

Abstract
Purpose
A major mechanism of resistance to methylating agents, including temozolomide, is the DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT). Preclinical data indicates that defective DNA mismatch repair (MMR) results in tolerance to temozolomide regardless of AGT activity. The purpose of this study was to determine the role of MMR deficiency in mediating resistance in samples from patients with both newly diagnosed malignant gliomas and those who have failed temozolomide therapy.
Experimental Design
The roles of AGT and MMR deficiency in mediating resistance in glioblastoma multiforme were assessed by immunohistochemistry and microsatellite instability (MSI), respectively. The mutation status of the MSH6 gene, a proposed correlate of temozolomide resistance, was determined by direct sequencing and compared with data from immunofluorescent detection of MSH6 protein and reverse transcription-PCR amplification of MSH6 RNA.
Results
Seventy percent of newly diagnosed and 78 % of failed-therapy glioblastoma multiforme samples expressed nuclear AGT protein in ≥20% of cells analyzed, suggesting alternate means of resistance in 20% to 30% of cases. Single loci MSI was observed in 3% of patient samples; no sample showed the presence of high MSI. MSI was not shown to correlate with MSH6 mutation or loss of MSH6 protein expression.
Conclusions
Although high AGT levels may mediate resistance in a portion of these samples, MMR deficiency does not seem to be responsible for mediating temozolomide resistance in adult malignant glioma. Accordingly, the presence of a fraction of samples exhibiting both lowAGT expression and MMR proficiency suggests that additional mechanisms of temozolomide resistance are operational in the clinic.
The global standard of care for the treatment of glioblastoma multiforme is maximal surgical debulking, followed by temozolomide (Temodar; Schering-Plough Corporation) plus external beam radiation therapy (XRT; ref. 1). Despite the 14.6-month median survival produced by this therapy, the prognosis for glioblastoma multiforme is bleak, with the majority of patients surviving <2 years (1, 2). Temozolomide, an imidazotetrazine similar to dacarbazine, is spontaneously degraded to 5(3-methyl-1-triazeno)imidazole-4-carboxamide, and subsequently, to methylhydrazine at physiologic pH. Methylhydrazine actively methylates DNA producing three main adducts, N7-methylguanine, N3-methyladenine, and O6-methylguanine, the primary cytotoxic lesion. During replication, O6-methylguanine is incorrectly paired with thymine by DNA polymerase. Recognition of the incorrect O6-methylguanine/T coupling by the DNA mismatch repair (MMR) system leads to the activation of an apoptotic response (3, 4).
De novo and acquired mechanisms of resistance contribute to the tumor progression and low survival characteristic of glioblastoma multiforme. Many investigators have accordingly focused their work on the modulation of these mechanisms, with the goal of increasing the efficacy of temozolomide as well as other regimens. The DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT) is the primary defense against O6-methylguanine adducts and is a recognized mechanism of temozolomide resistance (5-16). AGT directly removes the O6-methyl adduct from the O6 position of guanine in a suicide reaction that reduces the toxicity of temozolomide. Although the combination of temozolomide and O6-benzylguanine (a pseudosubstrate inactivator of AGT) increases sensitivity to temozolomide in vitro and in vivo, the less than complete response to this combination suggests that additional mechanisms are operational in mediating clinical resistance (5, 6).
Defects in MMR result in tolerance to methylators, regardless of AGT protein level (7). Although the role of MMR deficiency in mediating resistance to DNA-methylating agents has been explored experimentally in preclinical studies (3, 8), the degree to which this pathway mediates clinical resistance has not been fully characterized. The few clinical reports addressing MMR deficiency in glioma used clinically derived cell lines and newly diagnosed clinical samples, indicating that these studies were unable to address the role of selective pressure and adaptive mutation on clinical resistance. A recent report showed somatic MSH6 mutations in two recurrent patients who previously received a combination of radiation and chemotherapy [temozolomide and/or 1,3-bis(2-chloroethyl)-1-nitrosourea]. Although the MSH6 mutations did not translate to detectable microsatellite instability (MSI) with the use of the NIH-NCI MSI panel plus seven additional dinucleotide markers, the MSH6 mutations did contribute to a mutator phenotype as characterized by the mutation of multiple protein kinases (9, 10). In addition, MSH6 mutations were not detected in the DNA samples analyzed from one matched patient sample before radiation and chemotherapy, nor in samples analyzed from patients who were untreated, which suggests that the MSH6 mutation was selected as a result of therapy (10). In this report, we address the role of MMR deficiency in mediating resistance to temozolomide in clinical samples by using a mononucleotide MSI assay as a surrogate marker. In addition, we screened for potential MMR deficiencies caused by MSH6 mutations or by lack of MSH6 protein expression, both in newly diagnosed tumor samples and in samples from patients who failed temozolomide therapy.
Materials and Methods
Brain tumor samples
Malignant gliomas were resected at Duke University Hospital. Informed consent was obtained from patients prior to resection in accordance with Duke Institutional Review Board stipulations. Tissue was frozen in Tissue-Tek optimal cutting temperature compound (Sakura Finetek) and frozen at −140°C. Slides were cut from all tissue specimens and stained with H&E to confirm the composition and homogeneity of all samples used; tissue blocks used for these experiments were 90% to 100% tumor cell – positive. Immunohistochemistry was done with a diagnostic block of tumor.
AGT nuclear staining by immunohistochemistry
Immunohistochememistry was done as described previously (11, 12); this method was selected for its strong correlation with AGT protein activity as assessed by other means (17). Embedded tissue sections were deparaffinized in xylene for 4 h. Sections were washed in absolute alcohol, blocked in 1.85% H2O2/methanol, and then rehydrated in distilled water. Antigens were retrieved after heating in AR-10 buffer (Biogenex) for 10 min followed by cooling for 30 min. Slides were washed twice in PBS, blocked with 5% normal goat serum for 15 min, and then incubated overnight with anti-AGT antibody (mT3.1) or control mouse IgG1. Slides were washed twice in PBS, incubated with secondary antibody, and resolved by using a multilink horseradish peroxidase detection system developed with diaminobenzidine solution. Slides were counterstained with Harris modified hematoxylin. Nuclei from 1,000 tumor cells were quantitated by two independent examiners to determine the percentage of positive immunoreactive nuclei. The result was accepted if the difference between independent quantitations was <5%. Cytoplasmic-only reactivity and granular nuclear reactivity were regarded as negative.
