

Abstract
The role of estrogens in the increased risk of lung adenocarcinoma in women remains uncertain. We reported that lung adenocarcinoma cell lines from female, but not male, patients with non–small cell lung cancer respond proliferatively and transcriptionally to estradiol (E2), despite equal protein expression of estrogen receptors (ER) α and β. To test the hypothesis that nuclear localization of ERα corresponds to genomic E2 activity in lung adenocarcinoma cells from females, cell fractionation, immunoblot, and confocal immunohistochemical microscopy were performed. We report for the first time that E2 increases phospho-serine-118-ERα (P-ser118-ERα) and cyclin D1 (CCND1) nuclear colocalization in H1793, but not A549 lung adenocarcinoma cells, derived from a female and male patient, respectively. ERβ was primarily in the cytoplasm and mitochondria, independent of E2 treatment, and showed no difference between H1793 and A549 cells. E2 induced higher transcription of endogenous ERα-regulated CCND1 in H1793 than in A549 cells. Likewise, higher rapid, non-genomic E2-induced extracellular signal–regulated kinase 1/2 activation was detected in H1793 compared with A549 cells, linking extracellular signal–regulated kinase activation to increased P-ser118-ERα. Furthermore, E2 increased cyclin D1 and P-ser118-ERα nuclear localization in H1793, but not A549 cells. Together, our results indicate that nuclear localization of P-ser118-ERα provides one explanation for sex-dependent differences in E2-genomic responses in lung adenocarcinoma cell lines.
Keywords: estrogen receptor, cyclin D1, phosphorylation, non–small cell lung cancer, sex differences
CLINICAL RELEVANCE.
Women have higher incidence of lung adenocarcinoma, a type of non–small cell lung cancer (NSCLC), compared to men. The mechanisms underlying the sex difference in NSCLC risk are multifactorial and complex. Here we show that nuclear localization of phospho-serine-118–estrogen receptor-α provides one explanation for sex-dependent differences in estradiol-genomic responses in lung adenocarcinoma cell lines.
Lung cancer is the leading cause of cancer death in both women and men in the United States. However, the twofold-higher risk of lung cancer in female smokers or nonsmokers than in male smokers strongly suggests the involvement of sex-dependent factors in the etiology of lung cancer (1). In particular, women have higher incidence of lung adenocarcinoma, a type of non–small cell lung cancer (NSCLC), compared with men. The mechanisms underlying the sex difference in NSCLC risk are multifactorial, and include differences in phase I and II drug–metabolizing enzyme activities (1–3). The role of estrogens in lung adenocarcinoma is of particular research interest and debate (4, 5). A new clinical case study of 59 patients with NSCLC reported that 73% had higher estradiol (E2) concentrations in the carcinomas compared with the corresponding nonneoplastic lung tissues from the same patient, regardless of sex, which correlated with aromatase mRNA, but not estrogen receptor (ER) α or β staining (6). Thus, local estrogen production may play a role in NSCLC.
There are multiple mechanisms through which ER mediates estrogenic effects. Best characterized is the genomic action of ligand-activated ER mediated by the binding of ER directly to estrogen-responsive elements (EREs), or by tethering of the ER dimer to other transcription factors (e.g., AP-1 and Sp1) (7). These interactions recruit coregulators, chromatin-remodeling complexes, and RNA polymerase II to the target gene promoter (8). Serine phosphorylation is required for full activation of ERα (9), and phosphorylation of ERα on ser118 (phospho-serine-118-ERα [P-ser118-ERα]) activates N-terminal transcription activation function–1. Plasma membrane localization of ER activates rapid, membrane-initiated, “nongenomic” responses (e.g., activation of phosphatidylinositol-3-kinase [PI3K] and mitogen-activated protein kinase [MAPK]) (10). P13K and MAPK phosphorylate ERα ser167 and ser118, respectively, thus linking rapid E2 responses with later genomic actions (11). Approximately 20% of ERα in H23 lung adenocarcinoma cells was associated with the plasma membrane fraction, and E2 rapidly activated MAPK (5). ERα and ERβ have also been localized to mitochondria in human cell lines and tissues (12).
The expression and intracellular location of ERα and ERβ have been examined by immunohistochemical (IHC) staining in normal and neoplastic human lung (reviewed in Refs. 6, 13–15). ERβ was present in columnar epithelium, intermediate, basal, and smooth muscle lung cells, whereas ERα was expressed in basal and smooth muscle cells (16). One IHC study of tumors from 122 patients with NSCLC reported that ERα was primarily cytoplasmic, and ERα and epidermal growth factor receptor were correlated with poor prognosis, but not with sex (17). Nuclear ERα and ERβ staining was detected in 45 and 52% of 65 archival NSCLC tumors, respectively, whereas 75 and 69% showed “extranuclear” ERα and ERβ staining, respectively (18). This study also reported phosphorylation of nuclear ERα on ser118 and ser167 in the NSCLC tumors (18).
We reported that lung adenocarcinoma cell lines from female patients proliferated in response to E2, and that growth was blocked by antiestrogens (i.e., 4-hydroxytamoxifen [4-OHT] and ICI 182,780 [ICI]) (13). In contrast, lung adenocarcinoma cell lines from male patients were not responsive to E2 or antiestrogens (13). Similar results were obtained when the cell lines were transfected with an ERE-luciferase reporter and treated with E2 (i.e., cell lines from females, but not males), showing genomic activation of ERα (13). Despite these phenotypic differences, all cell lines had equal expression of ERα and ERβ at the mRNA and protein levels, with ERβ expression being approximately two-times higher than ERα (13). These data indicate that the differences in the expression of ERα and ERβ are not the mechanism accounting for the ability of adenocarcinoma cell lines from females to respond to E2 and antiestrogens.
To test the hypothesis that nuclear ERα localization in lung adenocarcinoma cells from females accounts for their E2-transcriptional responses, and to better understand the mechanism underlying the difference in the response of male- and female-derived lung adenocarcinoma cells to treatment with E2, 4-OHT, and ICI (13), we examined the intracellular location of ERα, P-ser118-ERα, and ERβ in normal lung fibroblasts and lung adenocarcinoma cells using confocal microscopy and subcellular fractionation, followed by Western blotting. Results were compared with MCF-7 human breast cancer cells as an established ERα/ERβ, estrogen-responsive cell type. Our data demonstrate differences in the nuclear localization of ERα and P-ser118-ERα in lung adenocarcinoma cells derived from females and males that correspond to reported differences in the genomic activity of ERα in these cells (13).
MATERIALS AND METHODS
Chemicals
E2, PD98059, 4-OHT, pertussis toxin (PTX), and methyl-β-cyclodextrin (β-CD) were from Sigma (St. Louis, MO). Src tyrosine kinase inhibitor PP2 and ICI were from Tocris (Ellisville, MO).
Cell Lines and Treatment
A549, NCI-H23, NCI-H1299, NCI-H1395, NCI-H1435, NCI-H1792, NCI-H1793, NCI-H1944, NCI-H2073, and MCF-7 cell lines were purchased from ATCC (Manassas, VA). Before treatment, cells were placed in phenol red–free media supplemented with 5% dextran-coated, charcoal-stripped FBS (DCC-FBS) for 48 hours. Ethanol (EtOH) was used as the vehicle control for all experiments. Lung adenocarcinoma cell line characteristics are listed in Table 1. NF1604, karyotyped as 46XY, were derived from human fetal lung tissue and immortalized by human telomerase (19).
TABLE 1.
CHARACTERISTICS OF THE CELL LINES USED IN THIS STUDY AND A SUMMARY OF ESTROGEN RECEPTOR α AND ESTROGEN RECEPTOR β SUBCELLULAR DISTRIBUTION BY CONFOCAL AND IMMUNOFLUORESCENT MICROSCOPY
| ERα Subcellular Distribution |
ERβ Subcellular Distribution |
|||||
|---|---|---|---|---|---|---|
| Cell Line | Sex of Patient | Smoker/NS | Control | + E2 | Control | + E2 |
| NF1604 normal lung fibroblasts | Male | NS | C | C | N, C, M | N, C, M |
| A549 | Male | Unknown | N < < C | C | N, C, M | N, C, M |
| NCI-H1792 | Male | Smoker | C | C | C, M | C, M |
| NCI-H23* | Male | Smoker | C | C | ||
| NCI-H1299* | Male | Smoker | C | N, C | ||
| NCI-H1793 | Female | NS | N, C, M | N, C, M | N > C, M | N > C, M |
| NCI-H1944 | Female | Smoker | N, C | N, C | C, M | N < C, M |
| NCI-H1395* | Female | Smoker | N, C | N < < C | ||
| NCI-H1435* | Female | NS | N, C | N, C | ||
| MCF-7 human breast cancer | Female | Unknown | N, C | N, C | N < < C, M | N < C > M |
Definition of abbreviations: C, cytoplasmic; E2, estradiol; ER, estrogen receptor; M, colocalization with MitoTracker Red (mitochondria); N, nuclear; NS, nonsmoker.
