

Abstract
This study reports on the use of a new sensitive assay of cAMP-dependent protein kinase activity to examine the effect of cholecystokinin (CCK) on the cAMP second messenger cascade in rat pancreatic acini. Treatment of acini with both low (pM) and high (nM) concentrations of CCK was associated with an increase in cAMP-dependent protein kinase activity. The increases in kinase activity were detected in the absence of phosphodiesterase inhibition, a condition required to detect a measurable increase in cellular cAMP in these cells. Furthermore, the cAMP cascade was dissociated from the secretory effects of CCK, since the CCK analogue, OPE, mediates enzyme secretion but does not increase cellular cAMP levels or kinase activity.
Keywords: Pancreatic acini, Cholecystokinin, CCK receptor, Adenosine 3′, 5′ cyclic monophosphate, Protein kinase, Phosphodiesterase inhibitor
1. Introduction
The effects of hormone-receptor interaction on the production of Ca2+, inositol phosphate, or cyclic nucleotide second messengers has been extensively studied. In contrast, little is known about the subsequent coupling of these pathways to protein kinase activation. The complexity of interaction among different signal transduction pathways makes it difficult to quantitatively relate the generation of a particular intracellular signal to the activation of its effector kinase.
In this study, the relationship between cAMP and the activation of its physiologic effector, cAMP-dependent protein kinase, has been examined in rat pancreatic acini following stimulation with cholecystokinin (CCK). This system was chosen because it has served as a paradigm for the study of both CCK-regulated zymogen secretion and, most recently, intracellular proteolysis. The role of CCK in this system, however, is quite complex. Based on receptor binding assays, the pancreatic acinar cell exhibits at least two affinity states of the CCK receptor which are functionally coupled to different cellular responses. To better define the cellular mechanisms of CCK action, CCK analogues such as OPE have been developed that stimulate only a subset of the biological effects of CCK [1]. Two groups of biological responses to CCK receptor agonists have been observed. Low concentrations of CCK and OPE stimulate zymogen secretion while other effects, such as the inhibition of secretion and the stimulation of intracellular zymogen proteolysis, result only from stimulation by high concentrations of native CCK. The second messenger pathways that mediate these responses have not been fully defined. Although most efforts have focused on the role of the Ca2+ signalling pathway in the mediation of zymogen secretion, the role of the cAMP pathway in mediating some of the effects of CCK has not been well studied.
The present study describes a sensitive assay for cAMP-dependent protein kinase activity in pancreatic acini. This method has been used to directly examine the effect of CCK and OPE on the activity of cAMP-dependent protein kinase in rat pancreatic acini. In this system, we find that CCK activates cAMP-dependent protein kinase while OPE does not, apparently dissociating this activation from the secretory cascade. In addition, we find that the measurement of cAMP levels did not always correlate with total cAMP-dependent protein kinase activity in these cells.
2. Materials and Methods
Isolated pancreatic acini were prepared from fasted male Sprague–Dawley rats by collagenase digestion and mechanical disruption as described [2]. Crude type 3 collagenase (Worthington Biochemical Co., Freehold, NJ) was purified by gel filtration chromatography prior to use [3].
Acini (90 μl), suspended in oxygenated medium A (98 mM NaCl, 4.8 mM KCl, 2.5 mM KH2PO4, 2 mM CaCl2, 1.2 mM MgCl2, 0.2% BSA, 0.05% DTT, and 25 mM HEPES, pH 7.4), were equilibrated at 37°C prior to stimulation. Acinar suspensions used throughout this study contained ∼3 mg protein/ml.
Hormonal stimulation of acini utilized cholecystokinin octapeptide (CCK) from Squibb Diagnostics, New Brunswick, NJ and d-Tyr-Gly-[(Nle28,31)CCK-26-32]-phenethyl ester (OPE) [1]. Unstimulated (US) samples received an equivalent volume of medium alone. All secretagogue treatments were performed at 37°C under 100% oxygen in 100 μl final volumes.
