924;100:599–638. [Google Scholar]
- Sprague J, Doerry E, Douglas S, Westerfield M. The Zebrafish Information Network (ZFIN): a resource for genetic, genomic and developmental research. Nucleic Acids Research. 2001;29:87–90. doi: 10.1093/nar/29.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temal L, Dojat M, Kassel G, Gibaud B. Towards an ontology for sharing medical images and regions of interest in neuroimaging. Journal of Biomedical Informatics. 2008;41:766–778. doi: 10.1016/j.jbi.2008.03.002. [DOI] [PubMed] [Google Scholar]
- The Reference Genome Group of the Gene Ontology Consortium The Gene Ontology’s Reference Genome Project: A Unified Framework for Functional Annotation across Species. 2009. in press. [DOI] [PMC free article] [PubMed]
- Thoemke K, Yi W, Ross JM, Kim S, Reinke V, Zarkower D. Genome-wide analysis of sex-enriched gene expression during C. elegans larval development. Developmental Biology. 2005;284:500–508. doi: 10.1016/j.ydbio.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Tian W, Zhang LV, Taşan M, Gibbons FD, King OD, Park J, Wunderlich Z, Cherry JM, Roth FP. Combining guilt-by-association and guilt-by-profiling to predict Saccharomyces cerevisiae gene function. Genome Biology. 2008;9(Suppl 1):S7. doi: 10.1186/gb-2008-9-s1-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, Zhang H. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Research. 2009;37:D555–559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uschold M, Grüninger M. Ontologies: Principles, Methods and Applications. KNOWLEDGE ENGINEERING REVIEW. 1996;11:93–136. [Google Scholar]
- Vaahtomeri K, Ventelä E, Laajanen K, Katajisto P, Wipff P, Hinz B, Vallenius T, Tiainen M, Mäkelä TP. Lkb1 is required for TGFbeta-mediated myofibroblast differentiation. Journal of Cell Science. 2008;121:3531–3540. doi: 10.1242/jcs.032706. [DOI] [PubMed] [Google Scholar]
- Vaes BLT, Ducy P, Sijbers AM, Hendriks JMA, van Someren EP, de Jong NG, van den Heuvel ER, Olijve W, van Zoelen EJJ, Dechering KJ. Microarray analysis on Runx2-deficient mouse embryos reveals novel Runx2 functions and target genes during intramembranous and endochondral bone formation. Bone. 2006;39:724–738. doi: 10.1016/j.bone.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Veltmaat JM, Mailleux AA, Thiery JP, Bellusci S. Mouse embryonic mammogenesis as a model for the molecular regulation of pattern formation. Differentiation. Research in Biological Diversity. 2003;71:1–17. doi: 10.1046/j.1432-0436.2003.700601.x. [DOI] [PubMed] [Google Scholar]
- Wanderling S, Simen BB, Ostrovsky O, Ahmed NT, Vogen SM, Gidalevitz T, Argon Y. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Molecular Biology of the Cell. 2007;18:3764–3775. doi: 10.1091/mbc.E07-03-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang W, Song L, Zou J, Su Z, Wu W. Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiology. 2008;148:1201–1211. doi: 10.1104/pp.108.126375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetzel PL, Parkinson H, Causton HC, Fan L, Fostel J, Fragoso G, Game L, Heiskanen M, Morrison N, Rocca-Serra P, Sansone S, Taylor C, White J, et al. The MGED Ontology: a resource for semantics-based description of microarray experiments. Bioinformatics (Oxford, England) 2006;22:866–873. doi: 10.1093/bioinformatics/btl005. [DOI] [PubMed] [Google Scholar]
- Whittle CM, McClinic KN, Ercan S, Zhang X, Green RD, Kelly WG, Lieb JD. The genomic distribution and function of histone variant HTZ-1 during C. elegans embryogenesis. PLoS Genetics. 2008;4:e1000187. doi: 10.1371/journal.pgen.1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Santos RAS, Bader M, Alenina N. Alterations in gene expression in the testis of angiotensin-(1-7)-receptor Mas-deficient mice. Regulatory Peptides. 2007;138:51–55. doi: 10.1016/j.regpep.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Zammit PS. All muscle satellite cells are equal, but are some more equal than others? Journal of Cell Science. 2008;121:2975–2982. doi: 10.1242/jcs.019661. [DOI] [PubMed] [Google Scholar]
- Zannino DA, Appel B. Olig2+ precursors produce abducens motor neurons and oligodendrocytes in the zebrafish hindbrain. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2009;29:2322–2333. doi: 10.1523/JNEUROSCI.3755-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Moseley A, Jegga AG, Gupta A, Witte DP, Sartor M, Medvedovic M, Williams SS, Ley-Ebert C, Coolen LM, Egnaczyk G, Genter MB, Lehman M, et al. Neural system-enriched gene expression: relationship to biological pathways and neurological diseases. Physiological Genomics. 2004;18:167–183. doi: 10.1152/physiolgenomics.00220.2003. [DOI] [PubMed] [Google Scholar]
- Zhu H, Cabrera RM, Wlodarczyk BJ, Bozinov D, Wang D, Schwartz RJ, Finnell RH. Differentially expressed genes in embryonic cardiac tissues of mice lacking Folr1 gene activity. BMC Developmental Biology. 2007;7:128. doi: 10.1186/1471-213X-7-128. [DOI] [PMC free article] [PubMed] [Google Scholar]