

Abstract
Entry of herpes simplex virus (HSV) occurs either by fusion at the plasma membrane or by endocytosis and fusion with an endosome. Binding of glycoprotein D (gD) to a receptor such as nectin-1 is essential in both cases. We show that virion gD triggered the rapid down-regulation of nectin-1 with kinetics similar to those of virus entry. In contrast, nectin-1 was not constitutively recycled from the surface of uninfected cells. Both the nectin-1 α and β isoforms were internalized in response to gD despite having different cytoplasmic tails. However, deletion of the nectin-1 cytoplasmic tail slowed down-regulation of nectin-1 and internalization of virions. These data suggest that nectin-1 interaction with a cytoplasmic protein is not required for its down-regulation. Overall, this study shows that gD binding actively induces the rapid internalization of various forms of nectin-1. We suggest that HSV activates a nectin-1 internalization pathway to use for endocytic entry.
Keywords: herpes simplex virus, HSV, entry, nectin-1, glycoprotein, down-regulation, endocytosis, receptor
INTRODUCTION
Four glycoproteins in the herpes simplex virus (HSV) envelope are essential for virus entry and membrane fusion: gB, gD, and gH/gL (Heldwein and Krummenacher, 2008; Spear et al., 2000). Additionally, gC enhances the efficiency of entry by binding to heparan sulfate proteoglycans on the cell surface (Herold et al., 1991). Binding of gD to one of several cellular receptors initiates virus entry (Heldwein and Krummenacher, 2008; Spear et al., 2000). In addition, receptor binding induces a conformational change in gD that activates the fusion machinery comprised of gB and gH/gL (Fusco et al., 2005; Krummenacher et al., 2005; Lazear et al., 2008; Subramanian and Geraghty, 2007; Zago et al., 2004). Additionally, gB can interact with PILRα and gH can interact with αvβ3 integrins, but the role of these interactions in virus entry is unclear (Parry et al., 2005; Satoh et al., 2008).
Three unrelated cell surface molecules can independently serve as receptors for HSV-1: nectin-1, herpes virus entry mediator (HVEM), and 3-O-sulfated heparan sulfate (Geraghty et al., 1998; Krummenacher et al., 1998; Montgomery et al., 1996; Shukla et al., 1999; Whitbeck et al., 1997). Nectin-1, a member of the immunoglobulin superfamily, is the main HSV receptor on epithelial cells and neurons (Hung et al., 2002; Kopp et al., 2009; Simpson et al., 2005). It is an adhesion molecule found at adherens junctions in polarized epithelial cells, at synapses and varicosities in neurons, and at points of cell-cell contact in cultured cells (De Regge et al., 2006; Krummenacher et al., 2002; Sakisaka and Takai, 2004; Takahashi et al., 1999; Takai et al., 2008). Nectin-1 binding to a ligand, such as another nectin-1 or nectin-3, results in the accumulation of nectin-1 at contact sites (Krummenacher et al., 2002; Takahashi et al., 1999).
Multiple splice isoforms of nectin-1 (nectin-1α, β, and γ) are produced from the human PVRL1 gene (Lopez et al., 1995; Takai et al., 2008). The ectodomains of the three forms are identical, but nectin-1 α and β differ in the sequence of their transmembrane region and cytoplasmic tail, while nectin-1γ lacks a transmembrane region and is secreted (Cocchi et al., 1998; Geraghty et al., 1998; Lopez et al., 2001; Takai et al., 2008). Nectin-1α and β accumulate at cell contacts upon homophilic trans-interaction with a ligand and both promote cell adhesion. Nectin-1α recruits cytoplasmic afadin through a PDZ motif (WYV) in its cytoplasmic tail. In contrast, nectin-1β does not contain this motif (Cocchi et al., 1998; Takahashi et al., 1999; Takai et al., 2008). Afadin connects nectin-1α to the actin cytoskeleton and the catenin-cadherin complex to establish adherens junctions (Mandai et al., 1997; Takahashi et al., 1999). The role of nectin-1β in the organization of cell junctions is unclear. Both nectin-1α and β are functional receptors for HSV entry (Cocchi et al., 1998; Geraghty et al., 1998), and in fact, nectin-1α lacking its cytoplasmic tail still functions as an entry receptor (Subramanian et al., 2005).
In addition to interacting with gD during HSV entry, nectin-1α can interact with newly synthesized gD on the surface of HSV infected cells (Krummenacher et al., 2003). However, unlike nectin-nectin interactions, gD-nectin-1α interactions between cells result in down-regulation (internalization) of nectin-1α (Stiles et al., 2008). In this paper, down-regulation refers to changes in the level of protein expression at the cell surface rather than modulation of expression at the transcription/translation level. A mutant form of gD that cannot bind to nectin-1 (gD(A3C-Y38C)) (Connolly et al., 2005) does not induce nectin-1α down-regulation. The gD induced internalization of nectin-1 occurs in the absence of any other viral components. Although this effect on nectin-1α occurs in SY5Y, HeLa, and A431 cells, it does not occur in Vero cells. Interestingly, HSV entry into SY5Y, A431, and HeLa cells is by endocytosis, whereas entry into Vero cells is by direct fusion (Milne et al., 2005; Nicola et al., 2003; Stiles et al., 2008; Wittels and Spear, 1990).
This correlation between gD induced down-regulation and the pathway of HSV entry suggested that gD actually directs HSV into an endocytic pathway in certain cell types. To test this hypothesis, we compared the kinetics of HSV entry with those of HSV induced nectin-1 down-regulation. Indeed, we found that nectin-1 was rapidly internalized when exposed to virus with kinetics similar to those seen for HSV entry. In contrast, a gD-null virus had no effect on surface expression of nectin-1. We also observed colocalization between virus and receptor inside the cell during virus entry. Since the domain of nectin-1 that is required for down-regulation is unknown and the cytoplasmic tail of many surface proteins is involved in endocytosis (Le Roy and Wrana, 2005), we compared the kinetics of internalization of nectin-1α and β as well as forms of these two receptors lacking the cytoplasmic tail. We found that the cytoplasmic tail of nectin-1 had little influence on gD-induced down-regulation. These results suggest that down-regulation does not rely on the direct interaction of nectin-1 with cytoplasmic proteins, and highlights the importance of the nectin-1 ectodomain in this process. Lastly, we examined the rate of nectin-1α turnover in the absence or presence of gD expressing cells to determine whether gD takes advantage of the natural turnover of nectin-1 for down-regulation or actively triggers internalization. Nectin-1α was not naturally internalized in the absence of gD over the course of the experiment. In contrast, nectin-1α was rapidly internalized when gD was present. Collectively, our data show that binding of gD actively induces two isoforms of nectin-1 to be rapidly internalized.
RESULTS
HSV virions down-regulate nectin-1 with kinetics similar to virus entry
First, we examined whether nectin-1α was down-regulated from the cell surface early during endocytic entry of HSV (Fig. 1). We infected NGC12 cells, which express GFP-nectin-1α (Fig. 3A), with HSV KOS (MOI = 50). Virus was incubated with cells at 4°C for 45 min for attachment, and then incubated at 37°C for 30 min to allow entry. Cells were detached with versene and analyzed by FACS for surface nectin-1 expression. We used a pAb against GFP (ab290) to detect the tagged nectin-1α because the anti-nectin-1 antibodies suitable for FACS detection (mAbs CK6 and CK41) are blocked by gD binding to nectin-1 (Krummenacher et al., 2000). We found that infected cells expressed only 30% as much nectin-1α as mock infected cells (Fig. 1A). To demonstrate that down-regulation of nectin-1α was caused by virion gD, we inoculated cells with the gD null virus KOSgDβ at the equivalent MOI. This virus preparation was produced in Vero cells and lacked gD both genotypically and phenotypically (Dean et al., 1994). The level of surface nectin-1 expression in cells treated with KOSgDβ was the same as the level detected on the mock infected control cells (Fig. 1A). Thus, down-regulation of nectin-1α requires virion gD. We also observed that the amount of GFP-nectin-1α on the cell surface decreased as a function of the MOI of HSV (Fig. 1B). This decrease is not linear and appears to reach a plateau at about 40% of the initial amount of nectin-1. It is possible that this represents a more resistant population of nectin-1 buried at cell-cell contacts and less accessible to virus during the short time of the experiment.