Acinar homogenates were prepared, following secretagogue treatment, by adding 100 μl of ice cold medium B (100 mM NaCl, 20 mM Na pyrophosphate, 2 mM EDTA, 3 mM EGTA, and 25 mM HEPES, pH 7.4 containing 2.5 mg/ml of STI, 0.1 mM PMSF, and 1 mg/ml each of leupeptin, chymostatin, pepstatin, and aprotinin) to the acinar suspensions. The cells were then disrupted by 8 passes through a 30-gauge needle. Samples were centrifuged at 4°C at 1800 × g for 2 min and the supernatants, comprised almost exclusively of small membrane vesicles suspended in cytosol, were assayed for A-kinase activity.
cAMP-dependent protein kinase activity was assayed in vitro by measuring the cAMP-dependent protein kinase site-specific phosphorylation of an exogenous substrate, synapsin 1 [4,5], provided by A. Czernik, Rockefeller University, New York. In vitro phosphorylation of the synapsin 1 by endogenous acinar cell cAMP-dependent protein kinase was performed under calcium-free conditions with 50 mM HEPES, pH 7.4, 10 mM MgCl2, 1 mM EGTA, 20 mM ATP containing 0.1 mCi/ml [γ-32P]ATP (DuPont/NEN, Boston, MA), 0.01 mg/ml synapsin 1, and ∼0.3 mg/ml of acinar supernatant protein [6]. Phosphorylation reactions, performed for 2 min at 30°C, were terminated by adding 50 μl of boiling 3× Laemmli sample buffer.
The in vitro phosphorylated synapsin 1 was separated from other acinar proteins by SDS-PAGE [7] and identified by autoradiography. Total synapsin counts per minute (cpm) of 32P incorporated into the synapsin 1 bands were determined by Chrenkov counting. To quantitate the cAMP-dependent protein kinase component of total synapsin phosphorylation, the synapsin 1 band was excised and subjected to one-dimensional peptide mapping using Staphylococcus aureus V-8 protease (Boehringer Mannheim, Indianapolis, IN) as described [8]. Fig. 1a shows the V-8 phosphopeptide maps of synapsin 1 phosphorylation by homogenates from unstimulated acini. As reported [4], V-8 protease cleaves phosphorylated synapsin 1 into two phosphopeptides (32 kDa and 10 kDa). Phosphorylation of the 32 kDa peptide, which contains the synapsin 1 phosphorylation site for Ca2+/calmodulin-dependent protein kinase II, is enhanced by the addition of Ca2+ and calmodulin (lane 2). The addition of cAMP (lane 3) markedly enhances the phosphorylation of the 10 kDa peptide, which contains the synapsin 1 phosphorylation site for cAMP-dependent protein kinase. Although a small amount of Ca2+-dependent phosphorylation of the 10 kDa peptide, presumably by Ca2+/calmodulin-dependent protein kinase I, can be appreciated in lane 2, the assays performed in lane 3 and throughout the remainder of this study were performed under Ca2+-free conditions. Addition of the selective cAMP-dependent protein kinase inhibitor H-89 [9], supplied by H. Hidaka, Nagoya University, Nagoya, Japan, blocked the Ca2+-independent phosphorylation of the 10 kDa peptide (Fig. 1b). This finding supports the conclusion that phosphorylation of this lower MW peptide under Ca2+-free conditions represent the activity of cAMP-dependent protein kinase. In the current study, relative activity of the kinase in stimulated acini was normalized to that present in unstimulated controls.
Fig. 1.