MSH6 nuclear staining by immunofluorescent labeling
Slide-bound sections were deparaffinized in two washes of xylene for 5 min each. Slides were rehydrated in duplicate washes of 100% and 95% ethanol for 10 min each, followed by sequential 5-min washes in distilled water and PBS. Epitope recovery was done in a microwave. Slides were boiled in citrate buffer (pH 6.0; 0.1 mol/L citric acid, 0.5% Tween 20), heated for another 15 min at half power, and then cooled to room temperature for 30 min. Slides were washed twice in distilled water, followed by PBS, for 5 min each. Sections were blocked for 1 h in normal goat serum (2% goat serum, 1% bovine serum albumin, 0.1% Triton X-100, 0.05% Tween 20, 0.05% sodium azide, and 0.01 mol/L PBS; pH 7.2) at room temperature in a humidified chamber. Slides were incubated overnight in primary mouse anti-MSH6 antibody (clone 44; BD Biosciences) diluted 1:200 in normal goat serum, rinsed thrice in PBS for 10 min each, and then incubated with rhodamine-conjugated secondary antibody (Chemicon International) diluted in PBS for 1 h. Slides were rinsed with PBS thrice; counterstaining was done with Vectashield hard dry mount medium containing 4′,6-diamidino-2-phenylinodole stain (Vector Laboratories). Slides were analyzed for MSH6 expression by using a Nikon Eclipse TE2000-E inverted scope (Nikon) and Metamorph Imaging Software (Molecular Devices).
MSI assay
Genomic DNA was obtained from a 25 mg section of each tissue block by using a Puregene DNA purification kit (Gentra Systems) according to the instructions of the manufacturer. Tumor DNA was amplified by PCR using five quasimonomorphic mononucleotide loci. Primers to BAT-25 (c-kit), BAT-26 (hMSH2), NR-21 (SLC7A8), NR-22 (IMP1), and NR-24 (ZNF-2) were synthesized according to sequences previously published (13). The pentaplex PCR reaction volume was 30 μL and contained 5 ng of DNA, 200 μmol/L of each deoxyribonucleotide triphosphate, 1.5 mmol/L of MgCl2, 0.75 units of Taq DNA polymerase, and 1 to 4 μmol/L of each primer. The reaction conditions were identical to those described previously (13). PCR products diluted 1:10 in 3% GeneScan 500 Rox internal lane standard in highly deionized formamide (Applied Biosystems) were resolved by using an ABI PRISM 3100-Avant Genetic Analyzer (Applied Biosystems) and analyzed with GeneMarker software (Softgenetics). Instability in any given loci was determined as a shift of PCR product size (increase or decrease) greater than 3 bp. A particular sample was determined to be highly unstable if instability was observed at three or more loci. Samples with one to two loci showing instability were gauged as MSI low (L-MSI) and those showing no unstable loci were determined to be microsatellite stable (MSS).
MSH6 gene sequence analysis
Genomic DNA was obtained from a 25 mg section of each tissue block with a Puregene DNA purification kit (Gentra Systems) according to the instructions of the manufacturer. Each exon of the MSH6 gene was amplified by PCR using the primer sequences listed in Table 1. Exon 4 was amplified in eight sections. DNA was amplified in a final reaction volume of 30 μL containing 20 ng of DNA, 200 μmol/L of each deoxyribonucleotide triphosphate, 1.5 mmol/L of MgCl2, 0.75 units of Taq DNA polymerase, and 1 μmol/L of each primer. Reaction conditions were as follows: (a) 5 cycles at 94°C for 1 min, 56°C for 1 min, and 72°C for 2 min; (b) 5 cycles at 94°C for 1 min, 53°C for 1 min, and 72°C for 2 min; (c) 25 cycles at 94°C for 1 min, 50°C for 1 min, and 72°C for 2 min; and (d) 72°C for 5 min. The temperature of the touchdown reactions of 5, 5, and 25 cycles described above were adjusted if necessary for primers of differing melting temperatures (Tm). DNA was gel-purified by using a QIAquick gel extraction kit (Qiagen). Sequencing was done on an ABI PRISM 3100-Avant Genetic Analyzer (Applied Biosystems) and analyzed with Mutation Surveyor software (SoftGenetics).
Table 1.
Primer sequences used for amplification of MSH6 exons 1 to 10
| MSH6 | Primer sequences 5′ to 3′ |
|---|---|
| Exon 1 | F: TCCGTCCGACAGAACGGTTG |
| R: CAACCCCCTGTGCGAGCCTC | |
| Exon 2 | F: AACTAAGTTATGTATTTCCT |
| R: CCTGTCTGTCTGTTTCTCTCT | |
| Exon 3 | F: GGATTACAGTCGTGAGCCTCTG |
| R: GTTAATACACCCTCCCCCTCTT | |
| Exon 4a | F: GTCTTACATTATGGTTTCC |
| R: CCACACAGAGCCACCAATG | |
| Exon 4b | F: CGAAGGGTCATATCAGATTC |
| R: ATACCAAACAGTAGGGCGAC | |
| Exon 4c | F: CGTTSGTGGAGGTGGTGATG |
| R: ATGAATACCAGCCCCGTTC | |
| Exon 4d | F: CTGTACCACATGGATGCTCT |
| R: CTTCCTCTTTTTCTTTGAGG | |
| Exon 4e | F: CCTCTGAGAACTACAGTAAG |
| R: CCAAAACTGGGAGCCGGGTA | |
| Exon 4f | F: CTGTTCTCTTCAGGAAGGTC |
| R: AGCCATTGCTTTAGGAGCCG | |
| Exon 4g | F: ACTTGCCATACTCCTTTTGG |
| R: CCTGCTTTGGGAGTATAAG | |
| Exon 4h | F: GAAAGGCTCGAAAGACTGG |
| R: CCAAGGGCTACTAAGATAAAAGCGAG | |
| Exon 5 | F: AAAACCCCCAAACGATGAAG |
| R: CTTTCTGATACAAAACTTT | |
| Exon 6 | F: GTTTATGAAACTGTTACTACC |
| R: GCAAATATCTTTTATCACAT | |
| Exon 7 | F: GCCAATAATTGCATAGTCTCTTAATG |
| R: CGCCCATGTTTTTAAGATAGTCTTC | |
| Exon 8 | F: CCTTTTTTGTTTTAATTCCT |
| R: CAACAGAAGTGCCCTCTCAA | |
| Exon 9 | F: TTTTGAGAGGGCACTTCTGT |
| R: CCCCTTTTACTGTTTCTTTG | |
| Exon 10 | F: GGAAGGGATGATGCACATG |
| R: GTTTATTAGATCATAATGTT |
MSH6 reverse transcription-PCR
RNA was purified from 25 mg of frozen tissue or paraffin-embedded sections by using the Versagene RNA purification kit (Gentra Systems) or a RecoverAll total nucleic acid isolation kit (Ambion), respectively, according to the instructions of the manufacturer. cDNA was generated for samples by using the MSH6 forward primer 5′-GGTTACCCCTGGTGGCCTTG-3′ and reverse primer 5′-TCCTCCGGCTCTGAAGGCTC-3′. Amplifiable cDNA was assessed by using glyceraldehyde-3-phosphate dehydrogenase primers, forward primer 5′-ACAACAGCCTCAAGATCATCAG-3′ and reverse primer 5′-GGTCCACCACTGACACGTTG-3′. Each reaction was done in 50 μL of total volume using the Titanium One-Step RT-PCR kit (ABgene, Inc.) according to the instructions of the manufacturer. Reverse transcription-PCR (RT-PCR) was done at 47°C for 30 min. The cDNA amplification reaction is as follows: 94°C (5 min), 25 cycles of 94°C (30 s), 60°C (30 s), 72°C (1 min) followed by a 7-min extension at 72°C. Amplified products were separated on a 1.5% agarose gel and visualized by UV transillumination with a Kodak Image Station 440CF (Eastman Kodak).