Figure 1.

gD on HSV virions down-regulates nectin-1. Virus was added to NGC12 cells for 45 min at 4°C before shifting to 37°C for indicated time. Cells were then stained with anti-GFP pAb ab290 followed by anti-rabbit Ig-PE to detect GFP-nectin-1α and analyzed by FACS. The level of GFP-nectin-1α detected on the cell surface when no virus was added was set to 100%. Average of 3 experiments +/− SE. (A) HSV KOS MOI=50 or KOSgDβ equivalent of MOI=50 was added to cells for 30 min. (B) Various MOIs of HSV KOS were added cells for 30 min. (C) MOI=50 of HSV KOS was added to cells for various time points. Average of 3 experiments +/− SE. (D) Rate of HSV entry. HSV KOS (100 pfu/well) was added to cell monolayers and extracellular virus was acid inactivated at various timepoints. Cells were then overlaid and plaques were allowed to form (Highlander et al., 1989; Huang and Wagner, 1964; Milne et al., 2005; Milne et al., 2003). A representative experiment of 3 is shown. (E) Soluble gD down-regulates nectin-1. Soluble gD285t (Rux et al., 1998) (100 μg/ml) was added to NGC12 cells for 30 min at 37°C. Cells were stained for FACS as in A-C. The level of GFP-nectin-1α detected on the cell surface in the absence of gD was set to 100%. Data represent the average of 4 experiments +/− SE.
Figure 3.

Characterization of nectin-1α and β cell lines. (A) Diagram of nectin-1α and β constructs. Glycosylation sites are represented by lollipops. Cells line marked with * were previously described by Krummenacher et al. (2003). ‘G’ in the cell line name indicates a GFP tag. (B) Surface expression of nectin-1 was detected by FACS using mAb CK41-PE. Histogram of PE fluorescence intensity is shown. B78 are the nectin-1 negative parental cells.
To determine the kinetics of nectin-1 down-regulation by HSV, we inoculated NGC12 cells with HSV KOS (MOI=50) and allowed virus to bind at 4°C for 45 min. The temperature was then shifted to 37°C for 0, 2, 5, 15, 30, or 40 min. At each time point, the infected cells were chilled on ice to stop infection and analyzed by FACS. We found that down-regulation of nectin-1α from the cell surface occurred rapidly and greater than 50% of nectin-1α was internalized within the first 5 minutes (Fig. 1C). An additional 30% internalization occurred over the next 30 min.
To determine whether the rate of nectin-1 down-regulation correlates with the rate of virus entry into NGC12 cells, HSV KOS (100 pfu/well) was allowed to bind to cells at 4°C, followed by a shift to 37°C for 0, 2, 5, 15, 30, or 45 min. At each time point, extracellular virions were inactivated with citrate buffer pH 3.0, cells were overlaid with methylcellulose, and incubated 60 hr for plaque formation (Highlander et al., 1989; Huang and Wagner, 1964; Milne et al., 2005; Milne et al., 2003). We found that greater than 45% of the virions were internalized within 5 min of temperature shift (Fig. 1D). This is similar to the rate of entry shown previously for C10 cells (Milne et al., 2005), which express untagged nectin-1α instead of GFP-nectin-1α. Thus, nectin-1α down-regulation from the cell surface is rapid and has kinetics similar to those of HSV endocytosis (Fig. 1C).
We next wanted to determine if gD needed to be membrane bound, in the virus or on the cell surface, to cause nectin-1α down-regulation (Fig. 1E). We added 100 μg/ml soluble gD285t (Rux et al., 1998) to NGC12 cells for 30 min at 37°C. Nectin-1α expression was then analyzed by FACS. We found that the level of nectin-1 expression was reduced 30% as compared to the mock treated cells. Therefore, gD does not need to be membrane bound in order to induce nectin-1 internalization, although this may improve the efficiency.
Since the kinetics of nectin-1 down-regulation and virus entry were similar, we hypothesized that HSV induced nectin-1 internalization in order to access the endosomal pathway. If this is the case, we should be able to detect HSV and nectin-1 together inside the cell at short time points after entry initiation. To test this, we added HSV KOS (MOI=50) to NGC12 cells for 45 min at 4°C before raising the temperature to 37°C for 15 min. Cells were then fixed, permeabilized, and stained with anti-gD mAb MC5 (Atanasiu et al., 2007). We found numerous instances of colocalization between the gD stain of the virus and distinct punctate dots of GFP-nectin-1 inside the cell (white arrows, Fig. 2). Similar colocalization was also seen at 10 min and 30 min infection (data not shown). The colocalization of HSV with nectin-1 inside the cell shortly after entry suggests that nectin-1 internalization and virus entry are part of the same pathway.
Figure 2.

HSV and nectin-1 colocalize inside the cell. Virus was added to NGC12 cells for 45 min at 4°C before shifting to 37°C for 15 min. Cells were then fixed and permeabilized. GFP-nectin-1 is shown in green and virus stained with anti-gD mAb MC5 and Alexa 594 is shown in red. White arrowheads point to spots of colocalization (yellow) between nectin-1 and gD staining. A single confocal slice from a representative experiment of two is shown.
Expression of nectin-1 isoforms and truncations
The cytoplasmic tail of nectin-1α contains a PDZ motif that allows it to interact with intracellular afadin while that of nectin-1β lacks this motif. To determine if this motif plays a role in gD induced nectin-1 down-regulation, we compared several forms of nectin-1. First, we cloned the cDNA of the splice isoform, nectin-1β, from SY5Y human neuroblastoma cells. The splice site at amino acid 336 results in a different transmembrane region and cytoplasmic tail from that of nectin-1α (Cocchi et al., 1998; Geraghty et al., 1998) (Fig. 3A). Secondly, we truncated nectin-1α after amino acid 389 thereby eliminating most of its cytoplasmic tail (aa 390-517). We also truncated nectin-1β at amino acid 373 to remove its cytoplasmic tail (aa 374-458) (Fig. 3A). Each nectin-1 was tagged with GFP at either the N or C terminus. Previous studies determined that the position of the GFP tag had no effect on the function of nectin-1α in cell adhesion or HSV infection (Krummenacher et al., 2003).
We engineered stable cell lines, derived from nectin-1 negative B78H1 cells, each expressing one of the forms of nectin-1. These cell lines were called N1B, N1BG, NGC-389t, and N1BG-373t (Fig. 3A). Cell lines were screened by GFP fluorescence (not shown) and clones were selected based on the level of nectin-1 surface expression by FACS using mAb CK41. Nectin-1 was readily detected on each of the cell lines (Fig. 3B) and accumulated at sites of cell-cell contacts (not shown) similarly to nectin-1α (Krummenacher et al., 2003).
To determine if each form of nectin-1 could be used as an HSV receptor, each cell line was infected with HSV KOStk12, which contains the lacZ gene, and entry was measured by β-galactosidase expression at 6 hpi (Warner et al., 1998). As expected, HSV KOStk12 was unable to enter receptor negative B78H1 cells, but it entered each of the B78 derived nectin-1α or β cell lines (data not shown). Additionally, viral titers and plaque sizes on N1B, N1BG, NGC-389t, and N1BG-373t cells were equivalent to those on C10, NGC12, and CG23 cells expressing nectin-1α (not shown) (Krummenacher et al., 2003). These data confirmed that both the α and β isoforms of nectin-1 are receptors for HSV (Cocchi et al., 1998; Geraghty et al., 1998). Previously, Subramanian et al. (2005) found that the cytoplasmic tail of nectin-1α was not required for HSV entry into CHO cells. Our data indicate that the cytoplasmic tail of nectin-1 α and β is also not required for entry into B78H1 derived cells, which HSV enters through a different endocytic entry pathway that does not require endosomal acidification (Milne et al., 2005; Nicola et al., 2003; Nicola and Straus, 2004). Thus, these B78H1 derived cell lines are suitable for analyzing virus endocytosis and nectin-1 down-regulation.
