

Abstract
We present monozygotic twins discordant for the autosomal dominant disorder neurofibromatosis type 1 (NF1). The affected twin was diagnosed with NF1 at age 12, based upon accepted clinical criteria for the disorder. Both twins were re-examined at ages 35 and 57, at which times the unaffected twin continued to show no clinical manifestations of NF1. Short tandem repeat marker (STR) genotyping at 10 loci on chromosome 17 and 10 additional loci dispersed across the genome revealed identical genotypes for the twins, confirming their monozygosity. The affected twin has three children, two of whom also have NF1, while the unaffected twin has two children, both unaffected. Using lymphoblastoid, fibroblast, and buccal cell samples collected from both twins and from other family members in three generations, we discovered a pathogenic nonsense mutation in exon 40 of the NF1 gene. This mutation was found in all cell samples from the affected twin and her affected daughter, and in lymphoblastoid and buccal cells but not fibroblasts from the unaffected twin. We also found a novel non-synonymous change in exon 16 of the NF1 gene that was transmitted from the unaffected mother to both twins and co-segregated with the pathogenic mutation in the ensuing generation. All cells from the twins were heterozygous for this apparent exon 16 polymorphism and for single nucleotide polymorphisms (SNPs) within 2.5kb flanking the site of the exon 40 nonsense mutation. This suggests that the NF1 gene of the unaffected twin differed in the respective lymphoblastoid cells and fibroblasts only at the mutation site itself, making post-zygotic mutation leading to mosaicism the most likely mechanism of phenotypic discordance. Although the unaffected twin is a mosaic, the distribution of the mutant allele among different cells and tissues appears to be insufficient to cause overt clinical manifestations of NF1.
Keywords: Neurofibromatosis type 1, monozygotic twins, discordance, post-zygotic mutation
INTRODUCTION
Neurofibromatosis type 1 (NF1; OMIM 162200) is a progressive inherited neurocutaneous disorder affecting ~1/3500 individuals worldwide, though ~50% of cases are thought to represent de novo mutations [Huson et al., 1989; Littler and Morton, 1990]. Clinical features are extremely variable and include café-au-lait macules, skin fold freckling, benign peripheral nerve sheath tumors (primarily neurofibromas), Lisch nodules of the iris, optic gliomas, and bone manifestations and learning disabilities [Boyd et al., 2009; National Institutes of Health Consensus Development Conference, 1988]. Variation is considerable even within the same family [Riccardi, 1992] and irrespective of the causative genetic alterations, most of which are point mutations or small deletions, insertions or duplications [Kayes et al., 1994; Szudek et al., 2000]. The NF1 gene is located on chromosome 17q11.2, spans a region of 283 kb of genomic DNA and produces an 11–12.5 kb mRNA. Though a variety of splice forms are known, the longest, isoform 1, combines 58 exons to encode 2,839 amino acids. The NF1 protein, neurofibromin, is a tumor suppressor with a GAP domain that is involved in negative regulation of Ras family GTPases [Ballester et al., 1990; Xu et al., 1990]. The NF1 gene shows a remarkably high and as yet unexplained rate of spontaneous mutation: ~50% of patients have no family history of the disorder and appear to result from de novo germline mutations, although some may have a parent who is mosaic for a NF1 mutation [Kehrer-Sawatzki and Cooper 2008]. A number of reports have presented monozygotic twins who are both affected with NF1 but who differ markedly in their phenotypes [Akesson et al., 1983; Bauer et al., 1988; Detjen et al., 2007; Kelly et al., 1998; Koul et al., 2000]. None of these reports identified the causative mutation. The authors are unaware of any previous reports of monozygotic twins discordant for NF1.
