

Abstract
AIMS
To investigate the influence of genetic polymorphisms in the CYP3A5, CYP2C19 and ABCB1 genes on the population pharmacokinetics of cilostazol in healthy subjects.
METHODS
Subjects who participated in four separate cilostazol bioequivalence studies with the same protocols were included in this retrospective analysis. One hundred and four healthy Korean volunteers were orally administered a single 50- or 100-mg dose of cilostazol. We estimated the population pharmacokinetics of cilostazol using a nonlinear mixed effects modelling (nonmem) method and explored the possible influence of genetic polymorphisms in CYP3A (CYP3A5*3), CYP2C19 (CYP2C19*2 and CYP2C19*3) and ABCB1 (C1236T, G2677T/A and C3435T) on the population pharmacokinetics of cilostazol.
RESULTS
A two-compartment model with a first-order absorption and lag time described the cilostazol serum concentrations well. The apparent oral clearance (CL/F) was estimated to be 12.8 l h−1. The volumes of the central and the peripheral compartment were characterized as 20.5 l and 73.1 l, respectively. Intercompartmental clearance was estimated at 5.6 l h−1. Absorption rate constant was estimated at 0.24 h−1 and lag time was predicted at 0.57 h. The genetic polymorphisms of CYP3A5 had a significant (P < 0.001) influence on the CL/F of cilostazol. When CYP2C19 was evaluated, a significant difference (P < 0.01) was observed among the three genotypes (extensive metabolizers, intermediate metabolizers and poor metabolizers) for the CL/F. In addition, a combination of CYP3A5 and CYP2C19 genotypes was found to be associated with a significant difference (P < 0.005) in the CL/F. When including these genotypes, the interindividual variability of the CL/F was reduced from 34.1% in the base model to 27.3% in the final model. However, no significant differences between the ABCB1 genotypes and cilostazol pharmacokinetic parameters were observed.
CONCLUSIONS
The results of the present study indicate that CYP3A5 and CYP2C19 polymorphisms explain the substantial interindividual variability that occurs in the metabolism of cilostazol.
Keywords: cilostazol, NONMEM, pharmacogenetics, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The interindividual variability of the pharmacokinetic parameters of cilostazol is relatively large.
Cilostazol undergoes extensive hepatic metabolism via the P450 enzymes, primarily CYP3A and, to a lesser extent, CYP2C19.
Indeed, <1% of the administered dose of cilostazol is excreted unchanged in the urine.
WHAT THIS STUDY ADDS
A population pharmacokinetic analysis of cilostazol was conducted to evaluate the impact of CYP3A, CYP2C19 and ABCB1 polymorphisms on cilostazol disposition in vivo.
Genetic polymorphisms of CYP3A5 and CYP2C19 explain the substantial interindividual variability in the pharmacokinetics of cilostazol.
ABCB1 genotypes do not to appear to be associated with the disposition of cilostazol.
Introduction
Cilostazol (6-[4-(1-cyclohexyl-1H-tetrazol-5-yl)butoxyl]-3,4-dihydro-2(1H)-quinolinone) inhibits phosphodiesterase III (PDE III) and adenosine uptake, which results in an increase in intracellular 3′-5′ cyclic adenosine monophosphate (cAMP) and a reduction in calcium levels [1, 2]. PDE III inhibitors are known to inhibit platelet aggregation and stimulate vasodilation through the accumulation of cAMP. The primary pharmacological effects of cilostazol are antiplatelet aggregation and vasodilation for the treatment of intermittent claudication caused by peripheral arterial disease [1]. Cilostazol also effectively prevents cerebral infarction [2] as well as apoptotic and oxidative cell death [3]. Cilostazol is rapidly absorbed and reaches peak plasma concentrations about 2.4 h after oral administration. Most administered cilostazol binds with proteins (95–98%), primarily albumin [4, 5].
In a previous study, the interindividual variability in the pharmacokinetic (PK) parameters of cilostazol was found to be relatively large, with a coefficient variation of about 40–60% [4]. This variability can be explained by interindividual differences in metabolism. In vitro studies of human cytochrome P450 (CYP) preparations and clinical studies showed that cilostazol undergoes extensive hepatic metabolism via P450 enzymes. Specifically, metabolism of cilostazol primarily occurs via CYP3A and, to a lesser extent, CYP2C19, while <1% of the administered dose is excreted unchanged in the urine [6, 7]. Furthermore, we have reported that CYP2C19 polymorphisms play an important role in the metabolism of cilostazol for the CYP3A5*3/*3 group, which has low metabolic activity [8].