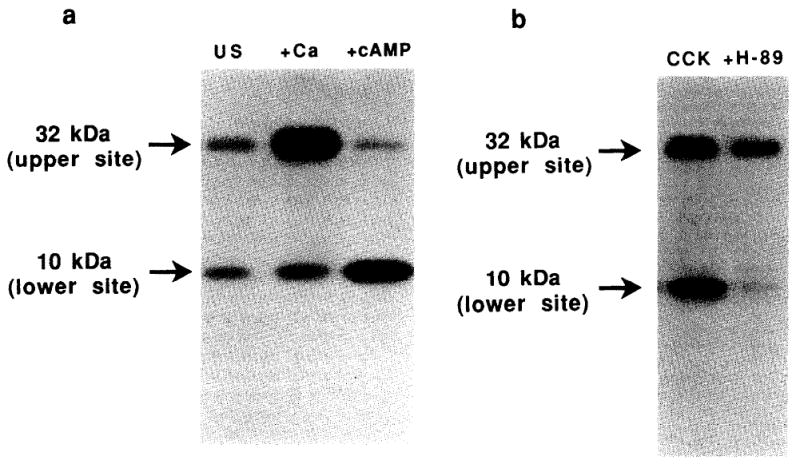
Autoradiograph of in vitro phosphorylated synapsin 1 following digestion with Staphylococcus aureus V-8 protease. Rat pancreatic acini were treated with 10−7 M CCK (CCK) or medium alone (US) for 10 min. Synapsin 1 phosphorylation and V-8 protease peptide mapping was then performed as described in section 2. (a) Effect of kinase cofactor additions on the site-specific phosphorylation of synapsin 1 in vitro. Basal kinase activity in the post-nuclear supernatant of unstimulated acinar cells is shown in the left panel (US). Effect of adding 1.5 mM CaCl2 (Ca) or 10 mM cAMP (cAMP) on the in vitro phosphorylation of synapsin 1 is shown in the subsequent panels. (b) Effect of 10 mM H-89 on the site-specific phosphorylation of synapsin 1 by acini treated with 10−7 M CCK.
Measurement of cAMP levels from rat pancreatic acini were determined using a competitive radio-binding assay (Diagnostic Products Corporation, Torrence, CA). In these studies, ∼400 mg of acinar cell protein was used for each determination. Some acini were pre-incubated for 15 min with 1 mM 3-isobutyl-1-methylxanthene (IBMX), from Aldrich Chemical Company, Milwaukee, WI, before treatment with secretagogue or medium.
Statistical analysis was applied to each experimental condition which was run in triplicate and expressed as the mean ± one S.E. of the triplicates. In some determinations (Figs. 4 and 5), triplicates from replicate experiments were combined for data analysis. Statistical probability was determined by the unpaired t-test.
Fig. 4.
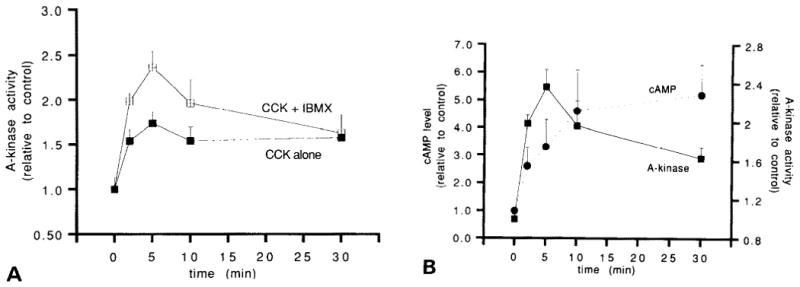
Time course of activation of the cAMP pathway by 10−7 M CCK. (A) Effect of IBMX on A-kinase activation. Closed squares, no IBMX pretreatment. Crossed squares, acini pretreated for 15 min with 1 mM IBMX. Analysis of 8 separate experiments demonstrates a significant increase in A-kinase activity over the unstimulated control at the T = 5 min time point (P < 0.001). (B) Relationship between cAMP levels and A-kinase activation in IBMX pretreated acini. Results are expressed as the mean ± one S.E. of each condition run in triplicate from a representative experiment.
Fig. 5.