Results
Patient and tumor characteristics
Fifty-two samples were identified from patients who underwent resection at Duke University Medical Center between September 2001 and January 2006, including five pairs of samples from patients before and after therapy. Twenty-five of the samples were procured from patients who had newly diagnosed disease (14 females, 11 males), and 27 samples were procured from patients (11 females, 16 males) who had failed temozolomide therapy in combination with radiotherapy. Thirteen patients had been treated with chemotherapeutic agents in addition to temozolomide and radiotherapy. The median age of patients in both the newly diagnosed and failed-therapy categories was 49 (range, 20-89). The tumor types of the newly diagnosed patients included 1 anaplastic oligodendroglioma (WHO grade 3), 5 high-grade anaplastic astrocytomas (WHO grade 3), and 19 glioblastoma multiforme (WHO grade 4). Tumors of patients experiencing therapy failures were categorized as 4 anaplastic oligodendrogliomas, 2 anaplastic astrocytomas, and 21 WHO grade 4 gliomas.
AGT protein expression
AGT protein expression was determined by immunohistochemistry as described above. The level of AGT protein expressed in glioma specimens was broad, spanning from 0% to 100% positively stained nuclei per sample (Table 2). Five matched before-and-after samples were available for immunohistochemistry analysis (samples from paired therapeutic failures were designated as “A”), in addition to single characterization samples, two samples were unavailable for AGT quantitation. AGT protein staining in the “high” category (≥20% nuclear AGT staining), in which patients have a marked likelihood of failing to respond to temozolomide therapy (11), was found in a similar percentages of both newly diagnosed (70%) and therapy failure (78%) samples. Additionally, within the paired samples, an increase in the proportion of AGT-expressing cells did not seem to be the potential cause of therapy failure, with the exception of sample 2, in which the percentage of AGT-expressing cells increased from 0.8% to 40% after therapy. In the therapy failure group, there was no observed correlation between the percentage of AGT-expressing cells and the interval between diagnosis and re-resection.
Table 2.
Microsatellite and AGT analysis in newly diagnosed and temozolomide-resistant tumors
| New diagnosis | |||||||
|---|---|---|---|---|---|---|---|
| ID* | Bat-25 | Bat-26 | NR-21 | NR-22 | NR-24 | MSI Status | AGT immunohistochemistry† |
| 1 | − | − | − | − | − | MSS | 0 |
| 2 | − | − | − | − | − | MSS | 0.8 |
| 3 | − | − | + | − | − | L-MSI | 1 |
| 4 | − | − | − | − | − | MSS | 2.5 |
| 5 | − | − | − | − | − | MSS | 5.3 |
| 6 | − | − | − | − | − | MSS | 7 |
| 7 | + | − | − | − | − | L-MSI | 10 |
| 8 | − | − | − | − | − | MSS | 20 |
| 9 | − | − | − | − | − | MSS | 20 |
| 10 | − | − | − | − | − | MSS | 24 |
| 11 | − | − | − | − | − | MSS | 24 |
| 12 | − | − | − | − | + | L-MSI | 30 |
| 13 | − | − | − | − | − | MSS | 35 |
| 14 | − | − | − | − | − | MSS | 39 |
| 15 | − | − | − | − | − | MSS | 45 |
| 16 | − | − | − | − | − | MSS | 56 |
| 17 | − | − | − | − | − | MSS | 60 |
| 18 | − | − | − | − | − | MSS | 60 |
| 19 | − | − | − | − | − | MSS | 60 |
| 20 | − | − | − | − | − | MSS | 65 |
| 21 | − | − | + | − | − | L-MSI | 70 |
| 22 | − | − | − | − | − | MSS | 73 |
| 23 | − | − | − | − | − | MSS | 85 |
| 24 | − | − | − | − | − | MSS | NA |
| 25 | − | − | − | − | − | MSS | NA |
| Therapeutic failures | |||||||
|---|---|---|---|---|---|---|---|
| ID* | Bat-25 | Bat-26 | NR-21 | NR-22 | NR-24 | MSI Status | AGT immunohistochemistry† |
| 5A | − | − | − | − | − | MSS | 1 |
| 26 | − | − | − | − | − | MSS | 1.7 |
| 27 | − | − | − | − | − | MSS | 5.5 |
| 28 | − | − | − | − | − | MSS | 10 |
| 29 | − | − | − | − | − | MSS | 11 |
| 30 | − | − | − | + | − | L-MSI | 14 |
| 31 | − | − | − | − | − | MSS | 20 |
| 24A | − | − | − | − | − | MSS | 25 |
| 8A | − | − | − | − | − | MSS | 30 |
| 2A | − | − | − | − | − | MSS | 40 |
| 32 | − | − | − | − | − | MSS | 40 |
| 33 | − | − | − | − | − | MSS | 40 |
| 34 | − | + | − | − | − | L-MSI | 42 |
| 35 | − | − | + | − | − | L-MSI | 46 |
| 22A | − | − | − | − | − | MSS | 47 |
| 36 | − | − | − | − | − | MSS | 51 |
| 37 | − | − | − | − | − | MSS | 56 |
| 38 | − | − | − | − | − | MSS | 60 |
| 39 | − | − | − | − | − | MSS | 60 |
| 40 | − | − | − | − | − | MSS | 60 |
| 41 | + | − | − | − | − | L-MSI | 63 |
| 42 | − | − | − | − | − | MSS | 66 |
| 43 | − | − | − | − | − | MSS | 68 |
| 44 | − | − | − | − | − | MSS | 70 |
| 45 | − | − | − | − | − | MSS | 80 |
| 46 | − | − | − | − | − | MSS | 90 |
| 47 | − | − | − | − | − | MSS | 100 |
NOTE: +, indicates the presence of MSI for respective marker; L-MSI, ≤2 unstable markers observed.
All samples from both newly diagnosed and temozolomide failure groups are displayed sequentially according to immunohistochemistry AGT expression. Newly diagnosed patients who received and failed subsequent temozolomide therapy are identified in the temozolomide failure column with the letter (A) following their newly diagnosed numerical designation.
Immunohistochemical AGT score = percentage of nuclei/sample stained positive with MT3.1 antibodies.