The CYP3A subfamily, which includes CYP3A4, CYP3A5, CYP3A7 and CYP3A43, is one of the most versatile enzymes of the biotransformation systems involved in the elimination of drugs [9, 10]. CYP3A (especially CYP3A4 and CYP3A5) accounts for approximately 30% of the total hepatic P450 activity [11]. The interindividual variation in the hepatic expression and activity of CYP3A isozymes is primarily due to PK variability in drugs that are CYP3A substrates [11]. It has also been reported that genetic polymorphisms in CYP3A4 are rare in Asians [12]. Conversely, the CYP3A5 polymorphism is more prevalent and shows marked differences in protein expression and catalytic activity between ethnic groups [13]. Among the CYP3A5 variant alleles (CYP3A5*3, *6 and *7) that are associated with CYP3A5 expression, CYP3A5*6 and *7 have low allelic frequencies in Asians [14, 15]. Recently, single nucleotide polymorphisms (SNPs) that cause alternative splicing and protein truncation were found in intron 3 (A to G; CYP3A5*3) and exon 7 (G to A; CYP3A5*6) of the CYP3A5 gene [13].
CYP2C19 is a highly polymorphic liver enzyme that belongs to the P450 superfamily. Two allelic variants of CYP2C19 have been observed as major mutations that contribute to poor metabolizers (PMs). The most common defect, CYP2C19*2, is a splice mutation in exon 5 that accounts for 75–83% of the defective alleles in PMs [16]. A second defect, CYP2C19*3, consists of a premature stop codon in exon 4. CYP2C19*3 is rarely found in Caucasians, but accounts for nearly all remaining mutant alleles in Asians [17, 18].
In addition to the CYP3A enzymes, P-glycoprotein (P-gp) could play an important role in the obscured metabolic profile of some CYP3A substrates [19]. Furthermore, it was previously reported that the ABCB1 polymorphism (C3435T) affects the intestinal expression of CYP3A4.
In the present study, we estimated the population PK parameters of cilostazol using a nonlinear mixed effects modelling (nonmem) method. We also evaluated the possible influence of genetic polymorphisms in CYP3A (CYP3A5*3), CYP2C19 (CYP2C19*2 and CYP2C19*3) and ABCB1 (C1236T, G2677T/A and C3435T) as covariates on the population PK of cilostazol in healthy Korean subjects to elucidate the interindividual variability in the PK of cilostazol.
Methods
Subjects
A total of 104 healthy Korean male volunteers participated in this study. The age of the subjects ranged from 19 to 28 years (mean ± SD, 23.7 ± 1.6 years), while their weight ranged from 44 to 83.5 kg (mean ± SD, 65.9 ± 7.7 kg), and their body surface area (BSA) ranged from 1.420 to 2.017 m2 (mean ± SD, 1.783 ± 0.124 m2). All subjects gave informed written consent prior to undergoing genotyping and PK evaluation. The Institutional Review Board of the Institute of Bioequivalence and Bridging Study at Chonnam National University (Gwangju, Korea) approved the study protocol. In addition, this study was conducted according to the revised Declaration of Helsinki for biomedical research involving human subjects and the rules of Good Clinical Practice. Each subject was physically normal and had no previous history of illness or hypersensitivity to any drugs. Furthermore, the health status of subjects was determined to be normal on the basis of a physical examination that included the screening of blood chemistry, a complete blood count and urinalysis prior to admission to the study. Finally, subjects were asked to refrain from taking any medications, including alcohol and other drugs, for at least 1 week prior to and throughout the study period.
Study design
Subjects who participated in four separate cilostazol bioequivalence (BE) studies with the same protocol design were included in this retrospective analysis. In addition, only data from the reference formulation were used for the current analysis. The BE studies were conducted as single-dose, randomized, two-way, open-label and crossover studies at the Institute of Bioequivalence and Bridging Study, College of Pharmacy, Chonnam National University. A total of 104 subjects were assigned to four groups (n= 26) that were subjected to four separate single-dose BE studies in which 50 or 100 mg of cilostazol was administered (n= 52, respectively). After an overnight fast, subjects in each study received a single oral dose of 50 or 100 mg cilostazol with 240 ml of water (Pletaal tablet in each study, 50 mg, lot no. PM404009; and 100 mg, lot no. PT110005P; Otsuka Pharmaceuticals Co., Ltd, Tokyo, Japan). Blood samples were collected in Vacutainer® tubes prior to dosing and at 1, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 24 and 48 h after administration of the drug. Following centrifugation (3000 g, 20 min, 4 °C), serum samples were transferred to polyethylene tubes and immediately stored at −70 °C until analysis. Concentrations of cilostazol in serum were analysed using a validated high-performance liquid chromatography method based on a previous report, with minor modification [6].