The data summarized here were taken from confocal images, including those shown in Figures 1–4.
Cell lines examined by immunofluorescent microscopy; where indicated as +E2, cells were treated with 10 nM E2 for 45 minutes.
Antibodies
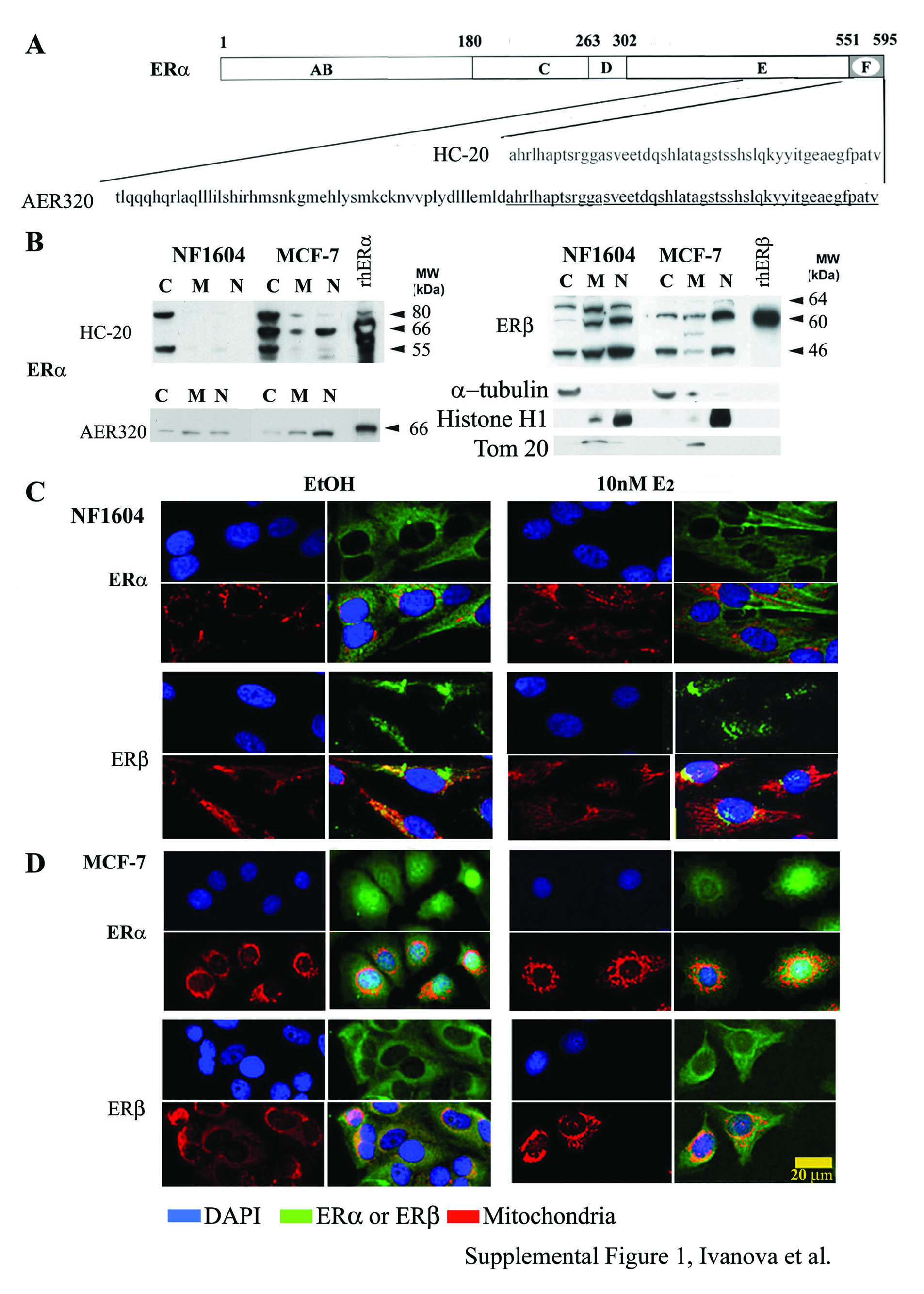
Polyclonal antibodies HC-20 for ERα and H150 for ERβ, and the HC-20 blocking peptide were from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal ERα antibody AER320 was from LabVision/Neomarkers (Thermo Fisher, Fremont, CA). HC-20 and AER320 were raised against peptides in the C terminus of ERα (aa 545–595 for HC-20 and aa 495–595 for AER320; see Figure E1A in the online supplement). HC-20 and H150 were demonstrated to be ER subtype–specific for IHC/confocal microscopy (20). HC-20 has been used for IHC staining of human lung tumors (14, 18), and AER320 has been used for IHC of human breast tumors (21, 22), but not, to our knowledge, for lung. The relative purity of subcellular fractions was analyzed by antibodies for α-tubulin (cytoplasm; LabVision), Tom20 (mitochondria, generously provided by Dr. Brian Wattenberg, University of Louisville), histone H1 or poly (ADP-ribose) polymerase (PARP) (nuclear; Santa Cruz Biotechnology). P-ser118-ERα antibody was from Cell Signaling (Danvers, MA). Cyclin D1 antibody was from Abcam (Cambridge, MA).
Western Blotting
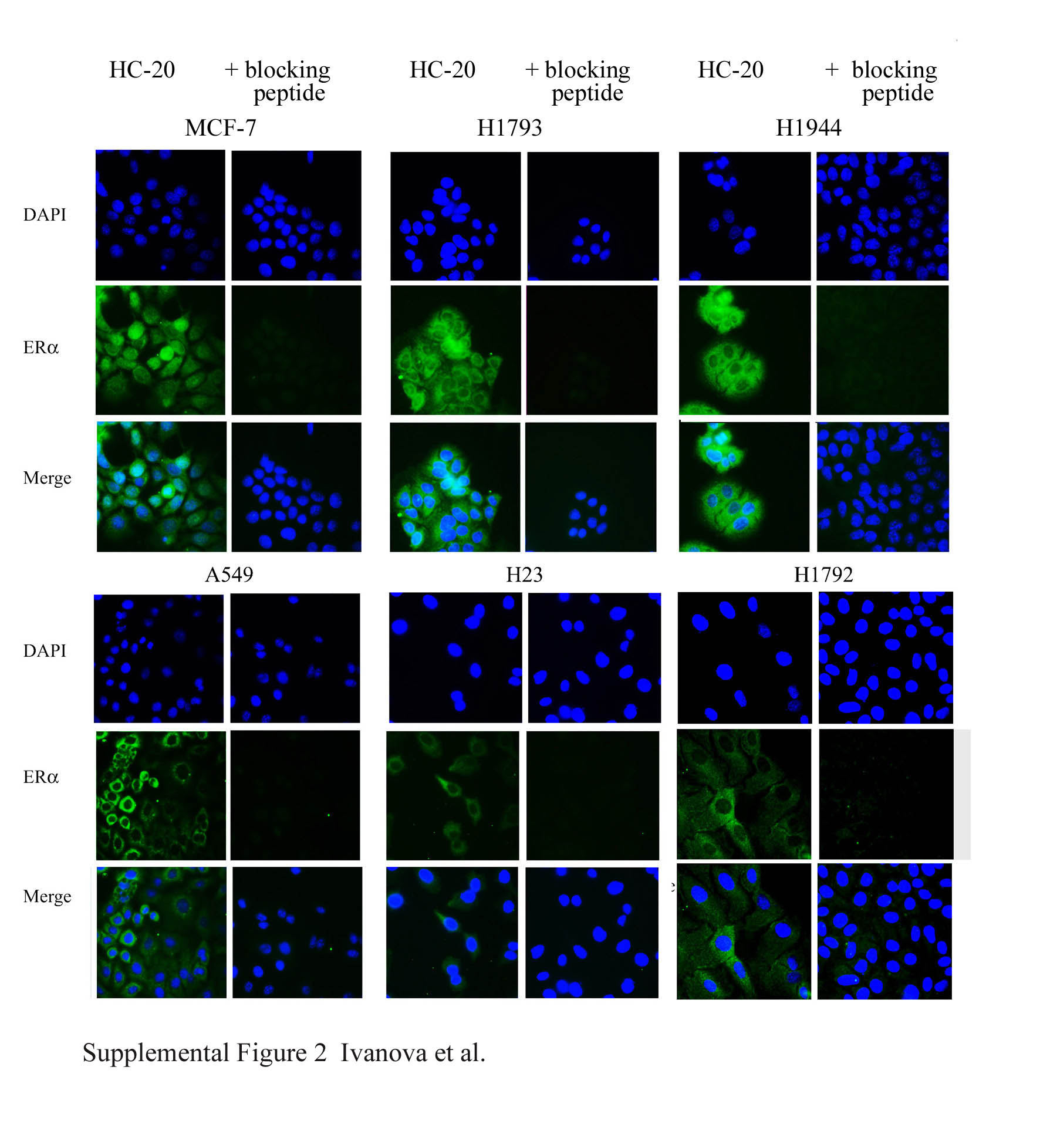
Subcellular fractions were separated according to the protocol provided in the Mitochondrial Isolation Kit for Mammalian Cells from Pierce (Rockford, IL). The indicated amounts of protein (see figures) of cytoplasmic, mitochondrial, and nuclear protein extracts (CE, ME, and NE, respectively) or whole-cell extract (WCE) were separated on 10% SDS-PAGE, transferred to polyvinylidene fluoride membranes, and blocked (13). The HC-20–blocking peptide was incubated at a 2:1 ratio with HC-20 antibody in 5% BSA–Tris-buffered saline–Tween for 1 hour at room temperature before incubation with the polyvinylidene fluoride membrane overnight at 4°C (Figure E2). Super Signal West Pico Chemiluminescent Substrate (Pierce) was used to detect protein bands as visualized on Kodak BioMaxML film (Eastman Kodak, Rochester, NY). Resulting immunoblots were scanned into Adobe Photoshop 7.0 (Adobe Systems, Inc., San Jose, CA) using a Microtek ScanMaker III scanner (Microtek, Carson, CA). Un-Scan-It 5.1 for Windows (Silk Scientific, Orem, UT) was used to quantitate the integrated optical densities for each band and analyze the relative amounts of immunoreactive protein, based on pixel density.