SUBJECTS AND METHODS
Clinical Evaluation
The pedigree shown in Figure 1 depicts a pair of monozygotic twins born at term to a G3P2 21-year-old mother (I-3) and her 23-year-old partner (I-2), at 3374 g (II-1) and 3487 g (II-2), respectively, following a pregnancy complicated by diagnostic X-ray exposure of the mother’s abdomen to confirm a twin pregnancy and initial breech presentation of one twin. The monozygotic twins shared a single placenta and spontaneous vaginal delivery was accomplished without complications following manual version of the breech twin. The postnatal course was uncomplicated. II-2 (the breech twin) was diagnosed with NF1 at age 12 based upon multiple café-au-lait macules, multiple cutaneous neurofibromas and a single plexiform neurofibroma involving the left lower extremity. Neither II-1 nor the parents showed signs of NF1. The twins were evaluated clinically at NIH at ages 35 and 57, with II-1 continuing to show no manifestations of the disorder. The NIH clinical evaluations included a full body skin examination, ophthalmologic examination, brain MR images, and X-rays of the skull, thoracic and lumbar spine, and long bones of upper and lower extremities. In addition to the cutaneous findings of NF1 mentioned above, II-2 also had Lisch nodules, axillary freckling and slight scalloping of some lumbar vertebra. Her skin color was darker and her facial features were coarser than those of her unaffected twin. She also differed from her unaffected twin in several other features: height (160.4 cm vs. 164 cm), occipitofrontal circumference (56.3 cm vs. 54.7 cm), dominant handedness (left vs. right) and eye findings (severe myopia vs. mild myopia/astigmatism). I-2 produced three children with the same mate, two of whom have been diagnosed with NF1. This study was approved by the Institutional Review Boards of the Massachusetts General Hospital and the National Cancer Institute.
Figure 1. Pedigree depicting this three generation NF1 family.
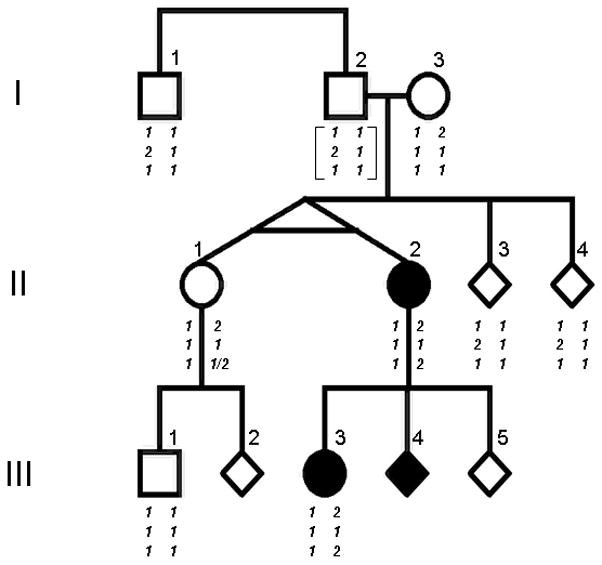
Symptomatic individuals are shown as filled symbols. Some symbols are given as diamonds to protect confidentiality. Genotypes are arranged as two phased haplotypes shown below each individual: top row, exon 16 change c.1776G>A (p.S592N) (1=G, 2=A); middle row, rs17882240 intron 39 8 base pair in/del (c.5813-663_654dupTGTGAACC; 1= 1 copy, 2= 2 copies); bottom row, exon 40 nonsense mutation c.5902C>T (p.R1968X) (1=C, 2=T). Individual II-2 shows the exon 40 nonsense mutation as a de novo event on the chromosome 17 inherited from her mother. II-1 shows either the mutation or wild-type sequence at this site, depending on the tissue source of DNA. Haplotypes in I-2 are shown in brackets, as the genotype was inferred.
MOLECULAR ANALYSIS
Lymphoblastoid cell lines were made from whole blood obtained from the twins and the extended family [Anderson and Gusella 1984] and fibroblast cells were cultured from skin punches. DNA and RNA were prepared from both types of cell lines [Owerbach et al., 1981] and DNA was also prepared from buccal cells. DNAs from the twins were genotyped using 10 highly informative multi-allele (STRs) markers across chromosome 17 (D17S784, D17S787, D17S791, D17S799, D17S802, D17S925, D17S928, D17S938, D17S945, D17S949), and an additional 10 markers across the genome (D2S337, D3S1267, D4S402, D4S414, D12S78, D12S86, D13S159, D13S170, D15S205, D22S283) to confirm the twins’ monozygosity. Array Comparative Genomic Hybridization (CGH) analysis of the twins’ DNA was performed using the Agilent 244K oligonucleotide array (G4411B) following the manufacturer’s instructions (Oligonucleotide Array-Based CGH for Genomic DNA Analysis protocol version 3 (Agilent Technologies, Palo Alto, CA, USA)). Images were captured by the Agilent DNA microarray scanner and quantified using Feature Extraction software v9.0 (Agilent Technologies, Palo Alto, CA, USA). CGH analytics software v3.4 (Agilent Technologies, Palo Alto, CA, USA) was subsequently used for data normalization, quality evaluation and data visualization. Copy number aberration was examined using the ADM-2 (Aberration Detection Method 2) algorithm.