Genotype analysis
Identification of CYP3A4*1B (−392A→G) and CYP3A5*3 (6986A→G)
The genotypes of each individual at the CYP3A4*1B and CYP3A5*3 alleles were determined using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) methods as previously described [20, 21]. CYP3A4*1B was amplified using the forward primer 5′-GGAATGAGGACAGCCATAGAGACAACTGCA-3′ and the reverse primer 5′-CCTTTCAGCTCTGTGTTGCTCTTTGCTG-3′. The sense primer was designed to generate a Pst I endonuclease restriction site where the −392G allele was incorporated. For CYP3A5*3, the forward primer was 5′-CATCAGTTAGTAGACAGATGA-3′ and the reverse primer was 5′-GGTCCAAACAGGGAAGAAATA-3′. The antisense primer was designed to generate a Ssp I endonuclease restriction site where the 6789A allele is incorporated. The PCR conditions consisted of denaturation at 94 °C for 1 min, annealing at 60 °C for CYP3A4*1B or 55 °C for CYP3A5*3 for 1 min and extension at 72 °C for 1 min. The PCR products (7.5 µl) were then digested with Pst I (CYP3A4*1B) and Ssp I (CYP3A5*3) in reaction mixtures with a total volume of 20 µl for at least 2 h at 37 °C, after which they were separated by electrophoresis on 3.5% agarose gel and visualized by ethidium bromide staining.
Identification of CYP2C19*2 (681 G→A) and CYP2C19*3 (636 G→A)
Two variant alleles for CYP2C19, CYP2C19*2 and CYP2C19*3, were identified using the PCR-RFLP methods described by de Morais et al., with minor modifications [16, 17]. Briefly, the following primers were created based on previously published sequences: 5′-AATTACAACCAGAGCTTGGC-3′ and 5′-TATCACTTTCCATAAAAGCAAG-3′ for CYP2C19*2 and 5′-AAATTGTTTCCAATCATTTAGCT-3′ and 5′-ACTTCAGGGCTTGGTCAATA-3′ for CYP2C19*3. The PCR conditions for CYP2C19*2 and CYP2C19*3 consisted of denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 1 min, 57 °C (CYP2C19*2) or 52 °C (CYP2C19*3) for 1 min, and 72 °C for 1 min, and 5 min of final extension at 72 °C. The PCR product (7.5 µl) was digested with Sma I (CYP2C19*2) or Bam HI (CYP2C19*3) in a total volume of 20 µl for at least 10 h at 25 °C (Sma I) or 37 °C (Bam HI) and subsequently separated on a 2.5% agarose gel and then visualized by ethidium bromide staining.
Identification of variants in ABCB1
Candidate SNPs (C1236T, G2677T/A and C3435T) in the ABCB1 gene were genotyped using PCR-RFLP, with minor modifications [12, 22]. The following primers were used to incorporate the indicated changes: C1236T forward, 5′-TATCCTGTGTCTGTGAATTGCC-3′ and reverse, 5′-CCTGACTCACCACACCAATG-3′; G2677A/T forward, 5′-TGCAGGCTATAGGTTCCAGG-3′ and reverse, 5′-TTTAGTTTGACT CACCTTCCCG-3′; C3435T forward, 5′-TGTTTTCAGCTGCTTGATGG-3′ and reverse, 5′-AAGGCATGTATGTTGGCCTC-3′. The primer sequences for C1236T and C3435T were obtained from published sequences, while the mismatch primer sets were designed for identification of the allelic variant, G2677T/A. The PCR conditions consisted of initial denaturation for 2 min at 94 °C, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 59.8 °C for 30 s, and extension at 72 °C for 30 s. The terminal extension was performed at 72 °C for 5 min. The PCR product was digested using the following specific restriction enzymes: Hae III for C1236T, Ban I/Rsa I for G3435T/A and Sau3A I for C3435T. The digested PCR products were then analysed on a 2.5% agarose gel and subsequently visualized by ethidium bromide staining. To control for the validity of genotyping, all genotype analyses were conducted three times and the results, which were 100% concordant, were used in the actual genotype run.
Population pharmacokinetic analysis
Population PK analysis was conducted using the nonlinear mixed effects modelling program, NONMEM (version VI, level 1.1; GloboMax, Hanover, MD, USA), to estimate the population mean parameters, interindividual (η), and residual (ε) random effects [23]. The intersubject variability of each of the structural parameters of the basic model was modelled using the following exponential error model:
where Pi is the value of the parameter in an ith individual, ηi is a random variable and the difference between Pi and PTV, which is the value of the parameter in a typical individual. It is assumed that the values of ηi are normally distributed with a mean of zero and a variance of ω2. The residual variability was evaluated (i.e. additive error, proportional error, combined additive and proportional error and log normal model) to describe the intrasubject variability. The combined additive and proportional error model based on the following equation best described the residual variability:
where Cij is the jth observed value in the ith subject, Cpred,ij is the jth predicted value in the ith subject, and εpro,ij and εadd,ij are the residual intrasubject variability with means of zero and variances of σpro2 and σadd2, respectively. The correlation between the random variables in the model was explored using the OMEGA BLOCK option. The PK model was estimated using the first-order conditional estimation (FOCE) method with η–ε interaction.