HSV enters B78 cells expressing nectin-1β and truncated forms of nectin-1 by endocytosis
HSV entry into B78 cells expressing nectin-1α (C10 cells) occurs only by endocytosis (Milne et al., 2005; Stiles et al., 2008). We used a protease protection assay (Milne et al., 2005) to determine whether the same route of entry is used when the receptor was nectin-1β or nectin-1 lacking the cytoplasmic tail. In this assay, if intact virions are internalized into endosomes before fusion, the viral glycoproteins will be protected from treatment of the cells with Proteinase K. Since entry does not occur at the plasma membrane of B78-nectin-1 cells (Milne et al., 2005), protection of virions is a good indication that productive infection proceeds through endocytosis. Virus was added to cells at 4°C for 45 min, and then shifted to 37°C for 15 min. Proteinase K was added to digest glycoproteins left on the cell surface, either in the envelope of extracellular virions or on the plasma membrane as a result of direct fusion of virus at the cell surface. Cell lysates were prepared and viral gB was immunoprecipitated and detected by western blot. When cells were maintained at 4°C (no virus entry), gB was completely digested by Proteinase K (Fig. 4, lane 1). As another control, infected cells were mock digested. In this case, equivalent amounts of gB were detected on each of the cell lines (Fig. 4, lane 2). We used B78 cells which do not express receptors and C10 cells which express nectin-1α as negative and positive controls for virus endocytosis (Milne et al., 2005; Stiles et al., 2008). When we exposed receptor negative B78 cells to virus, followed by proteinase K, gB was completely digested indicating that the virus remained on the surface. However, when C10 cells were infected with HSV, gB was protected from proteinase K digestion, as expected since HSV enters these cells by endocytosis (Milne et al., 2005). We found that a similar amount of virion gB was also protected from proteinase K treatment on cells expressing nectin-1β (N1B). gB was also protected in cells expressing truncated nectin-1α (NGC-389t) and truncated nectin-1β (N1BG-373t) (Fig. 4, lane 3). However, less gB was protected on these cells as compared to the cells expressing full length nectin-1 α and β, suggesting that less virus was endocytosed within 15 min on these cells. These results demonstrate that HSV is endocytosed into cells expressing either nectin-1α or nectin-1β. Endocytosis of HSV also occurred in cells expressing nectin-1α or β lacking its cytoplasmic tail, although it may occur less efficiently. Since B78H1 cells expressing nectin-1α do not allow HSV entry at the plasma membrane (Milne et al., 2005), we conclude that endocytosis of virions into B78H1 cells expressing nectin-1β or truncated nectin-1 also leads to productive infection of these cells.
Figure 4.
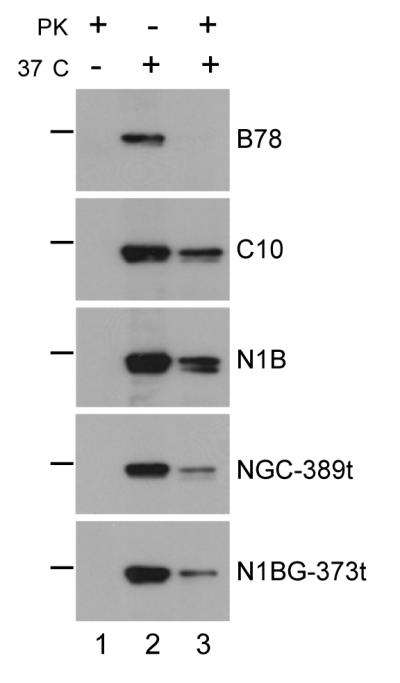
HSV is endocytosed during entry into nectin-1 cell lines. Virus was allowed to attach to cells at 4°C before infection was allowed to proceed for 15 min at 37°C. Cells were then treated with proteinase K (+) or mock digested (−). Cells were lysed in the presence of protease inhibitors and gB was detected by immunoprecipitation with mAb DL16 and western blot (pAb R69). The presence of gB in lane 3 indicates virion endocytosis. PK means proteinase K and the bar on the right indicates the position of the 115 kDa molecular weight marker.
Nectin-1β and truncated nectin-1α and β are down-regulated from the cell surface
We previously used a co-culture assay to show nectin-1α down-regulation after binding by gD expressed on an adjacent cell (Stiles et al., 2008). Here, we used the co-culture assay to determine if nectin-1β or truncated nectin-1α or β are also down-regulated. Each nectin-1 target cell line was co-cultured overnight with each of four effector cell lines: B78 cells (no gD), wild type gD (gDwt), gD(W294A) which is a mutant form of gD that binds to nectin-1 with higher affinity than gD(wt), or gD(A3C-Y38C) which is a mutant gD that cannot bind to nectin-1 but can bind HVEM (Connolly et al., 2005; Krummenacher et al., 2005). We found that all forms of nectin-1 were clearly detected by GFP fluorescence when cells were co-cultured with B78 cells (Fig. 5A) or with B78-gD(A3C-Y38C) cells (data not shown). However, when the cells were co-cultured with either B78-gD(wt) cells (Fig. 5B) or B78-gD(W294A) cells (data not shown), the amount of each form of nectin-1 was greatly reduced.
Figure 5.

Decreased expression of nectin-1α and β after co-culture with gD expressing cells. Cells expressing GFP tagged forms of nectin-1α (CG23, NGC12, NGC-389t) or nectin-1β (N1BG, N1BG-373t) were co-cultured with B78 cells (no gD) (A) or wild type gD expressing cells (B). After 16 hr of co-culture, cells were fixed and permeabilized. Nectin-1-GFP is shown in green and nuclei are stained with DAPI (blue). A representative experiment is shown. (C) Down-regulation of nectin-1 detected by FACS. Target cells were labeled with Qtracker655 and mixed with effector cells expressing the indicated forms of gD or with B78 cells at a target:effector ratio of 1:2. After overnight co-culture, cells were stained with mAb CK41-PE to detect nectin-1. Target nectin-1 cells were positively selected based on Qtracker655 fluorescence while unlabelled effector (gD expressing) cells were excluded. Bar graphs represent PE fluorescence of Qtracker655-positive target nectin-1 expressing cells as a percent of the PE fluorescence in co-cultures with B78 cells from the average of 3 experiments +/− SE.
We used FACS to quantitate this effect (Fig. 5C). Nectin-1 target cells were stained with Qtracker 655 prior to co-culture with effector cells. This facilitated selection of the target cell population for FACS analysis. We co-cultured nectin-1 target cells with gD expressing effector cells overnight, detached the cells, stained them with anti-nectin-1 mAb CK41, and used the Qtracker signal to gate on nectin-1 expressing cells for analysis. When each nectin-1 cell line was co-cultured with B78 cells, a high level of nectin-1 was detected and this was set at 100%. When each target cell line was co-cultured with B78-gD(A3C-Y38C) cells (Fig. 5C, black bar), we detected a similar level of nectin-1 as observed in co-cultures with B78 cells. However, when the nectin-1 cell lines were co-cultured with B78-gD(wt) cells, the level of nectin-1 detected on the cell surface was reduced to less than 30% of the control (Fig. 5C, white bars). Similarly, co-culture with B78-gD(W294A) cells resulted in reduction of nectin-1 levels to less than 15% of the control (Fig. 5C, gray bars). In fact, gD(W294A) down-regulated nectin-1 only slightly better than gD(wt) despite a 40 fold difference in affinity of these two forms of gD for nectin-1 (Krummenacher et al., 2005). Since binding of gD(wt) is sufficient to induce a high level of down-regulation, it is likely that the higher affinity of gD(W294A) offered little additional advantage for down-regulation. Moreover, the difference in affinity is almost entirely due to a difference in the rate of association; the stability of the complex (off rate) is the same for both forms of gD (Krummenacher et al., 2005). These data indicate that nectin-1β and truncated nectin-1α and β are down-regulated similarly to nectin-1α.