MSI assay
MSI was used as a surrogate marker for MMR deficiency. A panel of five mononucleotide loci, including Bat-25 and Bat-26, which are more sensitive than the standard National Cancer Institute panel (14), were used to assess MSI. High MSI was defined as instability in three of five loci. The results of microsatellite analysis in patient samples was compared with data generated with the previously described human xenografts D-245 MG and D-245 MG (PR), the latter of which shows instability in all five microsatellite loci analyzed (8). By these criteria, 0 of 52 tumor samples analyzed showed high MSI. Instability was found in a single locus in 4 of 25 newly diagnosed samples (ID: 3, 7, 12, and 21), which constituted ~3% of the 125 individual microsatellite markers; specifically, in markers Bat-25, NR-21, and NR-24 (Table 2). Four cases (ID: 30, 34, 35, 41) of single locus instability were observed in therapy failures or 3% of all markers. Although the microsatellite assay used herein was designed to be used without non – tumor controls, it has been suggested that simple changes in peak height or mild band offset of tumor sample relative to normal control can indicate MSI (15). Therefore, this MSI analysis assay was done on DNA isolated from peripheral blood mononuclear cells of seven normal controls (data not shown). MSI of normal controls confirmed that the few peak height differences and band offsets observed between samples were due to population heterogeneity, which further shows the validity of this assay in the absence of non – tumor DNA.
MSH6 sequence analysis
Recent literature suggests that prolonged exposure to alkylating agents may select for inactivating mutation of the MSH6 gene, resulting in a MMR defect that promotes tumor progression (9, 10). Although the MSI assay used herein should detect MMR defects, it has been shown that destabilizing mutations of MSH6 result in increased hMutShβ (MSH2/MSH3) complex formation. This complex may compensate for destabilized hMutSα and perform base/base and insertion/deletion loop repair (16, 18). These mutants would seem to have functional MMR and no presence of MSI. Therefore, all 10 exons of the MSH6 gene from patients who had failed XRT plus temozolomide therapy, plus the indicated additional interval therapy from time of initial resection, were analyzed by direct sequencing (Table 3). Fourteen of 27 (52%) samples contained one or more variations from the canonical DNA sequence, resulting in amino acid changes in MSH6. Two matched samples had detectable variations; however, these were present in both the pretreatment and posttreatment analyses (Table 3; bracketed samples). An additional three matched samples were determined to have the wild-type gene (analysis sample prior to therapy; data not shown). Multiple single nucleotide polymorphisms caused synonymous (silent) variations in the following amino acids: R62R, P92P, D180D, and E546E (data not shown). Previously uncharacterized silent mutations were observed in samples 32 and 43: T1189T and Y214Y, respectively. The homozygous variant G39E observed in many samples (ID: 26, 27, 32, 40, 42, 45, 24/24A, and 8/8A) is a previously characterized polymorphism (19). Sample 40 was shown to contain a heterozygous lysine-to-isoleucine mutation (K247I) at residue 247. Recent studies demonstrating MMR proficiency of a protease-resistant fragment of MSH6 (lacking the first 340 amino acids; ref. 20) suggest that this mutation may have little effect on the function of MMR.
Table 3.
MSH6 sequence analysis in posttreatment and pre/post-matched glioblastoma multiforme samples
| Sample | Treatment | MSH6 mutation |
|---|---|---|
| 26 | XRT + TMZ | Hom G39E |
| 27 | XRT + TMZ | Hom G39E |
| 29 | XRT, TWZ (200 mg/m2 × 12C D1-5Q28) | Het G971E |
| 30 | XRT, Temo/CPT-11, CPT-11 (125 mg/m2 × 4C), CCNU (70 mg/m2 × 2C), Hydrea/Gleevec × 2C, TMZ <150 mg/m2 × 4C) |
Hom E201Q, Hom, G932D, Het E487D |
| 32 | XRT + Gliadel, CCNU (100 mg/m2 × 2C), TMZ (150 mg/m2 × 2C) |
Hom G39E, Syn T1189T |
| 33 | XRT, CCNU (110 mg/m2 × 2C), TMZ (200 mg/m2 × 2C) | Het A1151V |
| 36 | XRT + TMZ | Het W365X |
| 38 | XRT, TMZ (150 mg/m2 × 4C D1-5Q28), CCNU (110 mg/m2 × 2C), TMZ (150 mg/m2 × 2C), CCNU (110 mg/m2 × 2C) |
Het S532A |
| 40 | IMRT + TMZ | Hom G39E, Het K247I |
| 42 | XRT + TMZ | Hom G39E |
| 43 | XRT + TMZ | Syn Y214Y |
| 44 | XRT + TMZ | Hom L109V |
| 45 | XRT, BCNU + TMZ × 3C, CPT-11/Thalidomide × 4C, Thalidomide × 6 mos., Gliadel + O6BG |
Hom G39E Het R58S Het Q835X |
| Hom G39E | ||
| 50mCi I-131, 10 mg murine 81C6, XRT, TMZ/CPT-11 × 4C |
Hom G39E | |
| Hom G39E | ||
| XRT + TMZ, TMZ (200 mg/m2 × 2C), CCNU (110 mg/m2) |
Hom G39E | |
| 5A | XRT + TMZ, TMZ (125 mg/m2, CCNU 70 mg/m2 | Wild-type |
| 28 | Gliadel + XRT, TMZ (200 mg/m2), Z (150 mg/m2 × 5C) |
Wild-type |
| 31 | XRT + TMZ | Wild-type |
| 2A | XRT + TMZ, TMZ (150 mg/m2) | Wild-type |
| 34 | XRT, TMZ (200 mg/m2 × 5C) | Wild-type |
| 35 | XRT, TMZ (200 mg/m2 × 2C) | Wild-type |
| 22A | XRT, TMZ (150 mg/m2), TMZ (200 mg/m2), Z/O6BG/CPT-11 × 6C |
Wild-type |
| 37 | XRT + TMZ, TMZ (200 mg/m2 × 2C), CCNU (110 mg/m2 × 2C), TMZ (200 mg/m2 × 2C) |
Wild-type |
| 39 | XRT, TMZ (150 mg/m2), TMZ (200 mg/m2), CCNU (110 mg/m2), TMZ/Topotecan × 6C |
Wild-type |
| 41 | XRT, TMZ QD × 6C, TMZ (200 mg/m2) | Wild-type |
| 46 | PCV × 6C, XRT + TMZ (150 mg/m2), Z (200 mg/m2 × 9C) |
Wild-type |
| 47 | XRT + TMZ, TMZ (200 mg/m2 × 2C) | Wild-type |
NOTE: TMZ failures are shown by the sequence (Table 2) based on ascending AGT expression. Samples with MSH6 mutations following direct sequencing are presented in the unshaded portion. Brackets indicate matched pretherapy/posttherapy glioblastoma multiforme samples, in which the posttherapy sample is designated by an “A” suffix. All other samples were obtained after the indicated interval therapy. MSH6 wild-type samples are shown in the shaded region. The number of therapy cycles received by each patient (>1) is indicated by a number and letter (C) following the therapy dose. Therapies lacking such a designation can be assumed to be a single cycle.
Abbreviations: BCNU, 1,3-bis(2-chloroethyl)-1-nitrosourea; CCNU, 1-(2-chloroethyl)-3-cyclohexyl-l-nitrosourea; PCV, procarbazine, CCNU, vincristine; TMZ, temozolomide.