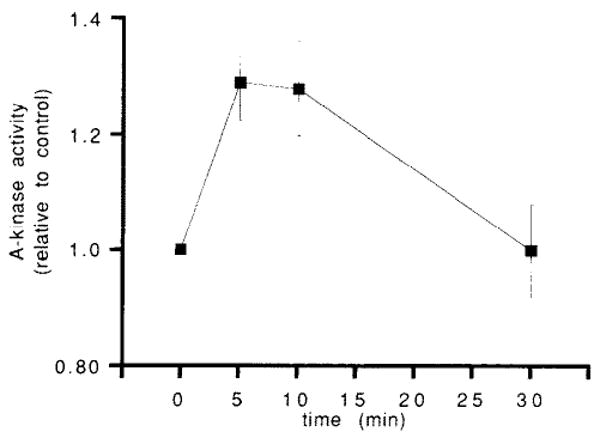
Time-course of A-kinase activation by 5 × 10−10 M CCK in acini not pretreated with IBMX. The data points represent the means ± one S.E. from 6 separate experiments, within which each time point was run in triplicate. Relative to the T = 0 min and T = 30 min time points, a significant increase in A-kinase activation is noted at 5 min (P < 0.001 for each comparison) and 10 min (P < 0.01 for each comparison).
3. Results
3.1. cAMP production
It has been well established in the literature that CCK does not produce a detectable increase in cellular cAMP levels in isolated pancreatic acini unless the acini are pretreated with a phosphodiesterase inhibitor [10–14]. Furthermore, in the presence of phosphodiesterase inhibition, a rise in cAMP levels can only be detected following stimulation by high (nM) concentrations of CCK. Studies in our laboratory confirm those findings (Fig. 2), demonstrating that rises in cAMP are detected only in IBMX pre-treated acini stimulated with high (greater than 10−9 M) concentrations of CCK.
Fig. 2.
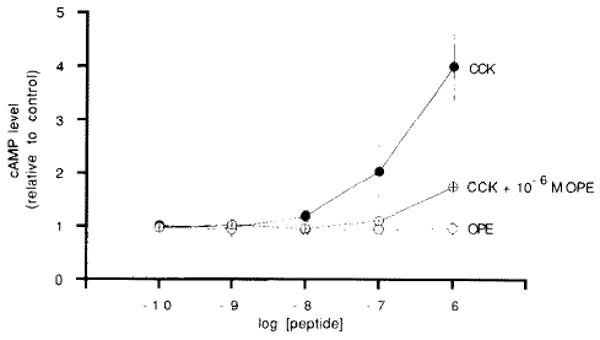
Dose effect of CCK and OPE on cAMP levels in acini pre-treated with 1 mM IBMX. Closed circles, CCK alone. Open circles, OPE alone. Crossed circles, CCK dose-response in the presence of 10−6 M OPE. Acini were treated with secretagogues for 10 min. Results are expressed as the mean ± one S.E. of each condition run in triplicate from a representative experiment.
To establish that this high-dose CCK effect is receptor mediated, the effect of the OPE analogue of CCK on cAMP production was examined. OPE competitively binds to the CCK receptor, stimulates enzyme secretion, and partially blocks the inhibition of enzyme secretion caused by high-dose CCK [1]. As demonstrated in Fig. 2, OPE is unable to raise cellular cAMP levels in IBMX pre-treated acini. Furthermore, 1 mM OPE partially blocks the accumulation of cAMP induced by high (nM) concentrations of CCK. These findings demonstrate that cAMP production by CCK results from a CCK receptor-mediated pathway that is not activated by OPE.
3.2. cAMP dependent protein kinase activity
Using the synapsin 1 phosphorylation assay system, the effect of CCK on the activation of cAMP-dependent protein kinase in rat pancreatic acini was examined. Initial studies were performed in acini that had not been pre-treated with IBMX (i.e. under conditions which do not increase cellular cAMP to measurable levels). Fig. 3 shows a representative concentration-dependence curve for CCK and cAMP-dependent protein kinase activity. Compared to unstimulated controls, a relative increase in cAMP-dependent protein kinase activity is most readily apparent following treatment with high (nM) concentrations of CCK. The OPE analogue of CCK, whether used in low or high concentrations, does not increase kinase activity.