Down-regulation of nectin-1 in response to gD occurs rapidly
Because the truncated forms of nectin-1α and β were down-regulated to the same extent as the full-length receptors after overnight co-culture, we wondered if a difference in the cytoplasmic tail would affect the rate of down-regulation. Therefore, we quantified the amount of nectin-1 that remained on the cell surface after 0, 20, 40, 60, 120, and 240 minutes of co-culture with gD expressing effector cells. In this experiment, we used a double referencing analysis. First, we expressed the level of nectin-1 on the target cells in co-cultures with gD relative to the nectin-1 level in the co-culture of target cells and B78 effector cells (100%) at each time point. Second, we normalized each value relative to the initial level of nectin-1 at time zero (100%). This enabled us to compare the kinetics of the various target cell lines incubated with two different effector cell lines (Suppl Table 1). For a negative control, we co-cultured each target cell line with B78-gD(A3C-Y38C) cells. In these cases, nectin-1 levels remained near 100% at all time points (not shown). When cells expressing either gD(wt) (Fig. 6 triangle) or gD(W294A) (Fig. 6 square) were co-cultured with any of the nectin-1 cell lines, the receptor was rapidly down-regulated from the cell surface (Fig. 6). In each case, cells co-cultured with the gD(W294A) high affinity mutant had lower levels of nectin-1 at each time point as compared with gD(wt), although this difference is greater in some cell lines (N1B, NGC-389t) than in others (Suppl. Table 1). The rate of nectin-1 down-regulation was comparable in C10, N1B and NGC12 cells, indicating that both the α and β isoforms acted similarly (Fig. 6A-C). Surprisingly, the addition of GFP at the C-terminus of wt nectin-1α and β (CG23 and N1BG cells respectively), accelerated down-regulation by 2-3 fold (Fig. 6D,E). Truncation of the cytoplasmic tails of nectin-1α and β (NGC-389t and N1BG-373t cells respectively, Fig. 6F,G) led to a slower rate of down-regulation compared to the corresponding full-length receptors (Fig. 6C,E). Together, these results show that although the cytoplasmic tail is not essential, it does contribute to down-regulation. However, this contribution does not rely on any connection between the cytoplasmic tail of nectin-1 and afadin since the α and β isoform have the same kinetics.
Figure 6.

Rate of nectin-1 down-regulation from the cell surface. Target cells were labeled with Qtracker655 and co-cultured with effector cells expressing the indicated forms of gD or with B78 cells (no gD) at a target:effector ratio of 1:2. At each time point, cells were stained with mAb CK41-PE to detect nectin-1 and analyzed by FACS. Target nectin-1 cells were positively selected based on Qtracker655 fluorescence. The level of nectin-1 surface expression (PE fluorescence) on cells co-cultured with B78 cells was set at 100% as in figure 5. For each target cell line, we normalized the level of fluorescence to that at time 0 for comparison between effector cells. An average of 3 experiments is shown +/− SE.
Nectin-1 internalization is induced by gD
Next, we investigated the mechanism by which gD redirects nectin-1 from the cell surface to the lysosomal pathway. Our previous data (Stiles et al., 2008) did not determine whether 1) gD takes advantage of the normal turnover of nectin-1 (i.e. constitutive endocytosis and recycling) from the cell surface and redirects it to a degradation pathway, or 2) whether gD actively induces the internalization of nectin-1 which would usually remain stably on the cell surface. To address this question, we biotinylated proteins on the surface of NGC12 cells, and then co-cultured these cells with either B78 or B78-gD(wt) expressing cells for 0, 15, 30, 60, 120, 180 minutes at 37°C (Fig. 7A). After co-culture, cell surface proteins were digested with Proteinase K, and any internalized biotinylated proteins were pulled down from lysates with neutravidin beads. Nectin-1α was then detected by western blot. No internalized nectin-1α was detected when NGC12 cells were co-cultured with B78 cells (Fig. 7B, left panel). However, when NGC12 cells were co-cultured with B78-gD(wt) cells, nectin-1α was detected inside the cells within 15 min, and the level of internalized nectin-1α increased up to 1 hr (Fig. 7B, middle panel). After 2 hr of co-culture, less nectin-1α was detected, likely because of degradation. These results show that nectin-1α does not usually turnover within 3 hr of cell culture and this contrasts with the rapid internalization of nectin-1α that occurs in response to gD.
Figure 7.

Nectin-1 internalization is triggered by gD. (A) Steps of nectin-1 internalization assay. (B) NGC12 cells were biotinylated and co-cultured with B78 (left panel) or B78-gD(wt) (middle panel) cells for various times before treatment with Proteinase K. Cells were lysed in the presence of protease inhibitors and internalized biotinylated GFP-nectin-1α was detected by pull-down with neutravidin beads and western blot (mAb CK8). Right panel shows control conditions: biotinylated, mock digested (lane 1); biotinylated, held at 4°C, digested with Proteinase K (lane 2); not biotinylated (lane 3). Arrowhead points to GFP-nectin-1α and the bar on the left indicates the 115 kDa molecular weight marker.
DISCUSSION
HSV enters most cells by endocytosis, even though it has the ability to fuse its envelope directly with the cell plasma membrane. The choice of entry pathway appears to be dependent on cell type (Milne et al., 2005; Nicola et al., 2005; Nicola et al., 2003; Nicola and Straus, 2004; Stiles et al., 2008). Although both HVEM and nectin-1 allow endocytic entry into CHO cells (Nicola et al., 2003), several pieces of data suggest that the interaction between gD and its receptor may play a role in virion endocytosis. In CHO cells, usage of nectin-1 or nectin-2 by HSV ANG leads to different entry pathways (Delboy et al., 2006). Binding of gD to nectin-1 causes down-regulation of the receptor only in cells where entry is endocytic and not in Vero cells where entry occurs at the cell surface (Stiles et al., 2008). In the present study, we correlated the kinetics of nectin-1 down-regulation with those of HSV entry and showed colocalization of internalized HSV with nectin-1 shortly after entry initiation. We also showed that the nectin-1 ectodomain rather than its cytoplasmic tail is critical for this activity. Through analysis of nectin-1 turnover, we showed that HSV gD actively induces internalization of nectin-1. Altogether, it appears that HSV gD efficiently activates an endocytic pathway for both nectin-1 internalization and HSV entry by interacting with two nectin-1 isoforms. This may explain the high efficiency of HSV entry into various cell types.
gD on virions induces nectin-1 down-regulation and virus entry
Our data show that gD on virions induces a rapid down-regulation of nectin-1 at a rate comparable to that of HSV entry. Nectin-1 down-regulation occurs when NGC12 cells are exposed to HSV virions at 37°C. In such cells derived from B78H1 cells, the virus itself is endocytosed. In contrast, nectin-1 was not down-regulated when cells were exposed to KOSgDβ virus which lacks gD. Therefore, nectin-1 internalization is driven by virion gD alone. In addition, the kinetics of gD-induced down-regulation of nectin-1 are comparable to those of virus entry; in both cases, 45% of the internalization occurs within 5 min. Indeed, we were able to visualize colocalization between nectin-1 and HSV inside cells at short time points after the initiation of entry. In addition, we did not see any increase in membrane protrusions or other indications of a generalized increase in phagocytic-like uptake as described by Clement et al (2006). Therefore, we suggest that HSV actively induces internalization of its receptor nectin-1, thereby promoting its own endocytosis into the cell during entry. Although, B78H1-derived cells do not require endosomal acidification for fusion, it is likely that the preceding step of viral internalization is driven in the same way in cells requiring low pH for endocytic entry.
Nectin-1 cytoplasmic tail is not required for down-regulation
Since nectin-1 is internalized from the cell surface, we reasoned that the cytoplasmic tail of the receptor might be important for the endocytic process. Adaptor proteins involved in the formation of clathrin coated pits or caveolae bind to the cytoplasmic tails of cell surface receptors that are endocytosed upon ligand binding (Le Roy and Wrana, 2005). Using the nectin-1β isoform and truncated forms of nectin-1α and nectin-1β, we found that the nectin-1 cytoplasmic tail is not required for its down-regulation from the cell surface. Previous studies with nectin-1 chimeras showed that nectin-1 with a heterogenous cytoplasmic tail mediated entry or cell-cell fusion (Gianni et al., 2004; Subramanian et al., 2005). The same studies also showed that nectin-1 lacking its cytoplasmic tail functioned as a receptor. In this study, we looked specifically at the effect of the cytoplasmic tail on the efficiency and the route of entry as well as nectin-1 internalization. These new results suggest that a change in or absence of the cytoplasmic tail does not alter the entry pathway into B78H1 derived cells, although it may affect entry efficiency. Indeed, the ability of HSV to use two isoforms of nectin-1 with different cytoplasmic tails and potentially different expression patterns may enhance the tropism of the virus. Furthermore, the endocytic process does not require binding of cytoplasmic proteins directly to the tail of nectin-1. However, it is still possible that the nectin-1 ectodomain interacts with other surface proteins that play a role in its down-regulation.