Seven samples contained nonsynonymous mutations and are shown relative to the resolved structure of MSH6 in Fig. 1. The human MSH6 protein can be divided into five domains; an NH2-terminal mismatch binding domain, a connector region, two α-helical regions separated by a clamp region, and finally, the COOH-terminal proximal domain containing the ATP hydrolytic centers (ABC-ATPase domain; ref. 20). Sample 36 contained a heterozygous mutation changing tryptophan (W365X) to a stop codon located in the mismatch binding domain adjacent to two helices (residues 366-369 and 371-373; ref. 20). Sample 30 contained a heterozygous glutamic acid-to-aspartic acid mutation at the highly conserved residue (E487D) between a sheet and a helix in the mismatch binding domain. The consistent small, polar nature of these residues suggests that substitution would not be deleterious to protein stability (21). Sample 38 contained a heterozygous mutation of serine to alanine at position 532 (S532A) located in the connector domain, five residues from a nine-residue sheet; however, there is a 95% confidence that this substitution will not affect protein conformation or function. Mutation of the α-helical lever domain were observed in samples 30 and 45, the former showing a homozygous mutation of glycine to aspartic acid at the conserved residue 932 (G932D) and the latter showing a heterozygous mutation of glutamine to a stop codon at position 835 (Q835X). Mutation in the clamp domain in sample 29 (G971E) is flanked by a region of residues important in DNA binding in association with region 1 (mismatch binding domain) in tertiary and quaternary protein structure (20). Lastly, a single mutation was found in sample 33 in the ATPase domain of MSH6, in a 16-residue helix, which caused a heterozygous shift of alanine to valine at position 1151 (A1151V).
Fig. 1.
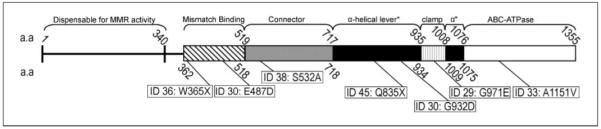
Resolved structure of the MSH6 protein showing domains with amino acid (a.a.) stop and start numerical designations. Mutations other than DNA polymorphisms and synonymous mutations from Table 3 at their corresponding locations.
MSH6 immunofluorescence and RT-PCR
We next sought to determine if any of the mutations observed were associated with a decrease in protein stability and, hence, a decrease in expression of protein as observed by immunofluorescence. Twenty-five posttherapy samples were available for analysis complemented by an additional five before-therapy matched samples. Samples were qualitatively scored according to the nuclear presence of MSH6 protein. Figure 2A shows MSH6 staining (ID: 47, top middle) and the MSH6 stain colocalized with nuclear 4′,6-diamidino-2-phenylinodole counterstain (ID: 47, top right), indicating nuclear localization of the MSH6 protein. Conversely, background fluorescence was observed (ID: 22A; bottom) in the MSH6-stained panel for a sample without detectable nuclear MSH6. Three (12%) of 25 posttherapy samples (5A, 22A, and 38), lacked detectable nuclear MSH6 protein. However, MSH6 protein was also not detectable in the matched pretherapy specimens from samples 5A and 22A, which suggests that loss of protein was not a result of therapy. Additionally, only one sample (ID: 38), which showed a missense mutation by direct sequencing, was devoid of protein as ascertained by immunofluorescence, even though a wild-type MSH6 allele was present. Interestingly, the two samples that contained heterozygous stop codon mutations (ID: 36 and 45) displayed very clear nuclear MSH6 staining, which further supports the concept that a single functional allele is sufficient for abundant protein generation. The other two sample pairs previously mentioned which lacked detectable MSH6 protein, samples 5 and 5A and samples 22 and 22A, were wild-type for the exon sequences of the MSH6 gene.
Fig. 2.
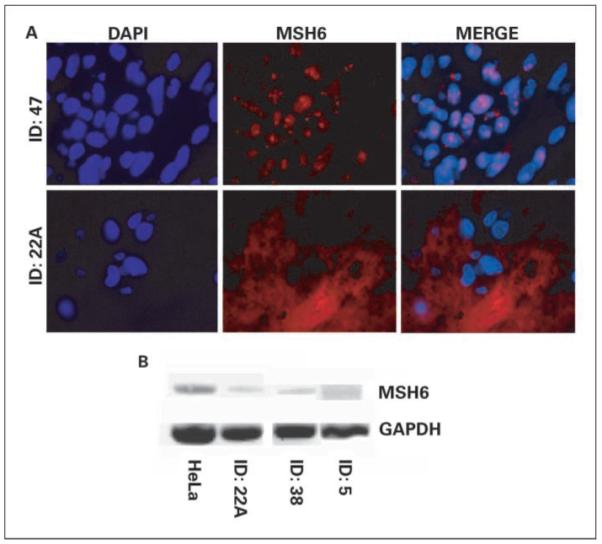
MSH6 immunofluorescence and RT-PCR. A, sample ID: 47 showing nuclear 4′,6-diamidino-2-phenylinodole staining, MSH6 staining, and colocalized merge (top); MSH6-deficient sample (ID: 22A). B, MSH6 RT-PCR compared with HeLa control; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) used as an internal loading control.
We then did RT-PCR on samples with undetectable MSH6 protein for which RNA was available, to determine if the MSH6 transcript was present. MLH1 has been shown to be epigenetically regulated by CpG island methylation. However, the TATAA and CAAT-less promoter of MSH6 seems to be regulated by SP1 binding sites, which are largely unaffected by methylation (22); this is, however, still controversial (23). Additionally, whereas mutations could affect the recruitment and stabilization of transcription machinery, it seems that mutations which can decrease MSH6 promoter activity to less than one-half the normal value are not abundant in the Caucasian population (24). Because the rate of overall primary brain tumor incidence is statistically higher in the Caucasian population than in Hispanic or African-American populations, these data suggest that this population represents the majority of patients who would be treated annually with radiotherapy and chemotherapy. Figure 2B shows the amplification of MSH6 cDNA from samples 22A, 38, and 5 in comparison with HeLa cells using glyceraldehyde-3-phosphate dehydrogenase as an internal control. mRNA was detectable to varying degrees in all samples analyzed. This suggests that protein would most likely be present in these samples but may be below the limit of detection of the immunofluorescence assay used.
Discussion
Combined therapy with surgery, radiotherapy, and temozolomide is the current global standard of care for patients with malignant glioma (2, 11, 25, 26). Unfortunately, the majority of these patients display progressive disease and subsequent death. Recurrence or progression is characterized by an increasing number of genetic tumor insults, including chromosomal rearrangements, amplifications, and deletions affecting gene expression (27). Given the histologic and genetic heterogeneity of glioma, this would suggest that a tumor subpopulation with increasingly robust resistance is selected by either intrinsic or acquired mechanisms. In many instances, recurrent tumors seem to be more resistant to a previously used therapy. In vitro, repetitive, high-level drug exposure creates resistant cell lines which often harbor identifiable mutator phenotypes (8, 28, 29). However, the clinical relevance of those identified mechanisms has not been established. Thus, in the current report, we address the clinical role of both AGT and MMR in mediating resistance to temozolomide.