Immunofluorescence Staining
Cells were grown on coverslips, and were incubated with 200 nM MitoTracker Red CMXRos (Molecular Probes, Eugene, OR) for 45 minutes, washed with PBS, then fixed with cold methanol:acetone (1:1) for 5 minutes and washed twice with cold PBS. After blocking with 1% goat serum and 0.3% Triton X-100 in PBS for 30 minutes, primary antibody (ERα, HC-20; ERβ, H150) was added at a 1:300 dilution for 1 hour. After removing primary antibodies, the cells were stained with secondary antibodies labeled with Quantum Dot 525 (Quantum Dot Corp., Hayward, CA). The immunofluorescence analysis of P-ser118-ERα and cyclin D1 used combinations: (1) monoclonal (anti-mouse) P-ser118-ERα and anti-rabbit cyclin D1 antibodies; and (2) polyclonal (anti-rabbit) P-ser118-ERα and AER320 ERα monoclonal (anti-mouse) antibodies. The H1793 and A549 cells were grown on coverslips. Before fixation, the cells were incubated in phenol red–free media supplemented with 5% DCC-FBS for 48 hours (serum starvation) and treated with 10 nM E2 for 6 hours. Cells were washed with PBS, fixed with cold methanol:acetone (1:1) for 5 minutes, and washed twice with cold PBS. After blocking with 1% goat serum and 0.3% Triton X-100 in PBS for 30 minutes, primary antibodies (cyclin D1, P-ser118-ERα, or AER320) were added at a 1:300 and 1:1,000 dilution, respectively, for a 1-hour incubation. After removing primary antibodies, secondary antibody was added. The secondary anti-mouse antibody was labeled with Zenon Alexa Fluor 488 (green color), and the secondary anti-rabbit antibody was labeled with Zenon Alexa Fluor 594 (red color) labeling kits from Molecular Probes. Cells were then incubated with ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (Molecular Probes). Images were obtained using a confocal microscopy (Axiovert 100; Zeiss, Jena, Germany) and LSM510 software (Zeiss), or by immunofluorescence imaging using a Zeiss Axiovert 200 inverted microscope with a 63× objective lens using AxioVision Release 4.3 software.
To examine the specific staining of HC-20 and H150 antibodies, A549, H1792, H1793, and H1944 cells were incubated with MitoTracker Red, stained with 4′,6-diamidino-2-phenylindole, and incubated with the FITC secondary antibody without primary antibodies (data not shown). Figures E1C and E1D show the effect of E2 on ERα and ERβ subcellular distribution on MCF-7 cells, with normal lung fibroblast cells (NF1604) used as controls. In addition, the specificity of the ERα immunofluorescence was demonstrated by ablation of any green fluorescence signal by the HC-20–blocking peptide (Figure E2).
Cell Proliferation/Viability Assays
Cell proliferation/viability was determined using the Cell Titer 96 AQueous One MTT assay from Promega (Madison, WI), according to the manufacturer's instructions. Cells were plated in 96-well plates in growth medium and allowed to adhere for 24 hours, after which the medium was replaced with phenol red–free Improved Minimum Essential Medium supplemented with 5% DCC-FBS for 24 hours before treatment with vehicle control (i.e., EtOH), E2, 4-OHT, or ICI, alone or in combination, for 4 days, with treatments and medium replenished after 48 hours. Where indicated, cells were preincubated with 50 μM PD98059 (mitogen-activated protein kinase 1 [MEK1] inhibitor, 1 h) before adding the indicated cotreatments. MTT assays were performed on Day 4.
FACS of Cell Cycle Phase Using Propidium Iodide
A549 and H1793 cells were grown in six-well plates with 1 × 106 cells per well, and treated with EtOH (vehicle control) or 10 nM E2 for 6 hours. For cell cycle analysis, cells were harvested, fixed in 70% EtOH, and incubated for 30 minutes in propidium iodide staining buffer (0.1 M PIPES [pH 6.8], 1.5 M NaCl, 0.5 M MgCl2, and 0.1% Triton X100) containing 2.5 μg/ml RNase DNA-free (Roche) and 10 μg/ml propidium iodide (Roche). Cellular DNA content was analyzed by flow cytometry using a MoFlo Flow Cytometer (Beckman Coulter, Hialeah, FL) equipped with an argon laser tuned at 488 nm. BD CellQuest Pro Software (BD Bioscience, San Jose, CA) was used for acquisition and analysis of each cell sample (number of cells, 10,000) in the J. G. Brown Cancer Center Cell Sorting Core Facility (n = 5 replicate experiments).
RNA Isolation, RT-PCR, and Real-Time Quantitative RT-PCR
RNA was isolated from cells using the RNeasy Mini kit (Qiagen, Gaithersburg, MD). Oligo(dT)12–18 primers (Promega) were annealed to 2 μg of total RNA and reverse transcribed with the High Capacity cDNA Archive kit (PE Applied Biosystems, Foster City, CA) using the manufacturer's protocol. The cDNA generated was used as a template for PCR with primers specific for ERα splice variants, Δ2 and Δ7 (23). Taqman primers and probes for cyclin D1 (CCND1) and 18S rRNA were purchased as Assays-on-Demand Gene Expression Products (PE Applied Biosystems). The expression of each target gene was determined in quadruplicate in two separate experiments and normalized using 18S. Real-time qRT-PCR was performed in the ABI PRISM 7900 SDS 2.1 (PE Applied Biosystems) using relative quantification real-time qRT-PCR for CCND1, and 18S was performed using absolute quantification. Analysis and fold differences were determined using the comparative CT method, and data are presented as relative to EtOH-treated cells.
P-Extracellular Signal–Regulated Kinase 1/2 and Extracellular Signal–Regulated Kinase 1/2 ELISA
A549 and H1793 cells were preincubated with 50 μM PD98059 (MEK1 inhibitor, 1 h), 100 ng/ml PTX (a Gα inhibitor, 17 h), 5 mM β-CD (a lipid raft disruptor, 30 min), or 10 μM PP2 (Src kinase inhibitor, 30 min) before E2 treatment. Cells were treated with 10 nM E2 for 0–30 minutes and 1, 2, 4, 6, 8, and 24 hours. After treatment, the cells were fixed with fresh 4% formaldehyde in PBS and assayed as described in the manufacturer's protocol (FACE extracellular signal–regulated kinase [ERK] 1/2 ELISA kit; Active Motif, Carlsbad, CA). Each treatment was run in triplicate wells. Values were averaged, and three experiments performed.
Statistical Analysis
Statistical analyses were performed using Student's t test or one-way ANOVA, followed by Student-Newman-Keuls or Dunnett's post hoc tests using GraphPad Prism (GraphPad Software, Inc., San Diego, CA).