In many aspects, the observations on HSV usage of nectin-1 are reminiscent of reovirus entry. Reovirus follows an endocytic entry pathway and uses a cell adhesion protein, JAM-A, as its receptor (Campbell et al., 2005). However, β1 integrin, which interacts with the JAM-A ectodomain, is required for internalization of virions into the endocytic compartment necessary for viral uncoating (Maginnis et al., 2006). Interestingly, the cytoplasmic tail of JAM-A was not necessary for reovirus infection, however, the NPXY internalization motifs in the cytoplasmic tail of β1 integrin were essential for endocytosis (Maginnis et al., 2008). The similarity between the circumstances for reovirus entry and those of HSV are reinforced by the fact that nectin-1 can form a cis-interaction with αvβ3 integrin through its ectodomain (Sakamoto et al., 2006). Interestingly, the equine herpesvirus type 1 (EHV-1) entry pathway, which is also cell-type dependent, requires an interaction between gD and αvβ5 integrin for endocytosis (Frampton et al., 2007; Hasebe et al., 2009; Van de Walle et al., 2008). However, HSV gD lacks the RSD motif found in EHV-1 gD and therefore cannot interact directly with integrins (Van de Walle et al., 2008).
Factors influencing the rate of nectin-1 down-regulation
The nectin-1α and β cytoplasmic tails and transmembrane domains are completely unrelated and do not contain identifiable common motifs. Since both isoforms were down-regulated at similar rates, we conclude that interaction with proteins containing a PDZ-domain (e.g. afadin, PAR-3) is not needed for nectin-1 down-regulation. Although the nectin-1 cytoplasmic tail was not required for its down-regulation, we found that the rate of down-regulation differed among the various forms of nectin-1. In the truncated nectin-1α (NGC-389t) and nectin-1β (N1BG-373t) cells, the absence of the cytoplasmic tail decreased the rate of receptor internalization. This could be due to the lack of binding sites for proteins or motifs that increase the efficiency of endocytosis. Alternatively, the absence of the cytoplasmic tail may alter the stability of the receptor in the membrane. Interestingly, we saw an enhanced rate of down-regulation of either nectin-1α or β with GFP on the C-terminus. This may be due to a stabilizing effect of the tag or slight changes in density of the receptors. Indeed, in some cases, aggregation or dimerization of cell surface receptors upon ligand binding is a mechanism inducing endocytosis. It is possible that the cytoplasmic tail of nectin-1 influences its ability to cluster and that the C-terminal GFP increases this propensity, thus favoring internalization. Interestingly, a soluble form of gD (gD285t) induces nectin-1 down-regulation with a relatively low efficiency. Although an accurate comparison between soluble concentrations and levels of cell or viral surface expression is difficult to achieve, it is possible that soluble gD may not cluster nectin-1 as efficiently as membrane bound gD to induce rapid and robust endocytosis.
Nectin-1 does not turnover naturally but internalization is induced by gD
In NGC12 cells, the interaction of gD with nectin-1 results in endocytosis of the virus and nectin-1. Two mechanisms could account for this: either the virus takes advantage of constitutive recycling of nectin-1 from the plasma membrane or binding of gD induces endocytosis of the receptor to the advantage of the virus. Since little is known about the turnover of nectin-1, we tested the stability of nectin-1 at the cell surface in the absence or presence of gD. We showed that nectin-1α is not internalized within 3 hr of co-culture of NGC12 cells with B78 cells. However, when NGC12 cells are co-cultured with gD expressing cells, nectin-1 is rapidly endocytosed. Furthermore, the internalized nectin-1 is degraded in a low pH-dependent manner in lysosomes (Stiles et al., 2008). This indicates that gD actively triggers an endocytic pathway which redirects the receptor to lysosomes for degradation. In contrast to nectin-1, E-cadherin, another cell adhesion molecule found at adherens junctions, undergoes constitutive endocytosis and recycling back to the plasma membrane (Le et al., 1999). This cycling is minimal when cells are confluent, but increases when cells begin to move apart and junctions are disassembled (Kamei et al., 1999; Le et al., 1999). Interestingly, under certain specialized conditions, such as epithelial-to-mesenchymal transition, E-cadherin is targeted to lysosomes for degradation (Fujita et al., 2002; Hicke and Dunn, 2003). Little is currently known about the regulation of nectin-1, but it is possible that nectin-1, like E-cadherin, is also subject to a number of levels of regulation, and that gD binding reactivates a dormant pathway for the benefit of the virus.
Interestingly, HSV is not the only pathogen that causes nectin-1 to be down-regulated from the cell surface. Chlamydia infected cells also show an 85% reduction in cell surface nectin-1 at 48 hpi. This down-regulation was shown to be at the level of protein degradation and involves the chlamydial protease-like activity factor (CPAF) (Sun et al., 2008; Sun and Schoborg, 2009). In this case, nectin-1 down-regulation is thought to cause disruption of adherens junctions allowing for better chlamydial dissemination and pathogenesis (Sun et al., 2008). In addition to allowing virus endocytosis, the down-regulation of nectin-1 may aid in HSV dissemination in a similar manner.
The new data presented here demonstrate the effects of gD on the natural turnover of nectin-1 and support the model that gD activates the internalization of nectin-1 to drive virions to an endocytic pathway. We found that the kinetics of nectin-1 down-regulation induced by virion gD were similar to those of virus entry and that virus and receptor colocalized inside of cells shortly after entry initiation. HSV gD triggers a rapid endocytosis of nectin-1, which is influenced by the nectin-1 cytoplasmic tail but does not require direct interaction of cytoplasmic proteins with the receptor. We suggest that another cell surface protein, such as an integrin, interacts with nectin-1 through its ectodomain and plays a crucial role in nectin-1 internalization by linking the receptor to the endocytosis machinery in the cytoplasm. Our data clearly show that the cellular response triggered by gD is very different from that which occurs when nectin-1 binds to a natural ligand. Although, we do not yet understand what causes this difference, it is possible that gD binding destabilizes nectin-1 cis dimers or induces nectin-1 association with other transmembrane proteins that then lead to internalization. These possibilities will be explored in future studies to better understand how HSV exploits nectin-1 to gain entry into cells.
MATERIALS AND METHODS
Viruses, antibodies and proteins
i) Viruses
HSV-1 KOS and KOS tk12 were grown and titered on Vero cells and purified as described (Handler et al., 1996; Montgomery et al., 1996). KOS gDβ was obtained from Dr. P.G. Spear (Dean et al., 1994). Non-complemented KOSgDβ was produced in Vero cells and complemented viruses were produced on VD60 cells (Dean et al., 1994; Ligas and Johnson, 1988). Partial purification of KOSgDβ was done as described by Stiles et al. (2008). Titration of KOSgDβ was performed on VD60 cells. Equivalent amount of capsid was present in supernatant for the virus produced in VD60 and that produced in Vero cells as determined by western blot.
ii) Antibodies
Anti-nectin-1 monoclonal antibodies (mAbs) CK8 and CK41 were described previously (Krummenacher et al., 2000). For FACS, CK41 was directly coupled with phycoerythrin (PE) at Molecular Probes/Invitrogen. Anti-HSV gB antibodies were as follows: pAb R69 (Eisenberg et al., 1987) and mAb DL16 (Bender et al., 2005). Anti-GFP pAb ab290 was purchased from Abcam. Secondary anti-IgG coupled with PE, Alexa 594, or Alexa 488 were purchased from Molecular Probes/Invitrogen.
iii) Cell lines
Murine melanoma B78H1 cells were grown in DMEM with 5% FCS and penicillin/streptomycin. Previously described B78H1 transfected cell lines B78-C10, B78-NGC12, B78-CG23 (abbreviated here as C10, CG23, NGC12 respectively), and B78H1-control-16 (abbreviated here B78) (Krummenacher et al., 2003) and B78-gD(wt), B78-gD(W294A) and B78-gD(A3C-Y38C) (Stiles et al., 2008) were grown in the same medium supplemented with 500 μg/ml G418.