Fig. 3.
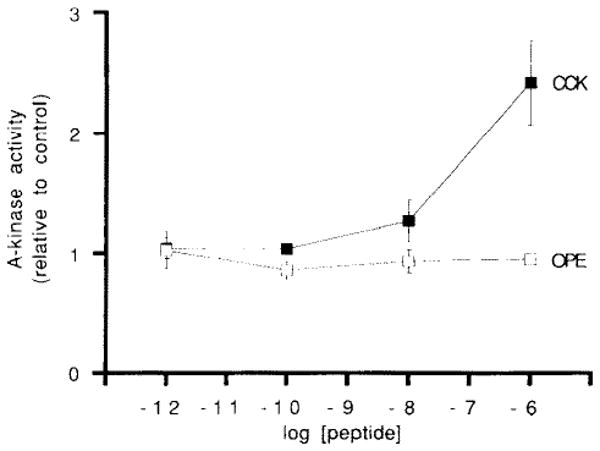
Dose effect of CCK-8 and OPE on A-kinase activity in acini not pretreated with IBMX. Acini were treated with secretagogues for 10 min. Results are expressed as the mean ± one S.E. of each condition run in triplicate from a representative experiment.
The temporal relationship between high-dose CCK, cAMP, and cAMP-dependent protein kinase activity was next examined (Fig. 4). Based on the concentration dependence curve in Fig. 3, 10−7 M CCK was chosen as the high concentration of CCK to be studied in greater detail. In acini not pre-treated with IBMX, cAMP-dependent protein kinase activity rose rapidly following exposure to 10−7 M CCK, peaked within 10 min, and remained above background levels for exposures as long as 30 min (closed squares, Fig. 4a). This figure is representative of the pattern of cAMP-dependent protein kinase activation by high-dose CCK. Replicate determinations at the 5-min time point (8 separate experiments, each run in triplicate) have demonstrated that 10−7 M CCK increases cAM-dependent protein kinase activity to 1.58 times that of unstimulated controls (S.E. = 0.04, P < 0.001 compared to controls). Pre-treatment of acini with IBMX, which raises total cellular cAMP to detectable levels, increased the magnitude of kinase activation by high-dose CCK without appreciably altering the time course of activation (open squares, Fig. 4a). To further characterize the relationship between cAMP and cAMP-dependent protein kinase activity, the temporal relationship between cAMP and its effector kinase was examined in IBMX-pretreated acini (Fig. 4b). At early time points, kinase activity closely followed the rise in cellular cAMP. At later time points, kinase activity closely followed the rise in cellular cAMP. At later time points, however, kinase activity decreased while cAMP levels continued to rise.
Finally, the effect of a low dose of CCK on cAMP-dependent protein kinase activity was examined. In our laboratory, 5 × 10−10 M CCK maximally stimulates amylase release in rat pancreatic acini. This concentration of CCK, however, does not increase cAMP to measurable levels, even in the presence of phosphodiesterase inhibition. Fig. 5 shows the time course of cAMP-dependent protein kinase activation by 5 × 10−10 M CCK in acini not exposed to IBMX. A small but statistically significant 24% increase in kinase activity is detected 5 min after treatment with this low concentration of CCK (for 7 separate experiments, each time point run in triplicate, the relative activity = 1.24 ± 0.045 S.E., P < 0.01). This increase in kinase activity was of lesser magnitude and of shorter duration than that seen following high-dose CCK treatment. Furthermore, this low-dose response occurred under conditions which do not ellicit a measurable increase in cellular cAMP levels [10–12], even in acini pre-treated with IBMX (data not shown).