AGT-mediated resistance to temozolomide and other alkylating or chloroethylating agents has been well-characterized in vitro (30-33) and clinically (34-36). Depletion of AGT activity using the substrate analogue O6-benzylguanine (37, 38) further validated AGT’s role in alkyl/methylator resistance, which prompted the exploration of temozolomide and O6-benzylguanine combination therapy clinically (5). Both AGT (39) and the MMR gene, MLH1 (40), have been shown to be regulated epigenetically; methylation of either gene promoter leads to loss of protein expression. Logically, epigenetic depletion of the former seems beneficial to successful temozolomide therapy (41), whereas epigenetic depletion of the latter is deleterious (40). Regardless of the mechanism regulating the expression of AGT, we show in this report the immunohistochemical detection of AGT protein in ≥20% of nuclei in ~70% to 78% of newly diagnosed and failed-therapy tumor samples analyzed. We have previously shown that ≥20% staining of tumor cells for AGT is correlated with temozolomide resistance (11). These data suggest that whereas therapeutic resistance may be attributed to reduced temozolomide toxicity mediated by AGT in many tumors, there is a substantial subset of cases in which resistance may be mediated by an alternate mechanism.
O6-methylguanine adducts not corrected by AGT can lead to a O6-methylguanine/T mismatch upon replication, which is recognized by the DNA MMR protein complex, MutSα (MSH2/MSH6 heterodimer). In response to chemotherapeutic insult, the MMR pathway, like other genome repair mechanisms, will induce checkpoint activation. Failure to repair mismatches in an MMR-deficient tumor results in the tolerance of DNA lesions’ increasing chemotherapy resistance and increasing mutagenesis. The MutSa complex processes a large portion of base/base mispairs and insertion/deletion loops within a cell, which suggests that mutation in both alleles of either heterodimer component may produce a mutator phenotype. Alternatively, functional redundancy in the MMR system can compensate for MSH6 depletion by shifting MSH2 dimerization to MSH3, forming the MutShβ complex (MSH2/MSH3). This alternate complex may be capable of repairing the single base pair mutations/substitutions normally addressed by the MutSα complex. However, if the mutation in MSH6 results in MSH6 loss of function without protein depletion, MMR proficiency may be greatly reduced. In this case, nonfunctional MutSα bound at sites of damage may hinder the binding of a functional, and in this context, compensatory, MutShβ complex; this may contribute to increased mutagenicity upon replication. Faulty MMR proteins will not properly trigger repair for microsatellite DNA sequences, whose replication is affected by slippage of DNA polymerase within the repeat sequence, producing an altered number of DNA repeats. This change in the DNA repeats has been termed MSI and is a recognized surrogate biomarker for loss of MMR activity (18).
Analysis of MSI in glioma and astrocytoma has yielded a disparate picture for the frequency of high-level MSI (42-45). The more recent studies suggest that high levels of MSI are common in pediatric glioma (45), but are relatively rare in adult glioma (44). Our results are consistent with these findings, as we observed incidences of instability in 3% of all loci, but no incidence of multiple loci instability indicative of MMR deficiency in individual tumor samples, either from newly diagnosed patients or from those whose tumors were no longer responsive to temozolomide therapy.
To complement these findings, we have also observed a low correlation between MSI data and MSH6 mutations or loss of MSH6 protein expression, as measured by immunofluorescence. The observed MSH6 mutations were not associated with a defined range of AGT protein expression as one might expect to find in low AGT – expressing tumors exposed to the selective pressures of what some are suggesting to be potentially mutagenic therapy. DNA sequence variations, other than the G39E polymorphism, were observed in 10 of 27 samples procured from patients who had received prior therapy, two of which did not change the amino acid sequence of the protein. Additionally, only a single sample (ID: 30) showed the presence of MSI in one locus (NR-22) concurrent with DNA mutations; the two mutations observed in this sample occurred in the mismatch and α-helical functional domains of the MSH6 protein. These mutations, however, did not deplete the MSH6 protein, nor did they result in MSI in the locus most closely related to MSH6 mutation, Bat-26, which suggests that functionality is maintained for MMR activity (46). Sample 38, which lacked detectable MSH6 protein, was found to have a wild-type MSH6 gene and no detectable MSI; RT-PCR analysis revealed the presence of a MSH6 transcript, suggesting that although it is below the limit of detection for immunofluorescence, MSH6 protein is most likely present and was sufficient to maintain functional MMR. Stop codon mutations W365X and Q835X found in samples 36 and 45, respectively, were both heterozygous and did not seem to diminish the expression of MSH6 or contribute to MSI. It has been reported that reconstitution of MSH6-deficient cells (HCT15) with a functional MSH6 gene that yielded MSH6 protein expression at 20% of MSH2 expression alleviated MSI and restored the G2 checkpoint in response to methyl/alkylating agents, yet did not restore methylator sensitivity (47).
Clearly, MSH6 mutations are present in some patients who have received temozolomide therapy and ultimately failed that therapy. The majority of identified mutations described herein were heterozygous, which allowed for the generation of detectable wild-type MSH6 protein from at least a single allele. At last check, 47 single nucleotide polymorphisms were recognized and well documented in the MSH6 gene; this may suggest that a significant proportion of the mutations that we have shown are also population variances, undocumented until now, that result in little consequence to MMR. The presence of variances in before-and-after matched samples supports this notion. These mutations did not result in MSI, the clinical significance of which is the presence of functional MMR, regardless of mutation status, at the time the samples were procured and the ability to recognize the characteristic base/base mispair lesions induced by temozolomide.
Recent concerns that MSH6 mutations predispose to a mutator phenotype (9, 10) prompted us to take a further look at these studies in relation to the data presented here. Interestingly, in these previous studies, samples with clear MSH6 mutations, resulting in MSH6 protein depletion, showed no sign of MMR defects in mononucleotide repeats (Bat-25 and Bat-26). The data did, however, show what we would describe as high MSI in dinucleotide repeats; it is important to note that whereas our group would define these results as such, this is not the interpretation offered by the authors. Regardless of this apparent contradiction, the data do point to two related explanations for the observation that MSH6 deficiency may be associated with an increased frequency of mutation throughout the remainder of the cell. First, MSH2 protein requires stabilization by another protein for its persistence; ~80% to 90% of the MSH2 present in a cell is normally complexed with MSH6. Upon loss of MSH6 protein expression, formation of compensatory MutShβ (MSH2/MSH3) occurs. MSH3, typically expressed at a lower level as compared with MSH6, stabilizes a small portion of MSH2, and therefore, a smaller amount of MutShβ is formed. Although the MutShβ may be able to address those mutations formed by temozolomide and not alleviated by AGT, additional insertion/deletion loops and larger multiple base mispairs typically addressed by MutShβ may be left unresolved. This alone may contribute to the mutator phenotype, as has been suggested previously (48). Furthermore, dominant MSH6 missense mutations in both mice and yeast have shown that the functions of MutSα, particularly MMR and induction of the apoptosis response, may be separated according to the type and location of the mutation. These data suggest that the relationship of MSH6 with additional proteins in the cell may be weakened whereas its function for MMR remains intact, or that mutation of the MSH6 gene or reduction of MSH6 may produce a mutator phenotype only in the context of a concomitant mutation in another protein. Work is currently under way to address the role of suboptimal, yet functional, MSH6 expression in mediating methylator resistance without observable MMR deficiency.