For mutation identification, we performed RT-PCR and then amplified and sequenced the NF1 cDNA (primer sequences and PCR conditions available upon request). All variations are presented in base pair and codon positions based upon NF1 variant 1 in the NCBI database (accession number NM_001042492) (http://www.ncbi.nlm.nih.gov/) and the exon numbering is based upon the matching of this sequence to the genome in the UCSC Genome Browser (http://genome.ucsc.edu hg18 assembly). Sequencing was performed after PCR clean-up using the Qiagen MiniElute PCR purification kit (Valencia, CA), on an ABI 3730 DNA Analyzer, and the results analyzed using Sequencher 4.8 software (Gene Codes, Ann Arbor, MI). The cDNA sequence variations were confirmed in genomic DNA. The exon 16 variation was assessed by SNP analysis in 100 unrelated human DNAs from the CEPH collection, and was found in 0/200 alleles. Polymorphisms in the regions flanking exon 40 were found while examining the cause of the pathogenic mutation by methodical step-wise sequencing from either side of the exon. Primers were overlapping and sequencing ceased once a heterozygous SNP was found proximal and distal to the mutation (primer sequences available upon request).
RESULTS
STR analysis revealed that genotypes for 20 highly informative multi-allele markers (10 on chromosome 17 and 10 elsewhere in the genome) were identical in the twins, confirming that they are indeed monozygotic. Array CGH analysis of both the lymphoblastoid and fibroblast DNAs revealed no differences in copy number variants between the twins, although comparison across cell types revealed the expected deletion in the light and heavy chain immunoglobulin gene regions in the lymphoblastoid cell DNA. Sequencing the entire NF1 cDNA coding region from lymphoblastoid cells of the affected twin uncovered two novel changes. Using GenBank accession number NM_001042492 (http://www.ncbi.nlm.nih.gov/Genbank/) as our reference sequence, we found a non-synonymous alteration (S592N) in exon 16 and a nonsense alteration (R1968X) in exon 40 predicted to cause a truncated NF1 protein. Neither change was present on 200 control chromosomes. The S592N variant has not been reported previously from extensive mutation analyses of NF1 patients. This variant is predicted by analysis using PolyPhen (http://genetics.bwh.harvard.edu/pph/) [Ramensky et al., 2002; Sunyaev et al., 2000; Sunyaev et al., 2001] to be benign suggesting that it represents a rare polymorphism. In contrast, the R1968X alteration has been reported previously as a recurring pathogenic NF1 mutation (sometimes referred to as R1947X based upon neurofibromin isoform 2, GenBank NM_000267) [Cawthon et al., 1990; De Luca et al., 2004; Fahsold et al., 2000; Upadhyaya et al., 1994]. Interestingly and unexpectedly, both alterations were also found in the lymphoblastoid cells from the unaffected twin.
We next examined DNAs from other available family members and detected the S592N polymorphism in the unaffected mother of the twins (I-3) and in the affected daughter (III-3) of the affected twin (II-2) (Fig 1 and Table I). This helped us to phase the R1968X mutation, since it was transmitted together with the S592N variant to III-3, and to deduce that the mutation had occurred on the maternal chromosome transmitted from I-3 to the twins. Sequencing the genomic DNA surrounding exon 40 in the twins and family members also revealed an 8 base pair duplication in intron 39 of the paternal uncle and the two siblings of the twins. This is a known polymorphism (rs17882240) that further permitted us to phase the chromosomes in the siblings of the twins and to infer the genotypes at these three locations for their father, who was unavailable for study.
Table 1.