RESULTS
Subcellular Localization of ERα and ERβ in Lung Adenocarcinoma Cell Lines
To test the hypothesis that nuclear localization of ER in lung adenocarcinoma cells from female, but not male, patients accounts for their observed E2-dependent transcriptional and proliferative responses, the intracellular localization of ERα and ERβ was examined (Figures 1–3 and summarized in Tables 1 and 2). Because all four adenocarcinoma cell lines from males, and all five cell lines from females, showed similar cell growth and transcriptional responses to E2 (17), we selected A549 and H1793 as representative male- and female-derived lung adenocarcinoma cells for biochemical fractionation studies (Figures 1A and 1B). HC-20 ERα antibody detected the 66-kD ERα in MCF-7 cells, but not in A549 or H1793 cells (Figure 2A and Figure E3). The absence of the 66-kD ERα in A549 and H1793 is identical to the findings in a report on A549 and other lung cancer cell lines (14). In contrast, AER320 detected the 66-kD ERα band in cellular subfractions in which HC-20 failed to detect the 66-kD ERα, and demonstrated higher nuclear ERα in lung adenocarcinoma cell lines from females than males (Figures E1B and E4).
Figure 1.

Subcellular localization of estrogen receptor (ER) α and β in lung adenocarcinoma cell lines. (A) Western blot analysis of subcellular fractions from MCF-7 breast cancer cells, A549 and H1793 lung adenocarcinoma cells. Nuclear (N), cytoplasmic (C), and mitochondrial (M) extracts. Loading of equal amounts of protein from each subcellular fraction allowed a direct comparison of the relative amount of each ERα and ERβ variant expressed in each cellular compartment and between cell lines. The membranes were stripped and reprobed for histone H1 and Tom20 as a measure of the relative purity of the nuclear and mitochondrial fractions, respectively. (B) The graphs show the quantitation of the percent of total immunoreactive ERα and ERβ proteins in nuclear (N), cytoplasmic (C), and mitochondrial (M) extracts. Values are the average of three to six separate experiments (±SEM). *P < 0.001, nuclear ERα values were significantly different. (C) Confocal microscopic imaging of lung adenocarcinoma cells: A549 and H1792 from male patients and H1793 and H1944 from female patients. As indicated, cells were treated with either ethanol (EtOH; vehicle) of 10 nM estradiol (E2) for 45 minutes. Each set of two side-by-side images shows ER (ERα or ERβ as indicated, green color) on the left and a merged image of 4′,6-diamidino-2-phenylindole (DAPI; blue), ER (green), and Mitotracker Red CMXRos (red) on the right (labeled at the top as ER and Merge, respectively). The yellow color indicates overlap of MitoTracker red and ERα or ERβ signaling in mitochondria. Scale bars, 20 μm. The yellow arrows indicate the absence or presence of ERα or ERβ in the nucleus, as described in the text.
Figure 2.

Immunofluorescence microscopic imaging of ERα and ERβ in lung adenocarcinoma cells. (A) Immunofluorescent staining with ERα antibody HC-20 (green) in untreated H23, H1299, H1395, and H1495 cell lines, counterstaining with DAPI (blue), and the merged images (Merge) are shown. (B) Fluorescence microscopic imaging of ERβ in lung adenocarcinoma cells. Immunofluorescent staining with ERβ antibody H150 (green) is shown for untreated H23, H1299, H1395, and H1435 lung adenocarcinoma cells. Cells were counterstained with DAPI (blue), and the merged images (Merge) are shown. All images were captured with same time exposure. Scale bars, 20 μm.
Figure 3.

Phospho-serine-118-ERα (P-ser118-ERα) in lung cancer cells. (A) Western blot of whole-cell extract from H1793 and A549 cells using P-ser118-ERα, ERα 66-kD using monoclonal antibody AER320, and β-actin antibodies. Note that AER320 detects full-length, wild-type (WT) ERα (Figure 1 and Ref. 13). The cells were treated for either 4 or 6 hours with EtOH or 10 nM E2 as indicated. (B) The relative levels of P-ser118-ERα/ERα were quantitated. Values are the average of three to six separate experiments (±SEM). The P-ser118-ERα/ERα 66-kD ratio in EtOH-treated H1793 cells was set to 1 within each experiment and at each time point (4 or 6 h) for comparison between experiments. Values are significantly different from the P-ser118-ERα/ERα 66-kD ratio in EtOH-treated H1793 cells:*P < 0.05, **P < 0.001. (C) Western blot of cytoplasmic (C) and nuclear (N) extracts from H1793 cells for P-ser118-ERα, and subfraction controls (histone H1, nuclear; α-tubulin, cytosol) in EtOH-treated H1793 cells. (D) Dual-label fluorescence immunostaining of control (EtOH)- and E2-treated H1793 and A549 cells. ERα AER320 antibody (green) and P-ser118-ERα antibody (red) were used as described in Materials and Methods. Images of nuclei (DAPI) and ERα, and the merged image were captured at identical times of exposure (16 and 230 ms, respectively) for each cell line. To examine differences in the level of P-ser118-ERα in EtOH- versus E2-treated cells, images of P-ser118-ERα were captured at 100, 200, and 300 ms of exposure, as indicated at the bottom of the photos. Scale bars, 20 μm.
TABLE 2.
SUBCELLULAR DISTRIBUTION OF ESTROGEN RECEPTOR α AND ESTROGEN RECEPTOR β SPLICE VARIANTS AS DETERMINED BY BIOCHEMICAL FRACTIONATION
| MCF-7 |
A549 |
H1793 |
||||
|---|---|---|---|---|---|---|
| Localization | ERα | ERβ | ERα | ERβ | ERα | ERβ |
| Cytoplasmic | 80, 66, 52–55 | 64, 60, 46, 36 | 80, 52–55 | 64, 60, 46, 36 | 80, 52–55 | 64, 46, 36 |
| Mitochondrial | 80, 66 | 64, 60, 46, 36 | 80, 52–55 | 64, 60, 46, 36 | 80, 52–55 | 64, 60, 46, 36 |
| Nucleus | 80, 66, 52–55 | 64, 60, 46, 36 | 64, 60, 46, 36 | 80, 52–55 | 64, 60, 46, 36 | |
Definition of abbreviation: ER, estrogen receptor.
MCF-7 human breast cancer cells, A549 and H1793 lung adenocarcinoma cells, from male and female patients respectively, were separated into cytosolic, nuclear, and mitochondrial extracts as described in Materials and Methods. Conclusions summarized in this table are from Western blots with HC-20 ERα and H150 ERβ antibodies including those shown in Figures 1 and 2. Bold numbers are highest expression; all values presented are expressed in kilodaltons.
ERαΔ7 in H1793 Lung Adenocarcinoma Cells
To examine the identity of the apparent 52- to 55-kD ERα protein(s) detected by HC-20 in H1793 cells, RT-PCR was performed using specific primers to detect the deletion (Δ) of exons 7 and 2 (23). As a control, MCF-7 cells were shown to express 319- and 482-bp products, corresponding to ERαΔ7 (52 kD) (23, 24) and full-length ERα. Notably, ERαΔ7 and wild-type (WT) ERα were detected in H1793 (Figure E5). The ERαΔ2 primers did not detect the expected 365-bp product for ERαΔ2 in H1793 cells, but, instead, detected a 292-bp product. MCF-7 did not express ERαΔ2 (Figure E5). These data indicate that H1793 cells express ERαΔ7, corresponding to the 52- to 55-kD ERα band detected by HC-20 (Figure 1A), and that a mutation(s) in exons 1 and/or 3 preclude amplification of WT ERα with the ERαΔ2 primers.
Subcellular Localization of ERβ in Lung Adenocarcinoma Cell Lines
ERβ1 was predominantly in the nuclear fraction (Figure 1A, Table 2). In addition, a 46-kD ERβ band was detected in all subcellular fractions from A549 and H1793 cells. The amount of 46-kD ERβ was reduced in the nuclear fraction and enriched in the mitochondrial fraction of H1793 cells (Figure 1A, Table 2). A 36-kD ERβ band was predominantly in the nuclear fraction of A549 and H1793 cells (Figure 1A, Table 2).
MCF-7 has proportionally more ERα and ERβ in the nucleus than either A549 or H1793 cells (Figure 1B). Within the nuclear fraction, A549 had significantly lower total ERα (∼6%) than H1793 (∼17%) cells (P = 0.0012). A549 cells had approximately 2.5 times more ERβ in the nuclear fraction compared with ERα. H1793 cells had similar amounts (∼17%) of total ERβ and ERα in the nuclear fraction. Differences in the relative expression of the ERβ isoforms in the cellular subfractions and between cell lines are summarized in Table 2.
Confocal Imaging of ERα and ERβ in Lung Adenocarcinoma Cells
ERα was cytoplasmic in both untreated and E2-treated A549 cells, results that are in agreement with biochemical fractionation and Western blot analysis (Figure 1C). E2 did not increase nuclear ERα in A549 cells (Figure 1C, yellow arrow). ERβ was located in both nuclear and cytoplasmic compartments, and overlapped with Mitotracker Red, indicating that ERβ is in the mitochondria in the perinuclear area of A549 cells (Figure 1C). These observations support the biochemical fractionation/Western data in Figure E1B. E2 did not alter ERβ localization, but increased colocalization of ERβ with Mitotracker Red (Figure 1C).