In summary, we have observed variable levels of AGT repair proteins, the primary mechanism of resistance to temozolomide, in samples from patients who failed temozolomide treatment for a malignant glioma. Given that a significant number of the prior-treatment patients received multiple cycles of temozolomide, it does not seem that prolonged alkylator therapy selected for increased AGT expression in these tumors. The presence of high MSI, an indicator of MMR deficiency, was not observed in these samples. Similarly, MSI in cervical cancer treated with the alkylator cisplatin does not seem to be a result of drug-induced selection (49). Although recent discrepancies have been observed in tumors described as low MSI by the National Cancer Institute panel (mixed mononucleotide and dinucleotide microsatellite repeats) and the presence of potentially mutagenic MSH6 mutations (10, 50) following alkylator therapy, these discrepancies have not been observed when using mononucleotide MSI markers (14, 51, 52). Using a set of five matched before-and-after therapy samples, we have shown the incidence of increased AGT posttherapy as a potential mechanism of resistance in only one pair of samples (samples 2 and 2A) and no incidence of MSI or MSH6-mediated MMR defect as a result of prolonged alkylator therapy. These data suggest that although AGT does play a major role in clinical resistance to temozolomide in adult malignant glioma, MMR deficiency does not. In contrast to recent studies linking prolonged alkylator therapy with selection of MSH6 mutation resulting in reduced MSH6 protein expression, we did not observe this phenotype in the current population of 27 patient samples previously given temozolomide therapy. Further work in progress is attempting to define additional clinically relevant mechanisms of resistance to temozolomide.
Acknowledgments
Grant support: Pediatric Brain Tumor Foundation Institute and NIH grants NS30245, 5P50CA108786, 5P50NS20023, and R37CA011898.
Footnotes
Disclosure of Potential Conflicts of Interest
H. Friedman is a recipient of a commercial research grant from Schering-Plough, and is an advisor to Keryx.
References
- 1.Cohen MH, Johnson JR, Pazdur R. Food and Drug Administration Drug approval summary: temozolomide plus radiation therapy for the treatment of newly diagnosed glioblastoma multiforme. Clin Cancer Res. 2005;11:6767–71. doi: 10.1158/1078-0432.CCR-05-0722. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Dietrich PY, Ostermann Kraljevic S, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–82. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 3.Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci U S A. 1993;90:6424–8. doi: 10.1073/pnas.90.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sibghat U, Day RS., III Incision at O6-methylguanine: thymine mispairs in DNA by extracts of human cells. Biochemistry. 1992;31:7998–8008. doi: 10.1021/bi00149a034. [DOI] [PubMed] [Google Scholar]
- 5.Quinn JA, Desjardins A, Weingart J, et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005;23:7178–87. doi: 10.1200/JCO.2005.06.502. [DOI] [PubMed] [Google Scholar]
- 6.Quinn JA, Pluda J, Dolan ME, et al. Phase II trial of carmustine plus O(6)-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002;20:2277–83. doi: 10.1200/JCO.2002.09.084. [DOI] [PubMed] [Google Scholar]
- 7.Wedge SR, Porteous JK, Newlands ES. 3-Amino-benzamide and/or O6-benzylguanine evaluated as an adjuvant to temozolomide or BCNU treatment in cell lines of variable mismatch repair status and O6-alkylguanine-DNA alkyltransferase activity. Br J Cancer. 1996;74:1030–6. doi: 10.1038/bjc.1996.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman HS, Johnson SP, Dong Q, et al. Methylator resistance mediated by mismatch repair deficiency in a glioblastoma multiforme xenograft. Cancer Res. 1997;57:2933–6. [PubMed] [Google Scholar]
- 9.Cahill DP, Levine KK, Betensky RA, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038–45. doi: 10.1158/1078-0432.CCR-06-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hunter C, Smith R, Cahill DP, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006;66:3987–91. doi: 10.1158/0008-5472.CAN-06-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman HS, McLendon RE, Kerby T, et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response toTemodal in newly diagnosed malignant glioma. J Clin Oncol. 1998;16:3851–7. doi: 10.1200/JCO.1998.16.12.3851. [DOI] [PubMed] [Google Scholar]
- 12.McLendon RE, Cleveland L, Pegram C, Bigner SH, Bigner DD, Friedman HS. Immunohistochemical detection of the DNA repair enzyme O6-methylguanine-DNA methyltransferase in formalin-fixed, paraffin-embedded astrocytomas. Lab Invest. 1998;78:643–4. [PubMed] [Google Scholar]
- 13.Suraweera N, Duval A, Reperant M, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123:1804–11. doi: 10.1053/gast.2002.37070. [DOI] [PubMed] [Google Scholar]
- 14.Xicola RM, Llor X, Pons E, et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007;99:244–52. doi: 10.1093/jnci/djk033. [DOI] [PubMed] [Google Scholar]
- 15.Oda S, Oki E, Maehara Y, Sugimachi K. Precise assessment of microsatellite instability using high resolution fluorescent microsatellite analysis. Nucleic Acids Res. 1997;25:3415–20. doi: 10.1093/nar/25.17.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummond JT, Genschel J, Wolf E, Modrich P. DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutSα/hMutShβ ratio and reduces the efficiency of base-base mismatch repair. Proc Natl Acad Sci U S A. 1997;94:10144–9. doi: 10.1073/pnas.94.19.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maxwell JA, Johnson SP, Quinn JA, et al. Quantitative analysis of O6-alkylguanine-DNA alkyltransferase in malignant glioma. Mol Cancer Ther. 2006;5:2531–9. doi: 10.1158/1535-7163.MCT-06-0106. [DOI] [PubMed] [Google Scholar]
- 18.Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148:1637–46. doi: 10.1093/genetics/148.4.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parc YR, Halling KC, Wang L, et al. HMSH6 alterations in patients with microsatellite instability-low colorectal cancer. Cancer Res. 2000;60:2225–31. [PubMed] [Google Scholar]
- 20.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSα DNA lesion recognition complex. Mol Cell. 2007;26:579–92. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Dayhoff MO, Schwartz RM, Orcutt BC. Natl Biomed Res Found. Vol. 5. Washington DC: 1978. A model of evolutionary change in proteins. Atlas of protein sequence and structure; pp. 345–52. [Google Scholar]