Genotypes for novel exon 16 polymorphism, intron 39 in/del polymorphism and exon 40 mutation
| ID | PHENOTYPE/RELATIONSHIP | SAMPLE SOURCE | Ex16 (Ser592Asn)* | Intron 39 in/del (rs17882240)* | Ex40 (Arg1968>X)* |
|---|---|---|---|---|---|
| I-1 | Unaffected uncle | Lymphoblast | 1,1 | 1,2 | 1,1 |
| I-3 | Unaffected mother | Lymphoblast | 1,2 | 1,1 | 1,1 |
| II-1 | Unaffected twin | Lymphoblast #1 | 1,2 | 1,1 | 1,2 |
| Lymphoblast #2 | 1,2 | 1,2 | |||
| Blood | 1,2 | 1,2 | |||
| Buccal #1 | 1,2 | 1,2 | |||
| Buccal #2 | 1,2 | 1,2 | |||
| Fibroblast #1 | 1,2 | 1,1 | 1,1 | ||
| Fibroblast #2 | 1,2 | 1,1 | 1,1 | ||
| II-2 | Affected twin | Lymphoblast #1 | 1,2 | 1,1 | 1,2 |
| Lymphoblast #2 | 1,2 | 1,2 | |||
| Blood | 1,2 | 1,2 | |||
| Buccal #1 | 1,2 | 1,2 | |||
| Buccal #2 | 1,2 | 1,2 | |||
| Fibroblast #1 | 1,2 | 1,1 | 1,2 | ||
| Fibroblast #2 | 1,2 | 1,1 | 1,2 | ||
| II-3 | Unaffected sibling | Lymphoblast | 1,1 | 1,2 | 1,1 |
| II-4 | Unaffected sibling | Lymphoblast | 1,1 | 1,2 | 1,1 |
| III-1 | Unaffected son of II-1 | Lymphoblast #1 | 1,1 | 1,1 | 1,1 |
| Lymphoblast #2 | 1,1 | 1,1 | 1,1 | ||
| III-3 | Affected daughter of II-2 | Lymphoblast | 1,2 | 1,2 | |
| Blood | 1,2 | 1,2 |
Allele assignments are the same in Table 1 as represented in Figure 1.
To explore the distribution of the pathogenic R1968X mutation in the cells of both twins, we obtained an independent set of lymphoblastoid cell lines (collected 5 years after the first set), fibroblast cultures and buccal cells (two independent collections 8 years apart) from them. Sequencing DNAs from buccal cells, whole blood and all lymphoblastoid cells from both twins confirmed the presence in them of the nonsense mutation, whereas sequencing DNA from two independent fibroblast cultures from each twin revealed the R1968X mutation only in the affected twin II-2 (see Fig 2). The unaffected twin, II-1, displayed only the wild-type sequence at this location in both fibroblast cultures #1 and #2. These findings suggested that either the R1968X mutation occurred as a post-zygotic event, leading to mosaicism in the twins, or that the mother was mosaic in her germline for the mutation, which reverted to wild type, possibly by gene conversion, during embryonic development in the unaffected twin. Reversion of an NF1 mutation to wild type has not yet been reported in NF1. Mosaicism for NF1 mutation has been reported; it is sometimes manifested as segmental neurofibromatosis, in which a portion or segment of the patient is affected, while the rest of the body remains unaffected [Hager et al., 1997; Listernick et al., 2003; Moss and Green 1994; Riccardi 1982; Schultz et al., 2002; Tinschert et al., 2000]. To explore the possibility of gene conversion, we sequenced the genomic region on either side of the site of the nonsense mutation. We found that although the twins differed in fibroblast DNA for the presence of the R1968X mutation, they were identically heterozygous for SNPs 2.5kb proximal (rs17880965) and 1.7kb distal (rs35370249) to this site, indicating that any putative gene conversion event would have spanned at most 4.2kb. While this does not entirely rule out a small gene conversion event, or reversion in II-1 of an NF1 mutation that occurred in the mother, the simplest explanation for the discordant manifestations of NF1 in the twins is post-zygotic mutation leading to mosaicism in the unaffected twin.
Figure 2. Sequence analysis of the R1968X mutation.