ERα was localized in the cytoplasm, and not in the nucleus, of H1792, another male-derived lung adenocarcinoma cell line, and did not change with E2 treatment (Figure 1D). ERβ was detected in the cytoplasm and mitochondria of H1792 cells, but not in the nucleus, and did not change with E2 (Figure 1D).
H1793 cells showed diffuse cytoplasmic ERα staining, and the overlap with Mitotracker Red indicates mitochondrial colocalization (Figure 1E). ERα was also detected in the nucleus, and distribution was unaffected by E2 (Figure 1E). These observations are congruent with the biochemical fractionation/Western data for H1793 (Figure E1B). ERβ was in both cytoplasm and nucleus, and E2 increased nuclear and perinuclear fluorescence (Figure 1E, yellow arrow). In both control and E2-treated H1793, ERβ colocalized with Mitotracker Red (Figure 1E).
ERα was diffusely distributed throughout the cytoplasm and nucleus in H1944, another female-derived lung adenocarcinoma cell line (Figure 1F). E2 increased ERα signal intensity in the perinuclear region, and resulted in a punctuate ERα signal within the nucleus (Figure 1F). ERβ was distributed throughout the cytoplasm, and not the nucleus, and overlapped with Mitotracker Red in the perinuclear region of H1944 cells (Figure 1F). E2 increased ERβ in the nucleus of some H1944 cells (see yellow arrow in Figure 1F). Together, biochemical fractionation/Western and confocal imaging data indicate that the two lung adenocarcinoma cell lines from females have more nuclear ERα compared with the cell lines from males (Tables 1 and 2). The subcellular localization of ERβ was independent of cell line and donor sex.
Fluorescence Imaging of ERα and ERβ in Lung Adenocarcinoma Cell Lines
Fluorescence microscopy was used to examine ERα and ERβ in other lung adenocarcinoma cell lines from males (H23 and H1299) and females (H1395 and H1435) (Figure 2,). ERα was in the cytoplasm of H23, H1299, H1395, and H1435 cells (not all data shown). Nuclear ERα was detected in H1395 and H1435 cells (Figure 2A). In H1299 cells, ERα was excluded from nucleoli (Figure 2A). ERβ was cytoplasmic in H23 cells, and showed higher cytoplasmic than nuclear signal intensity in H1299, H1395, and H1435 cells (Figure 2B). Each experiment was repeated using at least four independent cell cultures at different times, and are summarized in Table 1. Taken together with the confocal imaging and cellular subfraction studies, we conclude that lung adenocarcinoma cells from females have more nuclear ERα than those from males.
P-ser118-ERα in H1793 and A549 Cells
Because P-ser118-ERα was reported in NSCLC tumors (18) and is a marker of functional, ligand-dependent ERα signaling in breast tumors (25), we tested the hypothesis that P-ser118-ERα levels correspond to estrogenic responses in H1793 cells. Basal P-ser118-ERα was approximately 4.3-fold higher in H1793 compared with A549 cells, and E2 increased P-ser118-ERα in H1793, but not in A549 cells (Figures 3A and 3B). P-ser118-ERα was exclusively nuclear in H1793 cells (Figure 3C). Dual-label immunofluorescence staining for ERα (green) and P-ser118-ERα (red) revealed more P-ser118-ERα in H1793 than in A549 cells (Figure 3D and Figure E6), confirming conclusions from the subcellular fractionation/Westerns: H1793 cells have more nuclear P-ser118-ERα than A549 cells. ERα was primarily in the perinuclear region of H1793, and showed diffuse staining throughout the cytoplasm and nucleus of A549. The detection of nuclear ERα in A549 cells using antibody AER320 is different from the lack of ERα nuclear staining using antibody HC-20 (Figure 1C). In addition, E2 increased the intensity of bright nuclear punctuate “speckles” of ERα that overlapped with P-ser118-ERα in H1793, but not in A549 cells (see arrows in Figure E6).
Cyclin D1 in Lung Adenocarcinoma Cell Lines
If E2 in up-regulation of cyclin D1 (CCND1) transcription by genomic ERα activity allows G1-S cell cycle progression in lung adenocarcinoma cells, as reported for breast tumors (26), then E2 should stimulate higher CCND1 transcription in H1793 than in A549 cells. Indeed, E2 increased higher CCND1 in H1793 than in A549 cells, commensurate with our previous report of greater E2-transcriptional responsiveness of the female-derived cells (Figure 4A and Ref. 13). The maximum induction was detected at 3 hours of E2 treatment in H1793 cells, and preincubation with the MAPK inhibitor, PD98059, blocked E2-induced CCND1 transcription (Figure 4B). These data are in agreement with a report that MAPK activity is required for E2-induced cyclin D1 expression and cell cycle progression in MCF-7 cells (27).
Figure 4.

E2 induces higher CCND1 transcription in H1793 than in A549 lung adenocarcinoma cells. (A) A549 and H1793 cell lines were treated with EtOH or E2 for the indicated time, and CCND1 expression was determined by real-time qRT-PCR, and is expressed relative to time zero EtOH for each cell line, as described in Materials and Methods. Values are the means (±SEM) of three to four separate experiments in which each sample was run in triplicate. The maximum induction of CCND1 was detected after 3 hours of E2 treatment in H1793 cells. *P < 0.05, significantly different from EtOH time zero for that cell line. (B) A549 and H1793 cell lines were pretreated with 50 μM PD98059 for 1 hour and treated with EtOH or E2 for 3 hours. CCDN1 is expressed as fold relative to EtOH for each cell line. Values for PD98059 pretreatment are the average of triplicate determinations. (C) A549 and H1793 cells were treated as indicated with EtOH, 10 nM E2, 100 nM 4-hydroxytamoxifen (4-OHT), 100 nM ICI 182,780 (ICI) (alone or in combination). Where indicated, cells were pretreated with 50 μM PD98059 for 1 hour before adding the other indicated treatments. MTT assays were performed as described in Materials and Methods. Values are the means (±SEM) of three to six separate experiments in which each sample was run in quadruplicate. *P < 0.05, significantly different from EtOH time zero for that cell line; **P < 0.05, significantly different from E2 alone value for that cell line.
To determine if pharmacological inhibition of E2-stimulated CCND1 transcription and P-ser118-ERα and cyclin D1 nuclear localization by PD98059 (Figure E7) affected cell proliferation, we compared the effect of ER ligands in the presence or absence of PD98059 pretreatment on A549 and H1793 cell viability/proliferation using the MTT assay (Figure 4C). E2 selectively increased H1793, but not A549, cell proliferation, and concomitant treatment with 4-OHT or ICI blocked the E2 stimulation, indicating that the E2-induced increase in cell proliferation was ER mediated. These data are in agreement with previous bromodeoxyuridine incorporation data on these cell lines (13). Pretreatment with PD98059 reduced cell viability of both A549 and H1793, although the effect was approximately 20% greater in H1793 than in A549, and blocked E2 stimulation of H1793 cell proliferation.
Nuclear localization of cyclin D1 is important for cell cycle progression (28), and cyclin D1 is an ERα coactivator (29). Dual-immunofluorescence staining detected nuclear cyclin D1 and P-ser118-ERα colocalization in H1793, but not in the A549 cells (Figure 5A). The intensity of nuclear cyclin D1 and P-ser118-ERα was highest in E2-treated H1793 cells in the prometaphase or metaphase stages (Figure 5B). In agreement with our report that E2 increases proliferation of H1793 cells, but not A549 cells (13), E2 increased the number of H1793 cells in metaphase, and these cells appeared to have higher levels of cyclin D1 and P-ser118-ERα around the mitotic plate (Figure 5). FACS was used to quantify cell cycle distribution after 6-hour treatment of H1793 and A549 cells with vehicle control (EtOH) or 10 nM E2 (Table 3). E2 specifically increased the proportion of H1793 cells in the S and G2/M phases of the cell cycle, but had no effect on cell-phase distribution of A549 cells (Table 3). Together, these data indicate that E2 stimulates cell cycle progression in H1793 cells, and not in A549 cells.
Figure 5.

E2 increases nuclear P-ser118-ERα and cyclin D1 in H1793 lung adenocarcinoma cells. (A) Immunofluorescence staining of control (EtOH)- and E2-treated H1793 (top two rows) and A549 (bottom two rows) cells for P-ser118-ERα (green) and cyclin D1 (red). Cells were counterstained with DAPI (blue) to image nuclei. Scale bar, 80 μm. The red and green scales with Max 5 and Min 1 indicate variation in staining intensity with P-ser118-ERα and cyclin D1 antibodies. (B) Examination of P-ser118-ERα (green) and cyclin D1 (red) in individual cells, counterstained with DAPI (blue) during the indicated phases of the cell cycle after 6 hours of treatment with 10 nM E2. Images were captured using identical exposure times. Scale bar, 20 μm.