Construction of nectin-1β and truncated nectin-1α constructs
i) Truncated nectin-1α construct
Nectin-1α truncated at amino acid 389 with GFP at the N-terminus was derived from plasmid pCK495 (Krummenacher et al., 2003) using forward primer HveC389tF (5′-CAAGGGTGACTAGAGCACCAAGAAGC) and reverse primer HveC389tR (5′-GCTTCTTGGTGCTCTAGTCACCCTTG) to insert a stop codon at amino acid 390 using the Quickchange Mutagenesis Kit (Stratagene). The resulting plasmid was named pCK613.
ii) Nectin-1β constructs
Total RNA was extracted from 7×106 SY5Y human neuroblastoma cells using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions including the optional DNase digestion and homogenization steps. cDNA was prepared using the Omniscript Reverse Transcription Kit (Qiagen) with random primers (Roche). Forward primer FHC5HIND (5′-GCCCAAGCTTATGGCTCGGATGGGGCTTGCGG) and reverse primer 3HIgRStopR (5′-CGCGGATCCCTAGGGGCACTCTCCTCGAGG) were used to amplify the full-length nectin-1β ORF by PCR. The nectin-1β fragment was digested with restriction enzymes HindIII and BamHI and ligated into pcDNA3.1 to generate plasmid pKS655.
For GFP tagged nectin-1β constructs, the full-length nectin-1β sequence was PCR amplified using forward primer FHC5HIND and reverse primer 3HIgRNSR (5′-GGCGGATCCGGGGCACTCTCCTCGAGGTTCG), digested with HindIII and BamHI, and ligated into pcDNA3.1 to generate plasmid pKS646. Nectin-1β truncated at amino acid 373 was amplified using forward primer FHC5HIND and reverse primer HIgR373tNSR (5′-CGCGGATCCCTGCCGGTTGTACAGGAAGAAGAC), digested with HindIII and BamHI, and ligated into pcDNA3.1 to create plasmid pKS647. The enhanced GFP sequence from plasmid pEGFP (Clontech Laboratories Inc.) was amplified using forward primer 5′EGFP (5′-CGCGGATCCATGGTGAGCAAGGGCGAGGAGCTGTT) and reverse primer 3′GFP (5′-CGGTCTAGACTACTTGTACAGCTCGTCCAT) by PCR. The GFP fragment was digested with restriction enzymes BamHI and XbaI. The digested GFP fragment was ligated into plasmid pKS646 to generate plasmid pKS654 (full-length nectin-1β-GFP) and into plasmid pKS647 to generate plasmid pKS649 (nectin-1β-373t-GFP).
Cell lines expressing nectin-1 β and truncated nectin-1
i) Selection
B78H1 cells were transfected with plasmids pCK613 (GFP-nectin-1α-389t), pKS655 (nectin-1β), pKS654 (nectin-1β-GFP), or pKS649 (nectin-1β-373t-GFP) with Geneporter as recommended by the manufacturer. Clonal cell lines were selected by limiting dilution in the presence of 1 mg/ml G418 and further maintained in DMEM with 5% FCS, antibiotics and 500 μg/ml G418. Clones to be used in this study were selected by immunofluorescence and FACS for their level of nectin-1 expression. Clonal cell lines used in this study are: B78-NGC-389t #3, B78-N1B #21, B78-N1BG #19 and B78-N1BG-373t #7.
ii) Nectin-1 expression
For flow cytometry (FACS) experiments, cells were detached with 0.02% di-sodium EDTA (w/v) in PBS (Versene; Gibco-BRL) and resuspended in PBS containing 3% FCS, 0.01% sodium azide (PBS-FCS). Anti-nectin-1 mAb CK41-PE was diluted to 5μg/ml in cold PBS-FCS, 50 μl was added to 5×105 cells. Cells were incubated for 30 min on ice and then washed with 1 ml cold PBS-FCS before being fixed in 0.3 ml 3% paraformaldehyde (PFA) + 3% FCS in PBS.
Cell co-cultures
i) Immunofluorescence analysis
Target and effector cells were detached with trypsin, mixed at a 1:2 target-effector ratio and co-cultured for 16 hr on glass coverslips (3×105 cells per coverslip). The culture medium was DMEM supplemented with 5% FCS and antibiotics. Cells were fixed with 3% PFA in PBS for 1 hr at RT. Quenching, permeabilization and staining were performed as previously described (Krummenacher et al., 2003; Sodeik et al., 1997). In addition to GFP fluorescence, cells were stained with anti-gD mAb MC5 (Atanasiu et al., 2007) at 1μg/ml, followed by goat anti-mouse IgG coupled with Alexa 594 to detect gD on effector cells (not shown). Images were collected on a Nikon Eclipse E600 microscope equipped with a 20x objective and analyzed using Image-Pro Plus software (Media Cybernetics, Inc).
ii) FACS analysis
Target cells expressing nectin-1 were detached with trypsin, then counted and divided in aliquots at 5×106 cells/ml for labeling with Qdots using the Qtracker ™ 655 labeling kit (Quantum Dot Corp/Invitrogen, Hayward CA) for 1 hr at 37°C in DMEM supplemented with 5% FCS following the manufacturer’s instructions. Qdot labeling was done to identify target cells during FACS analysis. Target cells were washed 3 times with culture medium and mixed with twice as many unlabeled effector cells. A total of 1.5×106 cells were plated in each well of a 6-well plate and cultivated overnight in DMEM supplemented with 5% FCS and antibiotics. Target and effector cells were also cultivated separately as controls. For FACS analysis, cells were detached with Versene and resuspended in cold PBS-FCS. Labeling with anti-nectin-1 mAb CK41-PE (5 μg/ml) was performed on 0.5×106 cells (50 μl) for 30 min on ice. Mock stained cells were used as negative controls. Cells were washed with cold PBS-FCS and fixed with 3% PFA in PBS with 3% FCS. In order to gate target cells during FACS analysis, Qtracker655 was detected by excitation at 630 nm and reading at 660+/−15 nm. Qtracker655-positive target cells were positively selected for measurement of PE fluorescence.
iii) FACS kinetics analysis
Target nectin-1 cells were detached with trypsin and labeled at 5×106 cells/ml with Qdots using Qtracker ™ 655 labeling kit (Quantum Dot Corp/Invitrogen, Hayward CA) as described above for FACS analysis. Cells were washed 3x with culture medium and seeded at 0.5×106 cells per well of a six well plate. Target cells were incubated overnight at 37°C. 1×106 effector cells (2:1 effector: target ratio) were added per well in 2 ml of culture media. Cells were co-cultured at 37°C for 0, 20, 40, 60, 120, and 240 minutes. At each time point, cells were detached with versene and processed for FACS as described above. The PE geometric mean fluorescence (nectin-1 expression) in co-cultures with B78 cells (no gD) was used as a reference level and set at 100%. Levels of nectin-1 expression in each co-culture were represented as a percentage of this reference level. For each nectin-1 target cell line, the PE fluorescence (nectin-1 expression level) was normalized to the level at time 0 for comparison between effector cell lines.
Virus Infections
i) FACS analysis
0.75×106 NGC12 cells, expressing GFP-nectin-1α, were seeded in 12 well plates in DMEM with 5% FBS and antibiotics and allowed to attach 4 hours at 37°C. Media was removed and virus was added in 1ml chilled media and allowed to bind for 45 min at 4°C. The cells were then shifted to 37°C for 30 min. Cells were detached with versene and resuspended using cold PBS-FCS. Cells were labeled with anti-GFP pAb ab290 serum (1:400) in 50 μl PBS-FCS for 30 min on ice at 4°C. Cells were washed with cold PBS-FCS and labeled with goat anti-rabbit Ig coupled to PE (2 μg/ml) in 50 μl. Cells were then washed and fixed in 3% PFA in PBS with 3% FCS.
ii) FACS kinetics analysis
NGC12 cells were seeded and exposed to virus as described above for FACS analysis. After the 4°C incubation for virus attachment, the temperature was shifted to 37°C by placing the plates in a water bath for 0, 2, 5, 15, 30, 40, and 120 min. At each time point, cells were placed on ice, washed with citrate buffer pH 3 and detached with versene. Cells were labeled with anti-GFP pAb ab290 for FACS as described above. The level of nectin-1 internalization at 120 min was considered to be 100% of the possible internalization and levels of internalization were normalized to this point.