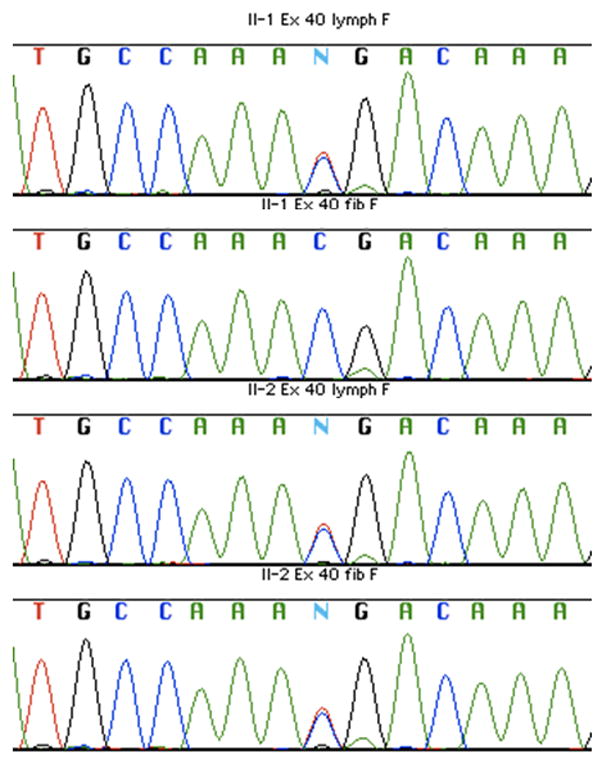
DNA sequence traces of the region of the R1968X (c.5902C>T) mutation are shown for pedigree member II-1 lymphoblastoid DNA (top panel), II-1 fibroblast DNA (second panel), II-2 lymphoblastoid DNA (third panel) and II-2 fibroblast DNA (bottom panel). Software-generated base calls are shown above the colored traces, with heterozygosity for the pathogenic mutation being designated by “N”. Only the II-1 fibroblast DNA fails to show evidence of the R1968X change.
DISCUSSION
Monozygotic twinning occurs in humans at a rate of 0.3–0.4% [Pison and D’Addato 2006] and is relatively constant across races and ethnic groups around the world. It is generally non-familial, as genetic factors are not thought to play a significant role in the global twinning rate. Monozygotic twins arise by division of an embryo to form identical copies from one fertilization event. Twinning can occur from as early as the two cell stage until after the formation of the ectoplacental cone. For ~1/3 of all monozygotic twins, the twinning event occurs early (dichorionic/diamnionic), prior to the formation of the two cell layers (inner-cell mass and trophectoderm) allowing discrete development of the extraembryonic and embryonic components of the blastocyst. The most common mechanism of monozygotic twinning, accounting for ~2/3 of all live twin births, occurs when the embryo divides at the blastocyst stage (diamnionic/monochorionic), after the formation of the two distinct cell layers. Rarely, the twinning event occurs very late and results in monoamnionic, monochorionic twins, or incomplete separation and conjoined twins [Scott, 2002]. The twinning event in the NF1 family reported here appears to have taken place after the early post-zygotic occurrence of a pathogenic R1968X mutation, leading to a shared placenta and mosaicism for the mutation in one twin. Indeed, it is conceivable that the effects of the NF1 mutation and consequent differences in microenvironment resulting from co-development of mutant and normal cells could have contributed to inducing the formation of two submasses, leading to the discordant monozygotic twins [Hall, 1996; Machin, 2009]. As monochorionic twins exchange their blood in utero through the single placenta, the appearance of the mutation in the blood elements of both twins might be expected even if the lesion had occurred after the twinning event. However, the presence of the mutation in two independent buccal samples (uncontaminated by blood) from the unaffected twin indicates that the gastrointestinal tract of the unaffected twin is also mosaic, as the mutation occurred before twinning. Interestingly, neither twin displays any abnormal gastrointestinal phenotype.