TABLE 3.
CELL CYCLE ANALYSIS OF ESTRADIOL-TREATED A549 AND H1793 LUNG ADENOCARCINOMA CELLS
| Cells and Treatment | G0/G1 | S | G2/M |
|---|---|---|---|
| A549 (EtOH control) | 79.9 ± 0.6 | 4.9 ± 0.1 | 12.1 ± 0.3 |
| A549 + 10 nM E2 | 80.2 ± 0.8 | 5.1 ± 0.1 | 11.6 ± 0.3 |
| H1793 (EtOH control) | 80.4 ± 3 | 5.1 ± 0.6 | 9.9 ± 1.6 |
| H1793 + 10 nM E2 | 74.2 ± 0.8* | 6.3 ± 0.2*† | 14.2 ± 0.5*‡ |
Definition of abbreviations: E2, estradiol; EtOH, ethanol.
Briefly, 1 × 106 cells were precultured for 48 hours in Ham's F12 for A549 or serum-free media with hydrocortisone, insulin, transferrin, estrogen, and selenium (HITES) for H1793 cells. Media were supplemented with 5% dextran-coated, charcoal-stripped FCS. Cells were treated with EtOH (vehicle control) or 10 nM E2 for 6 hours to examine cell cycle distribution at the same time used in immunohistochemical analyses of estrogen receptor α and cyclin D1 subcellular localization. After treatment, the cells were harvested, permeabilized, stained with propidium iodide, and analyzed by FACS as detailed in Materials and Methods. All phases are given in percentages; values are the means (±SEM) of five independent cultures.
*P < 0.005, significantly different between the same treatment between A549 and H1793.
P < 0.005, significantly different from EtOH in the same cell line.
P < 0.05, significantly different from EtOH in the same cell line.
E2 Rapidly Activated MAPK via Nongenomic Signaling in Lung Adenocarcinoma Cells
E2 rapidly (in 2 min) activates the phospho-p42/p44 MAPK (ERK1/2) pathway in H23 lung adenocarcinoma cells (5, 18), and MAPK phosphorylates ERα on ser118 (9). If the higher P-ser118-ERα in H1793 versus A549 cells (Figure 3) corresponds to differences in E2-induced ERK1/2 activation, then more P-ERK1/2 should be detected in E2-treated H1793 cells than A549 cells. Indeed, E2 increased the P-ERK1/2:ERK1/2 ratio in H1793 cells by approximately threefold in 5 minutes, which returned to basal levels after 24 hours (Figure 6A). A549 showed an approximately 1.9-fold increase in P-ERK1/2, peaking at 10 to 60 minutes. Basal P-ERK1/2 was not different in these cell lines (0.25 ± 0.03 in H1793 cells; 0.24 ± 0.02 in A549 cells). The higher E2-induced P-ERK1/2 in H1793 cells agrees with an E2-induced increase in P-ser118-ERα in H1793 cells, but not in A549 cells. Preincubation with PD98059 (a MEK1 inhibitor), PTX (a Gα inhibitor), β-CD (a lipid raft disruptor), or PP2 (a Src kinase inhibitor) before E2 treatment blocked E2-induced ERK1/2 phosphorylation (Figure 6B), indicating functional nongenomic ER-mediated signaling. The reason why PP2 stimulated basal ERK activity in H1793 is unknown, but PP2 increased basal MAPK in GH3/B6/F10 rat pituitary cell (30). In contrast, both β-CD and PD98059 inhibited basal ERK activation, results commensurate with the disruption of plasma membrane domains (e.g., caveolae) (5) and inhibition of MEK1 activity, respectively.
Figure 6.

E2 rapidly induces phosphorylation of extracellular signal–regulated kinase (ERK) 1/2 in lung adenocarcinoma cells. (A) H1793 and A549 cells were treated with EtOH (t0) or 10 nM E2 for the indicated times. (B) H1793 and A549 cells were pretreated with 50 μM PD98059 (MEK1 inhibitor, 1 h), 100 ng/ml pertussis toxin (PTX; a Gα inhibitor, 17 h), 5 mM methyl-β-cyclodextrin (βCD; a lipid raft disruptor, 30 min), or 10 μM PP2 (Src kinase inhibitor, 30 min), before EtOH (t0) or 10-minute E2 treatment. The values are the relative levels of P-ERK1/2:total ERK1/2 compared with the EtOH t0 for each cell line, and are the means (±SEM) from three experiments. *P < 0.05 compared with t0 values for that cell line; *P < 0.05 compared with t10 E2 values for that cell line.
Inhibition of MAPK Blocks E2-Induced Nuclear Accumulation of P-ser118-ERα and Cyclin D1 in H1793 Cells
If nuclear accumulation of P-ser118-ERα and cyclin D1 in E2-treated H1793 cells, but not A549 cells, is mediated by nongenomic E2 activation of the MAPK pathway, then pretreatment of H1793, but not A549, with MEK inhibitor, PD98059, should reduce nuclear P-ser118-ERα and cyclin D1. Indeed, this anticipated result was observed: PD98059 blocked the appearance of P-ser118-ERα and cyclin D1 in the nuclei of H1793 cells and not A549 cells (Figure 7 and Figure E7). These results indicate that nongenomic E2-signaling through the MAPK pathway leads to nuclear P-ser118-ERα, and thus genomic ER signaling in H1793 cells, but not in A549 cells.
Figure 7.

MEK1 inhibitor blocks nuclear accumulation of P-ser118-ERα and cyclin D1 in H1793 cells, but not in A549 cells. H1793 and A549 cells were pretreated with 50 μM PD98059 for 1 hour and treated with EtOH (t0) or 10 nM E2 for 6 hours. The cells were stained with P-ser118-ERα antibody (green) and cyclin D1 antibody (red) for dual-label fluorescence immunostaining, as described in Materials and Methods. Images of nuclei (DAPI), ERα, cyclin D1, and the merged image were captured at identical times of exposure for each cell line and treatment.
The Antiestrogens, ICI and 4-OHT, Block Nuclear Accumulation of P-ser118-ERα and Cyclin D1 in H1793 Cells
ICI and tamoxifen are well established antagonists of genomic ER that are clinically useful in the treatment of ER-positive breast tumors (31). ICI inhibits cyclin D1 activity, and 4-OHT decreases cyclin D1 expression (32). In contrast to the nuclear colocalization of P-ser118-ERα and cyclin D1 in E2-treated cells, dual-immunofluorescence staining detected diffuse intracellular distribution of cyclin D1 and P-ser118-ERα in H1793 cells treated with ICI alone or with E2, and 4-OHT alone or with E2 (Figure 8). Although the intensity of P-ser118-ERα staining in ICI-treated cells was similar to E2 treatment, no nuclear P-ser118-ERα was seen, even in cells treated with E2 and ICI (Figure 8). Although the combined treatment of H1793 cells with E2 and either ICI or 4-OHT did not completely inhibit nuclear localization of cyclin D1 in all cells, it is noteworthy that ICI or 4-OHT treatment blocked nuclear colocalization of cyclin D1 with P-ser118-ERα, as these two ER antagonists clearly blocked nuclear P-ser118-ERα accumulation.
Figure 8.

The antiestrogens, ICI and 4-OHT, block nuclear accumulation of P-ser118-ERα and cyclin D1 in H1793 lung adenocarcinoma cells. H1793 cells were pretreated with either EtOH or 100 nM ICI for 6 hours, and then treated with EtOH, 10 nM E2, ICI alone or with 10 nM E2, or 100 nM 4-OHT alone or with 10 nM E2 for 6 hours. The cells were stained with P-ser118-ERα antibody (green) and cyclin D1 antibody (red) as described in Materials and Methods. Images of nuclei (DAPI), ERα, cyclin D1, and the merged image were captured at identical times of exposure for each treatment. Scale bar, 20 μm.