Soluble gD
NGC12 cells were seeded as described above for the virus infections. Media was removed and 100 μg of soluble gD285t (Rux et al., 1998) was added in 1 ml of chilled media or cells were mock treated with media alone. Cells were placed at 4°C for 30 min and then shifted to 37°C for 30 min. At this time point, cells were detached with versene and cells were labeled with anti-GFP pAb ab290 for FACS as described for the virus infections.
Virus-receptor colocalization
NGC12 cells were seeded at 2×105 cells per glass coverslip. HSV KOS (MOI=50) was added in chilled DMEM + HEPES at 4°C for 45 min to allow for attachment. The temperature was raised to 37°C to initiate virus entry. At each time point (0, 5, 10, 15, 30 min), cells were fixed with 3% paraformaldehyde for 1hr at RT. Quenching, permeabilization and staining were performed as described (Krummenacher et al., 2003; Sodeik et al., 1997). In addition to GFP fluorescence of nectin-1, virus was stained with anti-gD mAb MC5 (Atanasiu et al., 2007) at 1μg/ml, followed by goat anti-mouse IgG coupled with Alexa 594 to detect gD on virions. Images were captured using a Nikon TE2000-U inverted microscope connected to a Perkin Elmer confocal imaging system.
Proteinase K protection assay
To detect virion internalization, proteinase K protection assays were carried out largely according to Milne et al. (2005). Confluent monolayers of cells in 60 mm petri dishes (3×106 cells/dish) were chilled on ice. Three dishes of each cell type were inoculated with sucrose gradient purified HSV-1 KOS (input multiplicity 20 pfu/cell in 5% DMEM containing 30mM HEPES). After 45 min adsorption at 4°C, two dishes were transferred to a 37°C water bath to initiate virus entry, while the third remained at 4°C. After 15 min, all three dishes were placed on ice. Cells were washed once with Hank’s balanced salt solution containing 30 mM HEPES (HBSS-HEPES), then treated or mock treated for 1 hr with proteinase K (50 μg/ml in HBSS-HEPES, 1 mM CaCl2). Cells were pelleted in a 1.5 ml tube and lysed on ice with Tris buffered saline (TBS) containing 1% Triton X-100 and 1mM PMSF. After 30 minutes, lysates were cleared by microcentrifugation. gB was immunoprecipitated with trimer specific mAb DL16, resolved on SDS-PAGE and detected by western blotting using rabbit serum R69 as previously described (Milne et al., 2005).
Rate of entry assay
The rate of virus entry was determined using a standard acid inactivation assay (Highlander et al., 1989; Huang and Wagner, 1964; Milne et al., 2005). Confluent monolayers of cells in a 6 well plate were inoculated with 100 pfu/well of KOS and incubated at 4°C for virus adsorption. The plates were then shifted to 37°C in a water bath. At various time intervals after temperature shift, the wells were washed with citrate buffer pH 3.0 to inactivate extracellular virus. At the end of the time course, the cells were overlaid with normal growth media containing 0.5% carboxymethyl cellulose and the cells were incubated at 37°C until plaques formed. Cells were stained with crystal violet and plaques were counted.
Nectin-1 internalization assay
Nectin-1 internalization was examined by biotinylating surface proteins on confluent monolayers of NGC12 cells (3×106 cells/ 60 mm dish) with 0.25 mg/ml EZ-link sulfo-NHS-SS biotin (Pierce) in PBS with CaCl2 and MgCl2 (Gibco) at 4°C followed by quenching with 50 mM Tris pH 8. Biotinylated NGC12 cells were co-cultured with 4.5×106 B78 (no gD) or B78-gD(wt) at 37°C. At various times after temperature shift, cells were plunged in ice, washed with HBSS-HEPES, and treated with Proteinase K for 1 hr at 4°C (100 μg/ml in HBSS-HEPES, 1 mM CaCl2) to digest extracellular proteins. Cells were pelleted in a 1.5 ml tube and lysed on ice with TBS containing 1% Triton X-100, 1mM PMSF, and complete protease inhibitors. After 30 minutes, lysates were cleared by microcentrifugation. Internalized biotinylated proteins were pulled down with neutravidin beads (Pierce) and resolved by SDS-PAGE. Nectin-1 was detected by western blotting with mAb CK8.
Supplementary Material
ACKNOWLEDGMENTS
This investigation was supported by Public Health Service grant AI-073384 to C.K. from the National Institute of Allergy and Infectious Diseases. K.M.S. was supported by NIH training grant T32-AI07324 and grants AI-056045 and AI-076231 to Roselyn J. Eisenberg and AI-18289 to Gary H. Cohen, University of Pennsylvania.
We are grateful to Patricia G. Spear for the C10 cells and gD-null KOSgDβ virus, David C. Johnson for VD60 cells, and Gary H. Cohen and Roselyn J. Eisenberg for numerous reagents, equipment, and suggestions. We thank Ali Zekavat and Bruce Shenker for FACS processing at the Flow Cytometry Facility of the University of Pennsylvania School of Dental Medicine. We appreciate the advice and helpful discussions from members of the Krummenacher and Cohen/Eisenberg laboratories.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 2005;79:11588–11597. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. Junctional adhesion molecule a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J. Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, Menotti L, Mirandola P, Lopez M, Campadelli-Fiume G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attribute of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998;72:9992–10002. doi: 10.1128/jvi.72.12.9992-10002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC, Cohen GH, Eisenberg RJ. Potential nectin-1 binding site on herpes simplex virus glycoprotein D. J. Virol. 2005;79:1282–1295. doi: 10.1128/JVI.79.2.1282-1295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Regge N, Nauwynck HJ, Geenen K, Krummenacher C, Cohen GH, Eisenberg RJ, Mettenleiter TC, Favoreel HW. Alpha-herpesvirus gD interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J. Cell Biol. 2006;174:267–275. doi: 10.1083/jcb.200510156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean HJ, Terhune SS, Shieh MT, Susmarski N, Spear PG. Single amino acid substitutions in gD of herpes simplex virus 1 confer resistance to gD-mediated interference and cause cell-type-dependent alterations in infectivity. Virology. 1994;199:67–80. doi: 10.1006/viro.1994.1098. [DOI] [PubMed] [Google Scholar]
- Delboy MG, Patterson JL, Hollander AM, Nicola AV. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol. J. 2006;3:105. doi: 10.1186/1743-422X-3-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg RJ, de Leon M. Ponce, Friedman HM, Fries LF, Frank MM, Hastings JC, Cohen GH. Complement component C3b binds directly to purified glycoprotein C of herpes simplex virus types 1 and 2. Microb. Pathog. 1987;3:423–435. doi: 10.1016/0882-4010(87)90012-x. [DOI] [PubMed] [Google Scholar]
- Frampton AR, Jr., Stolz DB, Uchida H, Goins WF, Cohen JB, Glorioso JC. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J. Virol. 2007;81:10879–10889. doi: 10.1128/JVI.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- Fusco D, Forghieri C, Campadelli-Fiume G. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9323–9328. doi: 10.1073/pnas.0503907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty RJ, Krummenacher C, Eisenberg RJ, Cohen GH, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- Gianni T, Campadelli-Fiume G, Menotti L. Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes. J. Virol. 2004;78:12268–12276. doi: 10.1128/JVI.78.22.12268-12276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler CG, Cohen GH, Eisenberg RJ. Crosslinking of glycoprotein oligomers during herpes simplex virus type 1 entry. J. Virol. 1996;70:6076–6082. doi: 10.1128/jvi.70.9.6076-6082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasebe R, Sasaki M, Sawa H, Wada R, Umemura T, Kimura T. Infectious entry of equine herpesvirus-1 into host cells through different endocytic pathways. Virology. 2009;393:198–209. doi: 10.1016/j.virol.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci. 2008;65:1653–1658. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold BC, WuDunn D, Soltys N, Spear PG. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991;65:1090–1098. doi: 10.1128/jvi.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Highlander SL, Dorney DJ, Gage PJ, Holland TC, Cai W, Person S, Levine M, Glorioso JC. Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration. J. Virol. 1989;63:730–738. doi: 10.1128/jvi.63.2.730-738.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Wagner R. Penetration of herpes simplex virus into human epidermoid cells. Proc. Soc. Exp. Biol. Med. 1964;116:863–869. doi: 10.3181/00379727-116-29392. [DOI] [PubMed] [Google Scholar]