The occurrence of mosaicism and post-zygotic mutations in NF1 are not new concepts; however, our report highlights the need to test a variety of accessible tissues from both patients and family members in situations where mosaicism is suspected or should be ruled out. Most patients documented to be mosaic for NF1 mutation have had whole gene deletions that have been found in varying levels in DNA from fibroblasts, buccal cells, and lymphocytes. This preponderance of whole gene deletions likely reflects an ascertainment bias due to the relative ease of detection of individual copies of such gross mutations by in situ hybridization techniques. If more subtle mutations, such as R1968X, are present in mosaics, they may not be detected using DNA from any of these sources using current DNA screening assays, leading to false negative results and underestimation of reproductive risks [Kehrer-Sawatzki et al., 2004]. Our ability to detect and characterize the mosaic status of twin II-1 depended on the testing of multiple tissues, on the high proportional difference of the mutant and wild-type alleles between fibroblasts and other tissues and on the ability to phase the allelic variants through genotyping of other family members. If the mutation had been more evenly distributed in the different tissues, we might have failed to detect it using standard DNA sequencing. This technological limitation may be overcome in the future through diagnostic implementation of next generation DNA sequencing, which allows ascertainment and counting of individual sequenced molecules rather than yielding only the predominant sequence in a mixture.
The pathogenic NF1 mutation found in this study (R1968X) is a recurrent variant present in 1–2% of patients with NF1 [Dublin et al., 1995; Krkljus et al., 1998] that results from alteration of a methylatable CpG dinucleotide which is thought to a hotspot for C to T transitions due to deamination of 5-methylcytosine [Andrews et al., 1996]. In our study, this mutated allele is of maternal origin; this is interesting because several studies have suggested that over 90% of spontaneous mutations in NF1 originate in the paternal genome [Jadayel et al., 1990; Wallace et al., 1991]. These authors have hypothesized that transition mutations occur at methylated CpG dinucleotides which are present in the male germline, but these sites remain unmethylated in female germ cells [Driscoll and Migeon, 1990]. Further, patterns of methylation may be disrupted during development of monozygotic twins as a single ovum supplies all nutrients until implantation, raising the possibility of early folate deficiency. In our study, the R1968X mutation occurred on the maternally derived chromosome 17, suggesting that the alteration occurred after methylation was re-established post-fertilization; this is consistent with the mosaic pattern that we observed in the unaffected twin’s cells.
The R1968X alteration has been reported previously in a mosaic individual who displayed segmental NF1 [Consoli et al., 2005]. In this woman, the mutation was found in a subset of the cells in affected areas, but not in any cells in unaffected areas. Interestingly, the mutation was present in her germline because it was transmitted from the patient to her daughter, who had a non-mosaic NF1 phenotype. The mosaic twin, whom we have described, displayed no signs of even segmental NF1. Experiments in mouse models have showed that variation in phenotypic manifestations of NF1 may be due to the micro-environment surrounding the target cells. In these models, tumor formation occurs following homozygous inactivation of the NF1 gene in the Schwann cells themselves, but only when the surrounding cells are heterozygous for an NF1 mutation and not if they are wild-type [Le and Parada, 2007]. Thus, the absence of manifestations of NF1 in the mosaic twin reported here result from too low a percentage of cells of neural crest origin with the mutation, from too low a percentage of cells with the mutation to create a micro-environment conducive to tumor formation, or to some combination of these factors. In any event, the unaffected twin described here demonstrates that some mosaic individuals may have no clinical evidence of NF1. Consequently, the frequency of NF1 mosaicism in the population may be underestimated.
Acknowledgments
The authors would like to thank the family members who have contributed samples and time in the clinic so that we could perform this research project. Also we thank Mary Anne Anderson and the CHGR Cell Culture Resource for making the stable cell lines, James Mull for technical assistance and Tammy Gillis and the CHGR Genomics Resource for DNA sequence analysis. Funded by grants from the NINDS (NS22224, NS24279), Neurofibromatosis Inc., Northeast, and the S. Sydney De Young Foundation.