DISCUSSION
There is increasing evidence that estrogens, including those produced locally by aromatase, stimulate lung adenocarcinoma cell proliferation (5, 6, 14, 33). Recently, the first mouse model demonstrating that E2 stimulates lung adenocarcinoma tumor formation in both ovex females and intact males was reported (34). Here, we demonstrate, for the first time, that nuclear P-ser118-ERα is higher in E2-transcriptionally responsive H1793 lung adenocarcinoma cells from females than in A549 non–E2-responsive lung adenocarcinoma cells from males. Phosphorylation of ERα on ser118 is required for transcriptional activity (9), and indicates an intact E2-dependent ERα signaling pathway in primary breast tumors in vivo (25). Ser-118-ERα phosphorylation is dependent on E2 activation of the Ras-Raf-MAPK (ERK) pathway (9). Here, we show that nuclear P-ser118-ERα corresponds to higher nongenomic E2-activation of MAPK activity in H1793 cells than A549 cells. Furthermore, we demonstrate that the MEK inhibitor, PD98059, blocked E2-induced nuclear localization of P-ser118-ERα and cyclin D1 in H1793 cells, while having no effect on ERα or cyclin D1 in A549 cells. The increase in nuclear P-ser118-ERα offers a mechanism to explain, in part, the observed differences in E2-induced ERE-luciferase activity in H1793 cells, but not in A549 cells, as we reported previously (13). Furthermore, data from the MTT assay, showing that pharmacologic inhibition of MAPK signaling with PD98059 inhibits E2-induced cell proliferation in H1793 cells, indicate a connection(s) between E2-induced MAPK signaling with ERα phosphorylation, CCND1 transcription, cyclin D1 nuclear localization, and cell proliferation in H1793 cells, but not in A549 cells. The inhibition of E2-activated ERK by PP2, PD98059, and β-CD agrees with previous reports of rapid E2-induced MAPK activation via nongenomic ER signaling (reviewed in Ref. 11) in H23 (5, 18) and 201T (33) NSCLC cell lines. Our H1793 data agree with a report showing that the maximum P-ser118-ERα levels occurred after 4–6 hours of E2 treatment in MCF-7 cells (35).
E2 increased higher levels of CCND1 gene transcription in H1793 cells compared with A549 cells, data corresponding with the higher E2-induced MAPK activation, nuclear P-ser118-ERα, and nuclear localization of cyclin D1 in H1793 cells. In addition, we detected colocalization of P-ser118-ERα and cyclin D1 in the nucleus of actively dividing H1793 cells, but not A549 cells, supporting a role for this interaction for cell replication (36, 37). Importantly, these data correspond to those of our previous report that E2 selectively, and in an ER-dependent manner, increased proliferation, ERE-driven reporter gene transcription, and induction of selected E2 target genes in H1793 cells and other lung adenocarcinoma cell lines from females, but not in A549 cells and other cell lines from males (13). These studies support a role for estrogens and other growth factors, the pathways of which intersect with ER signaling mechanisms in promoting the growth of NSCLC cells in female patients.
Cyclin D1 is a negative prognostic factor in lung cancer (38). Here, we demonstrate, for the first time, nuclear colocalization of cyclin D1 and P-ser118-ERα in female-derived H1793 cells, whereas cyclin D1 was perinuclear in male-derived A549 cells. Furthermore, A549 cells had significantly lower P-ser118-ERα compared with H1793 cells. These data indicate that a potential mechanism by which E2 selectively stimulates proliferation of H1793 and not A549 cells (as shown in Ref. 13) may involve cyclin D1 acting as a coactivator (29) for P-ser118-ERα activation of CCND1 (and other) genes involved in cell cycle progression.
Our findings, that NF1604 and male-derived lung adenocarcinoma A549 and H1792 cells have no or lower nuclear ERα compared with H1793, H1944, H1395, and H1435 lung adenocarcinoma cells from females and MCF-7 cells, indicate that proliferative and genomic activity of E2, which were specific for lung adenocarcinoma cell lines from females (13), correlate with more nuclear ERα in female-derived cells. Moreover, these observations also agree with higher nuclear ERα in NSCLC tumors from female than from male patients (49 and 35%, respectively) (18).
ERα splice variants have been detected in various normal and neoplastic tissues, including lung cancer (39). We observed discrepancies in ERα recognition between HC-20 and AER320 ERα antibodies. HC-20 revealed 80- and 52- to 55-kD ERα bands in Western blots, but did not recognize full-length (66-kD) ERα in NF1604 or the lung cancer cell lines. Band specificity was demonstrated (Figure E3). In contrast, as seen here in Western blots and reported previously (13), AER320 detected full-length ERα in WCE and NE of the lung adenocarcinoma cell lines and NF1604 lung fibroblasts. Discrepancies in ERα immunoreactivity in NSCLC have been attributed to variability in epitope recognition (reviewed in Ref. 6).
Similar to our findings, 80- and 52- 55-kD ERα, but no full-length ERα, were reported in WCE from lung tumor cell lines, including A549 and H23 cells using HC-20, and were attributed to ERα exons 6 and 7 duplication and exon 4 deletion (80 and 55 kD, respectively) (14). In agreement with reports that ERα Δ4 is cytoplasmic and unable to bind ligand or DNA (24), we observed a band corresponding to ERα Δ4 in the cytoplasm of A549 and H1793 cells, but not in the nucleus of H1793 or MCF-7 cells. HC-20 was reported to recognize ERα Δ2, Δ2–3, and Δ7 (58, 54, and 52 kD, respectively) (24). Here, PCR confirmed ERαΔ7 mRNA in H1793 and MCF-7 cells. Although not previously identified in lung adenocarcinomas, ERα Δ7 is the most common variant in breast and other cancers, and lacks ligand-dependent activation function–2, resulting in “dominant-negative activity” (24). ERα Δ7 is nuclear, binds EREs in vitro and in vivo, and may act as a monomer (40). Because ERα Δ7 competes with WT ERα, an increase in ERα Δ7 may result in an E2-resistant phenotype (40).
Although we have not excluded the possibility that the ERαΔ2 and ERαΔ2–3 splice variants may be in H1793 nuclei, these variants lack DNA-binding and dimerization domains, and are unable to affect WT ERα signaling (24). Therefore, ERα Δ2 or Δ2–3 would not be expected to affect ER genomic activity in the lung adenocarcinoma cells.
The 80-kD ERα protein detected in lung adenocarcinoma cell lines by HC-20 here, and as reported by others (14), has a duplication of exons 6 and 7, binds E2 or tamoxifen, and was originally identified in MCF-7 cells (24). We suggest that the 80-kD and Δ7 ERα splice variants may contribute to estrogen signaling in lung adenocarcinoma cells, but further studies will be necessary to probe the identity and parse the roles that ERα splice variants play in normal lung and in lung adenocarcinoma.
Because we detected full-length ERα using AER320, but not HC-20 (Figure 1), an alternative to epitope availability differences in ERα splice variants is that there may be post-translational modification of the F domain that precludes HC-20 interaction. The F domain contributes to ERα activity, is required for tamoxifen's agonist activity, and modulates ERα interaction with coregulators, including SRC-1 (41). Further studies will be required to identify post-translation modification(s) of the F domain of ERα in normal lung and in lung adenocarcinoma.
The data reported here demonstrate that, whereas ERβ is largely cytoplasmic, A549 cells had proportionally more ERβ in the nucleus than ERα when compared with H1793 and MCF-7 cells. These observations offer another possible mechanism for the lack of E2-induced reporter gene activity and gene transcription detected in the A549 cell line (13). Because heterodimerization of ERβ with ERα inhibits ERα-mediated transcriptional activity (42), an increase in ERβ relative to ERα could block ERα's transcriptional activity in lung adenocarcinoma cells. A new study reported that ERβ is cytoplasmic, and not phosphorylated in response to E2 in NSCLC cell lines, including 201T, Calu-6, and A549 cells, but did not examine ERα phosphorylation (43).
In conclusion, our results demonstrate nuclear localization of WT and splice variants of ERα in female-derived, but not male-derived, lung adenocarcinoma cells. E2-induced activation of ERK1/2 signaling results in higher nuclear P-ser118-ERα and cyclin D1 in E2-transcriptionally responsive H1793 compared with E2-nonresponsive A549 lung adenocarcinoma cells. Importantly, these data correlate with our previous report of intact E2-activated ERα genomic signaling in the H1793 female-derived, but not A549 male-derived, lung adenocarcinoma cells (13). Our findings imply that estrogens may affect lung adenocarcinoma cells through integration of nongenomic and genomic pathways in a cell line– and sex-dependent manner.
Supplementary Material
Acknowledgments
The authors thank Drs. Barbara J. Clark, Joan E. Magnusen, and Nalinie S. Wickramasinghe for their suggestions on this manuscript. They also thank Dr. Binks Wattenberg for the Tom20 antibody.