4. Discussion
In this study, we have described a sensitive method for assaying cAMP-dependent protein kinase activity in rat pancreatic acini. There are two reasons for developing this assay. First, physiologically significant changes in cAMP may be below the level of detection by currently available cAMP assay systems [12,15–17]. Second, the mechanism for the activation of some protein kinases has the potential to generate an enzymatic activity that persists even after second messenger levels decrease [6]. The assay used in this study takes advantage of the fact that the activation of kinase by cAMP results in the dissociation of the regulatory subunits from the holoenzyme, resulting in the liberation of cofactor-independent catalytic subunits. Thus, the activation of cAMP-dependent protein kinase following neurohumoral stimulation of intact cells can be subsequently determined by measuring in vitro the activity of its cofactor-independent catalytic subunit. Under the calcium-free conditions employed in this study, the in vitro phosphorylation on a specific region of synapsin 1 served as a specific assay for determining relative cAMP-dependent protein kinase activity. Although synapsin 1 was used as a substrate, it is likely that other cAMP-dependent protein kinase substrates may be utilized in similar assay systems. Furthermore, the methodology described in this study may be applicable to other dispersed cell systems in which the direct measurement of neurohumorally-stimulated cAMP-dependent protein kinase activity is desired.
Although it has been known that high concentrations of CCK can increase cellular cAMP levels in acini if they are pre-treated with IBMX [10–12], the effect of CCK treatment on cAMP-dependent protein kinase activity has not been previously examined. By measuring kinase activity directly, the current study was able to establish that cAMP-dependent protein kinase is activated by CCK in rat pancreatic acini over a broad range of concentrations and that this activation occurs in the absence of IBMB pre-treatment or a detectable increase in cellular cAMP. There are several potential explanations for this dissociation between second messenger levels and kinase activity. First, it is possible that the current methods for measuring cAMP are not sensitive enough to detect physiologically significant elevations in this intracellular signal. This possibility is supported by the data in Fig. 4a, which correlates the magnitude of early kinase activation with the amplification of a cAMP signal by IBMX. Thus, the activation of a small, yet functionally significant, pool of cAMP within the cell may go undetected when measuring total cellular cAMP levels. Secondly, it is possible that an increase in the metabolic cycling of cAMP (which is revealed under conditions of phosphodiesterase inhibition) is capable of activating this enzyme in the absence of a measurable increase in the total cellular level of cAMP. Such a metabolic cycling of cAMP has been reported to mediate early amylase secretion in the rat parotid gland [18]. Thirdly, since this assay measures net substrate phosphorylation, it is possible that mechanisms independent of cAMP-dependent protein kinase activity may influence total substrate phosphorylation. For example, CCK stimulation may indirectly increase net kinase activity by directly inhibiting an opposing phosphatase activity. Irrespective of the mechanism, the data presented in this study demonstrates that the absence of a measurable increase in cellular cAMP in response to secretagogues cannot be used to exclude a role for cAMP-dependent protein kinase in the effect of that secretagogue.
Another conclusion to be drawn from this study is that a dissociation can be made between CCK-induced enzyme secretion and the activation of cAMP-dependent protein kinase. This is illustrated by the OPE data, which demonstrates that this fully efficacious secretagogue, acting through occupation of the CCK receptor, does not activate cAMP-dependent protein kinase. Thus, enzyme secretion by CCK appears to be independent of the hormone's effect on cAMP-dependent protein kinase activity. Based on CCK dose-response data from this and other studies [12,19], it is tempting to speculate that cAMP-dependent protein kinase activation by CCK may be involved in the inhibition of secretion and/or the intracellular activation of zymogens.
Acknowledgments
The authors would like to thank Dr. Howard Rasmussen for his support and suggestions and Mr. Emanuel Lerner for assistance with statistical analysis. This research was supported by NIH grants DK01910 (C.R.M.), DK08216 (S.D.L.) and DK32878 (L.J.M.) as well as a Veterans Administration Merit Review Award (F.S.G.) and a Cystic Fibrosis Foundation New Investigator Award (C.R.M.).