- 22.Holler M, Westin G, Jiricny J, Schaffner W. Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes Dev. 1988;2:1127–35. doi: 10.1101/gad.2.9.1127. [DOI] [PubMed] [Google Scholar]
- 23.Szadkowski M, Jiricny J. Identification and functional characterization of the promoter region of the human MSH6 gene. Genes Chromosomes Cancer. 2002;33:36–46. doi: 10.1002/gcc.1211. [DOI] [PubMed] [Google Scholar]
- 24.Gazzoli I, Kolodner RD. Regulation of the human MSH6 gene by the Sp1 transcription factor and alteration of promoter activity and expression by polymorphisms. Mol Cell Biol. 2003;23:7992–8007. doi: 10.1128/MCB.23.22.7992-8007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinn JA, Reardon DA, Friedman AH, et al. Phase II trial of temozolomide in patients with progressive lowgrade glioma. J Clin Oncol. 2003;21:646–51. doi: 10.1200/JCO.2003.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Yung WK, Prados MD, Yaya-Tur R, et al. Temodal Brain Tumor Group Multicenter phase II trial of temozolomide in patients with anaplastic astrocytoma or anaplastic oligoastrocytoma at first relapse. J Clin Oncol. 1999;17:2762–71. doi: 10.1200/JCO.1999.17.9.2762. [DOI] [PubMed] [Google Scholar]
- 27.Rasheed BK, Wiltshire RN, Bigner SH, Bigner DD. Molecular pathogenesis of malignant gliomas. Curr Opin Oncol. 1999;11:162–7. doi: 10.1097/00001622-199905000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Bacolod MD, Johnson SP, Ali-Osman F, et al. Mechanisms of resistance to 1,3-bis(2-chloroethyl)-1-nitrosourea in human medulloblastoma and rhabdomyosarcoma. Mol CancerTher. 2002;1:727–36. [PubMed] [Google Scholar]
- 29.Bacolod MD, Johnson SP, Pegg AE, et al. Brain tumor cell lines resistant to O6-benzylguanine/1,3-bis(2-chloroethyl)-1-nitrosourea chemotherapy have O6-alkylguanine-DNA alkyltransferase mutations. Mol CancerTher. 2004;3:1127–35. [PubMed] [Google Scholar]
- 30.Friedman HS, Pegg AE, Johnson SP, et al. Modulation of cyclophosphamide activity by O6-alkylguanine-DNA alkyltransferase. Cancer Chemother Pharmacol. 1999;43:80–5. doi: 10.1007/s002800050866. [DOI] [PubMed] [Google Scholar]
- 31.He XM, Ostrowski LE, von Wronski MA, et al. Expression of O6-methylguanine-DNA methyltransferase in six human medulloblastoma cell lines. Cancer Res. 1992;52:1144–8. [PubMed] [Google Scholar]
- 32.Ostrowski LE, von Wronski MA, Bigner SH, et al. Expression of O6-methylguanine-DNA methyltransferase in malignant human glioma cell lines. Carcinogenesis. 1991;12:1739–44. doi: 10.1093/carcin/12.9.1739. [DOI] [PubMed] [Google Scholar]
- 33.Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119–29. [PubMed] [Google Scholar]
- 34.Belanich M, Pastor M, Randall T, et al. Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996;56:783–8. [PubMed] [Google Scholar]
- 35.Mineura K, Watanabe K, Yanagisawa T, Kowada M. Quantification of O6-methylguanine-DNA methyltransferase mRNA in human brain tumors. Biochim Biophys Acta. 1996;1289:105–9. doi: 10.1016/0304-4165(95)00123-9. [DOI] [PubMed] [Google Scholar]
- 36.Silber JR, Mueller BA, Ewers TG, Berger MS. Comparison of O6-methylguanine-DNA methyltransferase activity in brain tumors and adjacent normal brain. Cancer Res. 1993;53:3416–20. [PubMed] [Google Scholar]
- 37.Dolan ME, Moschel RC, Pegg AE. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci U S A. 1990;87:5368–72. doi: 10.1073/pnas.87.14.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domoradzki J, Pegg AE, Dolan ME, Maher VM, McCormick JJ. Depletion of O6-methylguanine-DNA-methyltransferase in human fibroblasts increases the mutagenic response to N-methyl-N’-nitro-N-nitrosoguanidine. Carcinogenesis. 1985;6:1823–6. doi: 10.1093/carcin/6.12.1823. [DOI] [PubMed] [Google Scholar]
- 39.Qian XC, Brent TP. Methylation hot spots in the 5′ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997;57:3672–7. [PubMed] [Google Scholar]
- 40.Bearzatto A, Szadkowski M, Macpherson P, Jiricny J, Karran P. Epigenetic regulation of the MGMT and hMSH6 DNA repair genes in cells resistant to methylating agents. Cancer Res. 2000;60:3262–70. [PubMed] [Google Scholar]
- 41.Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–4. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 42.Dams E, Van de Kelft EJ, Martin JJ, Verlooy J, Willems PJ. Instability of microsatellites in human gliomas. Cancer Res. 1995;55:1547–9. [PubMed] [Google Scholar]
- 43.Lundin DA, Blank A, Berger MS, Silber JR. Microsatellite instability is infrequent in sporadic adult gliomas. Oncol Res. 1998;10:421–8. [PubMed] [Google Scholar]
- 44.Martinez R, Schackert HK, Appelt H, Plaschke J, Baretton G, Schackert G. Low-level microsatellite instability phenotype in sporadic glioblastoma multiforme. J Cancer Res Clin Oncol. 2005;131:87–93. doi: 10.1007/s00432-004-0592-5. [DOI] [PubMed] [Google Scholar]
- 45.Szybka M, Bartkowiak J, Zakrzewski K, Polis L, Liberski P, Kordek R. Microsatellite instability and expression of DNA mismatch repair genes in malignant astrocytic tumors from adult and pediatric patients. Clin Neuropathol. 2003;22:180–6. [PubMed] [Google Scholar]
- 46.Papadopoulos N, Nicolaides NC, Liu B, et al. Mutations of GTBP in genetically unstable cells. Science. 1995;268:1915–7. doi: 10.1126/science.7604266. [DOI] [PubMed] [Google Scholar]
- 47.Lettieri T, Marra G, Aquilina G, et al. Effect of hMSH6 cDNA expression on the phenotype of mismatch repair-deficient colon cancer cell line HCT15. Carcinogenesis. 1999;20:373–82. doi: 10.1093/carcin/20.3.373. [DOI] [PubMed] [Google Scholar]
- 48.Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–20. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 49.Ercoli A, Ferrandina G, Genuardi M, et al. Microsatellite instability is not related to response to cisplatin-based chemotherapy in cervical cancer. Int J Gynecol Cancer. 2005;15:308–11. doi: 10.1111/j.1525-1438.2005.15221.x. [DOI] [PubMed] [Google Scholar]
- 50.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 51.Bacani J, Zwingerman R, Di Nicola N, et al. Tumor microsatellite instability in early onset gastric cancer. J Mol Diagn. 2005;7:465–77. doi: 10.1016/S1525-1578(10)60577-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong YF, Cheung TH, Lo KW, et al. Detection of microsatellite instability in endometrial cancer: advantages of a panel of five mononucleotide repeats over the National Cancer Institute panel of markers. Carcinogenesis. 2006;27:951–5. doi: 10.1093/carcin/bgi333. [DOI] [PubMed] [Google Scholar]