This work was supported by grants from Joan's Legacy Foundation, LUNGevity Foundation, the Kentucky Lung Cancer Research Program, and National Institutes of Health grant R01 DK53220 (C.M.K.).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2009-0059OC on June 25, 2009
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Patel JD. Lung cancer in women. J Clin Oncol 2005;23:3212–3218. [DOI] [PubMed] [Google Scholar]
- 2.Siegfried JM. Women and lung cancer: does oestrogen play a role? Lancet Oncol 2001;2:506–513. [DOI] [PubMed] [Google Scholar]
- 3.Patel JD, Bach PB, Kris MG. Lung cancer in us women: a contemporary epidemic. JAMA 2004;291:1763–1768. [DOI] [PubMed] [Google Scholar]
- 4.Dubey S, Siegfried JM, Traynor AM. Non–small-cell lung cancer and breast carcinoma: chemotherapy and beyond. Lancet Oncol 2006;7:416–424. [DOI] [PubMed] [Google Scholar]
- 5.Pietras RJ, Marquez DC, Chen HW, Tsai E, Weinberg O, Fishbein M. Estrogen and growth factor receptor interactions in human breast and non–small cell lung cancer cells. Steroids 2005;70:372–381. [DOI] [PubMed] [Google Scholar]
- 6.Niikawa H, Suzuki T, Miki Y, Suzuki S, Nagasaki S, Akahira J, Honma S, Evans DB, Hayashi S-i, Kondo T, et al. Intratumoral estrogens and estrogen receptors in human non–small cell lung carcinoma. Clin Cancer Res 2008;14:4417–4426. [DOI] [PubMed] [Google Scholar]
- 7.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 2001;29:2905–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klinge CM. Estrogen receptor interaction with co-activators and co-repressors. Steroids 2000;65:227–251. [DOI] [PubMed] [Google Scholar]
- 9.Lannigan DA. Estrogen receptor phosphorylation. Steroids 2003;68:1–9. [DOI] [PubMed] [Google Scholar]
- 10.Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-[alpha]: estrogens, xenoestrogens, and phytoestrogens. Steroids 2005;70:364–371. [DOI] [PubMed] [Google Scholar]
- 11.Watson CS, Alyea RA, Jeng YJ, Kochukov MY. Nongenomic actions of low concentration estrogens and xenoestrogens on multiple tissues. Mol Cell Endocrinol 2007;274:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klinge CM. Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem 2008;105:1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dougherty SM, Mazhawidza W, Bohn AR, Robinson KA, Mattingly KA, Blankenship KA, Huff MO, McGregor WG, Klinge CM. Gender difference in the activity but not expression of estrogen receptors alpha and beta in human lung adenocarcinoma cells. Endocr Relat Cancer 2006;13:113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stabile LP, Davis AL, Gubish CT, Hopkins TM, Luketich JD, Christie N, Finkelstein S, Siegfried JM. Human non–small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen. Cancer Res 2002;62:2141–2150. [PubMed] [Google Scholar]
- 15.Stabile LP, Siegfried JM. Sex and gender differences in lung cancer. J Gend Specif Med 2003;6:37–48. [PubMed] [Google Scholar]
- 16.Taylor AH, Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J Mol Endocrinol 2000;24:145–155. [DOI] [PubMed] [Google Scholar]
- 17.Kawai H, Ishii A, Washiya K, Konno T, Kon H, Yamaya C, Ono I, Ogawa J. Combined overexpression of egfr and estrogen receptor alpha correlates with a poor outcome in lung cancer. Anticancer Res 2005;25:4693–4698. [PubMed] [Google Scholar]
- 18.Marquez-Garban DC, Chen H-W, Fishbein MC, Goodglick L, Pietras RJ. Estrogen receptor signaling pathways in human non–small cell lung cancer. Steroids 2007;72:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum Mol Genet 2000;9:403–411. [DOI] [PubMed] [Google Scholar]
- 20.Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of eralpha and erbeta in human MCF7 cells. Am J Physiol Endocrinol Metab 2004;286:E1011–E1022. [DOI] [PubMed] [Google Scholar]
- 21.Huang A, Leygue E, Dotzlaw H, Murphy LC, Watson PH. Influence of estrogen receptor variants on the determination of ER status in human breast cancer. Breast Cancer Res Treat 1999;58:219–225. [DOI] [PubMed] [Google Scholar]
- 22.Pace MC, Chambliss KL, German Z, Yuhanna IS, Mendelsohn ME, Shaul PW. Establishment of an immortalized fetal intrapulmonary artery endothelial cell line. Am J Physiol 1999;277:L106–L112. [DOI] [PubMed] [Google Scholar]
- 23.Chaidarun SS, Klibanski A, Alexander JM. Tumor-specific expression of alternatively spliced estrogen receptor messenger ribonucleic acid variants in human pituitary adenomas. J Clin Endocrinol Metab 1997;82:1058–1065. [DOI] [PubMed] [Google Scholar]
- 24.Herynk MH, Fuqua SAW. Estrogen receptor mutations in human disease. Endocr Rev 2004;25:869–898. [DOI] [PubMed] [Google Scholar]
- 25.Weitsman GE, Li L, Skliris GP, Davie JR, Ung K, Niu Y, Curtis-Snell L, Tomes L, Watson PH, Murphy LC. Estrogen receptor-alpha phosphorylated at ser118 is present at the promoters of estrogen-regulated genes and is not altered due to HER-2 overexpression. Cancer Res 2006;66:10162–10170. [DOI] [PubMed] [Google Scholar]
- 26.Charpentier AH, Bednarek AK, Daniel RL, Hawkins KA, Laflin KJ, Gaddis S, MacLeod MC, Aldaz CM. Effects of estrogen on global gene expression: identification of novel targets of estrogen action. Cancer Res 2000;60:5977–5983. [PubMed] [Google Scholar]
- 27.Mawson A, Lai A, Carroll JS, Sergio CM, Mitchell CJ, Sarcevic B. Estrogen and insulin/IGF-1 cooperatively stimulate cell cycle progression in mcf-7 breast cancer cells through differential regulation of c-myc and cyclin d1. Mol Cell Endocrinol 2005;229:161–173. [DOI] [PubMed] [Google Scholar]
- 28.Lin DI, Lessie MD, Gladden AB, Bassing CH, Wagner KU, Diehl JA. Disruption of cyclin D1 nuclear export and proteolysis accelerates mammary carcinogenesis. Oncogene 2008;27:1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, Pestel RG, Hinds PW, Dowdy SF, Brown M, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of CDK4. Mol Cell Biol 1997;17:5338–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids 2004;69:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis-Wambi JS, Jordan VC. Treatment of postmenopausal breast cancer with selective estrogen receptor modulators (SERMs). Breast Dis 2005;24:93–105. [DOI] [PubMed] [Google Scholar]
- 32.Carroll JS, Prall OWJ, Musgrove EA, Sutherland RL. A pure estrogen antagonist inhibits cyclin E–CDK2 activity in MCF-7 breast cancer cells and induces accumulation of p130–e2f4 complexes characteristic of quiescence. J Biol Chem 2000;275:38221–38229. [DOI] [PubMed] [Google Scholar]
- 33.Stabile LP, Lyker JS, Gubish CT, Zhang W, Grandis JR, Siegfried JM. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non–small cell lung cancer shows enhanced antiproliferative effects. Cancer Res 2005;65:1459–1470. [DOI] [PubMed] [Google Scholar]
- 34.Hammoud Z, Tan B, Badve S, Bigsby RM. Estrogen promotes tumor progression in a genetically defined mouse model of lung adenocarcinoma. Endocr Relat Cancer 2008;15:475–483. [DOI] [PubMed] [Google Scholar]
- 35.Tang H-Y, Lin H-Y, Zhang S, Davis FB, Davis PJ. Thyroid hormone causes mitogen-activated protein kinase–dependent phosphorylation of the nuclear estrogen receptor. Endocrinology 2004;145:3265–3272. [DOI] [PubMed] [Google Scholar]
- 36.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997;88:405–415. [DOI] [PubMed] [Google Scholar]
- 37.Lamb J, Ladha MH, McMahon C, Sutherland RL, Ewen ME. Regulation of the functional interaction between cyclin D1 and the estrogen receptor. Mol Cell Biol 2000;20:8667–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Betticher DC, Heighway J, Hasleton PS, Altermatt HJ, Ryder WD, Cerny T, Thatcher N. Prognostic significance of CCND1 (cyclin D1) overexpression in primary resected non–small-cell lung cancer. Br J Cancer 1996;73:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fasco MJ, Hurteau GJ, Spivack SD. Gender-dependent expression of alpha and beta estrogen receptors in human nontumor and tumor lung tissue. Mol Cell Endocrinol 2002;188:125–140. [DOI] [PubMed] [Google Scholar]
- 40.Horvath G, Leser G, Helou K, Henriksson M. Function of the exon 7 deletion variant estrogen receptor alpha protein in an estradiol-resistant, tamoxifen-sensitive human endometrial adenocarcinoma grown in nude mice. Gynecol Oncol 2002;84:271–279. [DOI] [PubMed] [Google Scholar]
- 41.Skafar DF, Zhao C. The multifunctional estrogen receptor-alpha f domain. Endocrine 2008;33:1–8. [DOI] [PubMed] [Google Scholar]
- 42.Strom A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson J-A. Estrogen receptor {beta} inhibits 17{beta}-estradiol–stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA 2004;101:1566–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang G, Liu X, Farkas AM, Parwani AV, Lathrop KL, Lenzner D, Land SR, Srinivas H. Estrogen receptor {beta} functions through nongenomic mechanisms in lung cancer cells. Mol Endocrinol 2009;23:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}