Abbreviations
- CCK
cholecystokinin octapeptide
- OPE
d-Tyr-Gly-[(Nle28,31)CCK-26-32]-phenethyl ester
- cAMP
adenosine 3′,5′ cyclic monophosphate
- IBMX
3-isobutyl-l-methylxanthine
References
- 1.Gaisano HY, Kleuppelberg UG, Pinon DI, Pfenning MA, Powers SP, Miller LJ. J Clin Invest. 1989;83:321–325. doi: 10.1172/JCI113877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruzzone R, Halban PA, Gjinovci A, Trimble ER. Biochem J. 1985;226:621–624. doi: 10.1042/bj2260621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulz GS, Sarras MP, Gunther GR, Hull BE, Alicea HA, Gorelick FS, Jamieson JD. Exp Cell Res. 1980;130:49–62. doi: 10.1016/0014-4827(80)90041-5. [DOI] [PubMed] [Google Scholar]
- 4.Huttner WB, DeGennaro LJ, Greengard P. J Biol Chem. 1981;256:1482–1488. [PubMed] [Google Scholar]
- 5.Czernik AJ, Pang DT, Greengard P. Proc Natl Acad Sci USA. 1987;84:7518–7522. doi: 10.1073/pnas.84.21.7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorelick FS, Wang JKT, Lai Y, Nairn AC, Greengard P. J Biol Chem. 1988;263:17209–17212. [PubMed] [Google Scholar]
- 7.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 8.Cleveland DW, Fisher SG, Kirscher MW, Laemmli UK. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 9.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 10.Deschodt-Lanckman M, Robberecht P, DeNeef P, Labrie F, Christophe J. Gastroenterol. 1975;68:318–325. [PubMed] [Google Scholar]
- 11.Renckens BAM, Van Emst-DeVries SE, DePont JJHHM, Bonting SL. Biochim Biophys Acta. 1980;630:511–518. doi: 10.1016/0304-4165(80)90005-7. [DOI] [PubMed] [Google Scholar]
- 12.Gardner JD, Sutliff VE, Walker MD, Jensen RT. Am J Physiol. 1983;245:G676–G680. doi: 10.1152/ajpgi.1983.245.5.G676. [DOI] [PubMed] [Google Scholar]
- 13.Von Schrenck T, Jensen RT, Gardner JD. In: Gastrin and Cholecystokinin Chemistry, Physiology, and Pharmacology. Bali JP, Martinez J, editors. Elsevier; New York: 1987. pp. 49–56. [Google Scholar]
- 14.Willems PHGM, Fleuren-Jakobs AMM, De Pont JJHHM, Bonting SL. Biochim Biophys Acta. 1984;802:209–214. doi: 10.1016/0304-4165(84)90163-6. [DOI] [PubMed] [Google Scholar]
- 15.Corbin JD. Methods Enzymol. 1983;99:227–232. doi: 10.1016/0076-6879(83)99057-2. [DOI] [PubMed] [Google Scholar]
- 16.Lam SCT, Guccione MA, Packham MA, Mustard JF. Thromb Haemostasis. 1982;47:90–95. [PubMed] [Google Scholar]
- 17.Seiler S, Arnold AJ, Grove CA, Fifer CA, Keely SL, Stanton HC. J Pharmacol Exp Ther. 1987;243:767–774. [PubMed] [Google Scholar]
- 18.Deeg MA, Graeff RM, Walseth TF, Goldberg ND. Proc Natl Acad Sci USA. 1988;85:7867–7871. doi: 10.1073/pnas.85.21.7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leach SD, Modlin IM, Scheele GA, Gorelick FS. J Clin Invest. 1991;87:362–366. doi: 10.1172/JCI114995. [DOI] [PMC free article] [PubMed] [Google Scholar]