- Hung SL, Cheng YY, Wang YH, Chang KW, Chen YT. Expression and roles of herpesvirus entry mediators A and C in cells of oral origin. Oral Microbiol. Immunol. 2002;17:215–223. doi: 10.1034/j.1399-302x.2002.170403.x. [DOI] [PubMed] [Google Scholar]
- Kamei T, Matozaki T, Sakisaka T, Kodama A, Yokoyama S, Peng YF, Nakano K, Takaishi K, Takai Y. Coendocytosis of cadherin and c-Met coupled to disruption of cell-cell adhesion in MDCK cells--regulation by Rho, Rac and Rab small G proteins. Oncogene. 1999;18:6776–6784. doi: 10.1038/sj.onc.1203114. [DOI] [PubMed] [Google Scholar]
- Kopp SJ, Banisadr G, Glajch K, Maurer UE, Grunewald K, Miller RJ, Osten P, Spear PG. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. U. S. A. 2009;106:17916–17920. doi: 10.1073/pnas.0908892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Eisenberg RJ, Cohen GH. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J. Virol. 2003;77:8985–8999. doi: 10.1128/JVI.77.16.8985-8999.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, de Leon M. Ponce, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Localization of a binding site for herpes simplex virus glycoprotein D on the herpesvirus entry mediator C by using anti-receptor monoclonal antibodies. J. Virol. 2000;74:10863–10872. doi: 10.1128/jvi.74.23.10863-10872.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Sanzo JF, Cohen GH, Eisenberg RJ. Effects of herpes simplex virus on structure and function of nectin-1/HveC. J. Virol. 2002;76:2424–2433. doi: 10.1128/jvi.76.5.2424-2433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 1998;72:7064–7074. doi: 10.1128/jvi.72.9.7064-7074.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. Embo J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear E, Carfi A, Whitbeck JC, Cairns TM, Krummenacher C, Cohen GH, Eisenberg RJ. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 2008;82:700–709. doi: 10.1128/JVI.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy C, Wrana JL. Clathrin- and Non-clathrin-Mediated Endocytic Regulation of Cell Signaling. Nat. Rev. Mol. Cell Biol. 2005;6:112–126. doi: 10.1038/nrm1571. [DOI] [PubMed] [Google Scholar]
- Le TL, Yap AS, Stow JS. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol. 1999;146:219–232. [PMC free article] [PubMed] [Google Scholar]
- Ligas MW, Johnson DC. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M, Cocchi F, Avitabile E, Leclerc A, Adelaide J, Campadelli-Fiume G, Dubreuil P. Novel, soluble isoform of the herpes simplex virus (HSV) receptor nectin1 (or PRR1-HIgR-HveC) modulates positively and negatively susceptibility to HSV infection. J. Virol. 2001;75:5684–5691. doi: 10.1128/JVI.75.12.5684-5691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M, Eberlé F, Mattei M-G, Gabert J, Birg F, Bardin F, Maroc C, Dubreuil P. Complementary DNA characterization and chromosomal localization of a human gene related to the poliovirus receptor-encoding gene. Gene. 1995;155:261–265. doi: 10.1016/0378-1119(94)00842-g. [DOI] [PubMed] [Google Scholar]
- Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. Beta 1 Integrin mediates internalization of mammalian reovirus. J. Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Mainou BA, Derdowski A, Johnson EM, Zent R, Dermody TS. NPXY Motifs in the Beta 1 Integrin Cytoplasmic Tail are Required for Functional Reovirus Entry. J. Virol. 2008;82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, Itoh M, Mizoguchi A, Aoki T, Fujimoto T, Matsuda Y, Tsukita S, Takai Y. Afadin: A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J. Cell Biol. 1997;139:517–528. doi: 10.1083/jcb.139.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 2005;79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RSB, Hanna SL, Rux AH, Willis SH, Cohen GH, Eisenberg RJ. Function of herpes simplex virus type 1 gD mutants with different receptor-binding affinities in virus entry and fusion. J. Virol. 2003;77:8962–8972. doi: 10.1128/JVI.77.16.8962-8972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Nicola AV, Hou J, Major EO, Straus SE. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005;79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 2003;77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, Straus SE. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 2004;78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry C, Bell S, Minson T, Browne H. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J. Gen. Virol. 2005;86:7–10. doi: 10.1099/vir.0.80567-0. [DOI] [PubMed] [Google Scholar]
- Rux AH, Willis SH, Nicola AV, Hou W, Peng C, Lou H, Cohen GH, Eisenberg RJ. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpes virus entry mediator. J. Virol. 1998;72:7091–7098. doi: 10.1128/jvi.72.9.7091-7098.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y, Ogita H, Hirota T, Kawakatsu T, Fukuyama T, Yasumi M, Kanzaki N, Ozaki M, Takai Y. Interaction of integrin alpha(v)beta3 with nectin. Implication in cross-talk between cell-matrix and cell-cell junctions. J. Biol. Chem. 2006;281:19631–19644. doi: 10.1074/jbc.M600301200. [DOI] [PubMed] [Google Scholar]
- Sakisaka T, Takai Y. Biology and pathology of nectins and nectin-like molecules. Curr. Opin. Cell Biol. 2004;16:513–521. doi: 10.1016/j.ceb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell. 2008;132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J. Neurovirol. 2005;11:208–218. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear PG, Eisenberg RJ, Cohen GH. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 2000;275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- Stiles KM, Milne RS, Cohen GH, Eisenberg RJ, Krummenacher C. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology. 2008;373:98–111. doi: 10.1016/j.virol.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian RP, Dunn JE, Geraghty RJ. The nectin-1alpha transmembrane domain, but not the cytoplasmic tail, influences cell fusion induced by HSV-1 glycoproteins. Virology. 2005;339:176–191. doi: 10.1016/j.virol.2005.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian RP, Geraghty RJ. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. U. S. A. 2007;104:2903–2908. doi: 10.1073/pnas.0608374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Kintner J, Schoborg RV. The host adherens junction molecule nectin-1 is downregulated in Chlamydia trachomatis-infected genital epithelial cells. Microbiology. 2008;154:1290–1299. doi: 10.1099/mic.0.2007/015164-0. [DOI] [PubMed] [Google Scholar]
- Sun J, Schoborg RV. The host adherens junction molecule nectin-1 is degraded by chlamydial protease-like activity factor (CPAF) in Chlamydia trachomatis-infected genital epithelial cells. Microbes Infect. 2009;11:12–19. doi: 10.1016/j.micinf.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, Satoh A, Nishioka H, Aoki J, Nomoto A, Mizoguchi A, Takai Y. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with afadin, a PDZ domain-containing protein. J. Cell Biol. 1999;145:539–549. doi: 10.1083/jcb.145.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Ikeda W, Ogita H, Rikitake Y. The Immunoglobulin-Like Cell Adhesion Molecule Nectin and Its Associated Protein Afadin. Annu. Rev. Cell Dev. Biol. 2008;24:309–342. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- Van de Walle GR, Peters ST, VanderVen BC, O’Callaghan DJ, Osterrieder N. Equine herpesvirus 1 entry via endocytosis is facilitated by alphaV integrins and an RSD motif in glycoprotein D. J. Virol. 2008;82:11859–11868. doi: 10.1128/JVI.00868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner MS, Martinez W, Geraghty RJ, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by herpes simplex virus type 2, mutants of herpes simplex virus type 1 and pseudorabies virus. Virology. 1998;246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, de Leon M. Ponce, Peng T, Nicola AV, Montgomery RI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the TNFR superfamily and a mediator of HSV entry. J. Virol. 1997;71:6083–6093. doi: 10.1128/jvi.71.8.6083-6093.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittels M, Spear PG. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 1990;18:271–290. doi: 10.1016/0168-1702(91)90024-p. [DOI] [PubMed] [Google Scholar]
- Zago A, Jogger CR, Spear PG. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17498–17503. doi: 10.1073/pnas.0408186101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.