References
- Akesson HO, Axelsson R, Samuelsson B. Neurofibromatosis in monozygotic twins: a case report. Acta Genet Med Gemellol (Roma) 1983;32:245–249. doi: 10.1017/s0001566000005109. [DOI] [PubMed] [Google Scholar]
- Anderson MA, Gusella JF. Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–858. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]
- Andrews JD, Mancini DN, Singh SM, Rodenhiser DI. Site and sequence specific DNA methylation in the neurofibromatosis (NF1) gene includes C5839T: the site of the recurrent substitution mutation in exon 31. Hum Mol Genet. 1996;5:503–507. doi: 10.1093/hmg/5.4.503. [DOI] [PubMed] [Google Scholar]
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- Bauer M, Lubs H, Lubs ML. Variable expressivity of neurofibromatosis-1 in identical twins. Neurofibromatosis. 1988;1:323–329. [PubMed] [Google Scholar]
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1–14. doi: 10.1016/j.jaad.2008.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R, O’Connell P, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- Consoli C, Moss C, Green S, Balderson D, Cooper DN, Upadhyaya M. Gonosomal mosaicism for a nonsense mutation (R1947X) in the NF1 gene in segmental neurofibromatosis type 1. J Invest Dermatol. 2005;125:463–466. doi: 10.1111/j.0022-202X.2005.23834.x. [DOI] [PubMed] [Google Scholar]
- De Luca A, Schirinzi A, Buccino A, Bottillo I, Sinibaldi L, Torrente I, Ciavarella A, Dottorini T, Porciello R, Giustini S, Calvieri S, Dallapiccola B. Novel and recurrent mutations in the NF1 gene in Italian patients with neurofibromatosis type 1. Hum Mutat. 2004;23:629. doi: 10.1002/humu.9245. [DOI] [PubMed] [Google Scholar]
- Detjen AK, Tinschert S, Kaufmann D, Algermissen B, Nurnberg P, Schuelke M. Analysis of mitochondrial DNA in discordant monozygotic twins with neurofibromatosis type 1. Twin Res Hum Genet. 2007;10:486–495. doi: 10.1375/twin.10.3.486. [DOI] [PubMed] [Google Scholar]
- Driscoll DJ, Migeon BR. Sex difference in methylation of single-copy genes in human meiotic germ cells: implications for X chromosome inactivation, parental imprinting, and origin of CpG mutations. Somat Cell Mol Genet. 1990;16:267–282. doi: 10.1007/BF01233363. [DOI] [PubMed] [Google Scholar]
- Dublin S, Riccardi VM, Stephens K. Methods for rapid detection of a recurrent nonsense mutation and documentation of phenotypic features in neurofibromatosis type 1 patients. Hum Mutat. 1995;5:81–85. doi: 10.1002/humu.1380050111. [DOI] [PubMed] [Google Scholar]
- Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S, Nurnberg P. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. 2000;66:790–818. doi: 10.1086/302809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager CM, Cohen PR, Tschen JA. Segmental neurofibromatosis: case reports and review. J Am Acad Dermatol. 1997;37:864–869. doi: 10.1016/s0190-9622(97)80013-8. [DOI] [PubMed] [Google Scholar]
- Hall JG. Twinning: mechanisms and genetic implications. Curr Opin Genet Dev. 1996;6:343–7. doi: 10.1016/s0959-437x(96)80012-8. [DOI] [PubMed] [Google Scholar]
- Huson SM, Compston DA, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet. 1989;26:712–721. doi: 10.1136/jmg.26.11.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadayel D, Fain P, Upadhyaya M, Ponder MA, Huson SM, Carey J, Fryer A, Mathew CG, Barker DF, Ponder BA. Paternal origin of new mutations in von Recklinghausen neurofibromatosis. Nature. 1990;343:558–559. doi: 10.1038/343558a0. [DOI] [PubMed] [Google Scholar]
- Kayes LM, Burke W, Riccardi VM, Bennett R, Ehrlich P, Rubenstein A, Stephens K. Deletions spanning the neurofibromatosis 1 gene: identification and phenotype of five patients. Am J Hum Genet. 1994;54:424–36. [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN. Mosaicism in sporadic neurofibromatosis type 1: variations on a theme common to other hereditary cancer syndromes? J Med Genet. 2008;45:622–631. doi: 10.1136/jmg.2008.059329. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, Peyrl A, Jenne DE, Hansmann I, Mautner VF. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet. 2004;75:410–423. doi: 10.1086/423624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TE, Sproul GT, Huerta MG, Rogol AD. Discordant puberty in monozygotic twin sisters with neurofibromatosis type 1 (NF1) Clin Pediatr (Phila) 1998;37:301–304. doi: 10.1177/000992289803700504. [DOI] [PubMed] [Google Scholar]
- Koul RL, Chacko A, Leven HO. Dandy-Walker syndrome in association with neurofibromatosis in monozygotic twins. Saudi Med J. 2000;21:390–392. [PubMed] [Google Scholar]
- Krkljus S, Abernathy CR, Johnson JS, Williams CA, Driscoll DJ, Zori R, Stalker HJ, Rasmussen SA, Collins FS, Kousseff BG, Baumbach L, Wallace MR. Analysis of CpG C-to-T mutations in neurofibromatosis type 1. Mutations in brief no. 129. Online. Hum Mutat. 1998;11:411. doi: 10.1002/(SICI)1098-1004(1998)11:5<411::AID-HUMU11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–4616. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listernick R, Mancini AJ, Charrow J. Segmental neurofibromatosis in childhood. Am J Med Genet A. 2003;121A:132–135. doi: 10.1002/ajmg.a.20183. [DOI] [PubMed] [Google Scholar]
- Littler M, Morton NE. Segregation analysis of peripheral neurofibromatosis (NF1) J Med Genet. 1990;27:307–310. doi: 10.1136/jmg.27.5.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machin G. Non-identical monozygotic twins, intermediate twin types, zygosity testing, and the non-random nature of monozygotic twinning: a review. Am J Med Genet C Semin Med Genet. 2009;151C:110–127. doi: 10.1002/ajmg.c.30212. [DOI] [PubMed] [Google Scholar]
- Moss C, Green SH. What is segmental neurofibromatosis? Br J Dermatol. 1994;130:106–10. doi: 10.1111/j.1365-2133.1994.tb06893.x. [DOI] [PubMed] [Google Scholar]
- National Institutes of Health Consensus Development Conference. Neurofibromatosis Conference Statement. Arch Neurol. 1988;45:575–578. [PubMed] [Google Scholar]
- Owerbach D, Bell GI, Rutter WJ, Brown JA, Shows TB. The insulin gene is located on the short arm of chromosome 11 in humans. Diabetes. 1981;30:267–270. doi: 10.2337/diab.30.3.267. [DOI] [PubMed] [Google Scholar]
- Pison G, D’Addato AV. Frequency of twin births in developed countries. Twin Res Hum Genet. 2006;9:250–259. doi: 10.1375/183242706776382338. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi VM. Neurofibromatosis: clinical heterogeneity. Curr Probl Cancer. 1982;7:1–34. doi: 10.1016/s0147-0272(82)80016-0. [DOI] [PubMed] [Google Scholar]
- Riccardi VM. Neurofibromatosis: phenotype, natural history and pathogenesis. Baltimore: Johns Hopkins University Press; 1992. p. 498. [Google Scholar]
- Schultz ES, Kaufmann D, Tinschert S, Schell H, von den Driesch P, Schuler G. Segmental neurofibromatosis. Dermatology. 2002;204:296–297. doi: 10.1159/000063363. [DOI] [PubMed] [Google Scholar]
- Scott L. The origin of monozygotic twinning. Reprod Biomed Online. 2002;5:276–284. doi: 10.1016/s1472-6483(10)61833-0. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000;16:198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Szudek J, Birch P, Riccardi VM, Evans DG, Friedman JM. Associations of clinical features in neurofibromatosis 1 (NF1) Genet Epidemiol. 2000;19:429–39. doi: 10.1002/1098-2272(200012)19:4<429::AID-GEPI13>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Tinschert S, Naumann I, Stegmann E, Buske A, Kaufmann D, Thiel G, Jenne DE. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet. 2000;8:455–459. doi: 10.1038/sj.ejhg.5200493. [DOI] [PubMed] [Google Scholar]
- Upadhyaya M, Shaw DJ, Harper PS. Molecular basis of neurofibromatosis type 1 (NF1): mutation analysis and polymorphisms in the NF1 gene. Hum Mutat. 1994;4:83–101. doi: 10.1002/humu.1380040202. [DOI] [PubMed] [Google Scholar]
- Wallace MR, Andersen LB, Saulino AM, Gregory PE, Glover TW, Collins FS. A de novo Alu insertion results in neurofibromatosis type 1. Nature. 1991;353:864–866. doi: 10.1038/353864a0. [DOI] [PubMed] [Google Scholar]
- Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]