The base model, which includes key parameters and does not incorporate covariates, was selected based on goodness-of-fit plots, precision of estimates and the likelihood ratio test within NONMEM. The results were considered statistically significant if the decreases in the objective function value (OFV) of the two nested models were 3.84 units (P < 0.05, one degree of freedom) and 5.99 units (P < 0.05, two degrees of freedom). Each parameter was sequentially tested to determine if it should remain in the base model.
The two-compartment model with first-order absorption from the central compartment and lag time had a better fit than the one-compartment model. This model was implemented in the PREDPP library subroutine ADVAN4 and TRANS4 in NONMEM. Parameters from the two-compartment structural model included the apparent oral clearance and intercompartmental clearance (CL/F and Q/F), the apparent central and peripheral volume of distribution (Vc/F and Vp/F), where F is the bioavailability, the absorption rate constant (Ka), and the absorption lag time (Tlag).
Covariates analysis
The influence of each subject covariate on the PK was analysed. The covariates included in this analysis were limited to age, weight, BSA and genetic polymorphisms [CYP3A (CYP3A5*3), CYP2C19 (CYP2C19*2 and CYP2C19*3) and ABCB1 (exons 12, 21 and 26)], since all subjects were healthy volunteers. Generalized additive modelling (GAM) [24] implemented in Xpose version 4.0.4 [25] was used to identify important covariates. In addition, each covariate search was conducted using the automated procedure and the stepwise covariate method as implemented in Pearl-speaks-nonmem (PsN) Version 2.3.1 [26]. Continuous covariates were incorporated into the model using a linear function after centring on the median. For example, the continuous covariates relationship between the typical value of the oral clearance (CL/FTV) and BSA was modelled according to the following equation:
where θ1 is the estimate of the oral clearance in a typical individual with a median BSA and θ2 is the change in the oral clearance for each unit change in BSA.
For categorical covariates, we explored the possible influence of genetic polymorphisms in CYP3A (CYP3A5*3), CYP2C19 (CYP2C19*2 and CYP2C19*3) and ABCB1 (exons 12, 21 and 26) on cilostazol PK. Categorical covariates were included in the model as index variables. For example, the association between the typical value of the oral clearance (CL/FTV) and genetic polymorphisms in CYP3A (CYP3A5*3) was modelled according to the following equation:
where θ3 is the estimate of the oral clearance for the wild-type group, and θ4 or θ5 is the fractional change in the oral clearance for the hetero or mutant-type group. GHET was fixed to 1 for the hetero type and to 0 for the wild and mutant type. GMUT was fixed to 1 for the mutant type and to 0 for the wild and hetero type.
Model building was conducted using a stepwise approach, adding an additional covariate at each step. The effect of each covariate was evaluated based on changes in the OFV between the base model and the model including the covariate. The significance level was P < 0.05 during forward inclusion and P < 0.01 during backward deletion. Graphical diagnostics such as goodness of fit were obtained using Xpose [27].
Model evaluation
The accuracy and robustness of the final population model were evaluated using a simulation method and a bootstrap method. The simulation method was conducted by the visual predictive check (VPC) implemented in Xpose with the PsN. The predicted concentration–time profiles for 1000 datasets at each time point after each administration of 50 and 100 mg cilostazol were generated from the parameters and variances of the selected final model. For each dose, the estimated median and the 5th and 95th percentile of the concentrations predicted from the simulations were plotted against time with the observed cilostazol concentrations.
Nonparametric bootstrap analysis was conducted using the bootstrap option in the software package Wings for nonmem (version 614, August 2008, Auckland, New Zealand) [28]. The 1000 replicates were generated by repeated random sampling with replacement from the original dataset. The final model parameter estimates were compared with the median and 95% confidence intervals of the bootstrap replicates of the final model.
Results
Genetic analysis
For analysis of CYP3A4*1B, the 384-bp fragment amplified by PCR was digested with Pst I. Digestion of the PCR products containing −392A (CYP3A4*1) resulted in three bands (229, 122 and 33 bp), while the PCR products that contained −392G (CYP3A4*1B) yielded bands of 199, 122, 33 and 30 bp. For CYP3A5*3, digestion of the 293-bp PCR product with Ssp I yielded fragments of 168 and 125 bp, while digestion of CYP3A5*1 yielded fragments of 148, 125 and 20 bp. The CYP3A4*1B allele was not detected in any of the 104 Korean subjects. These results are consistent with those of Chowbay et al. [12]. The CYP3A5*3 allele frequency was 74.0%, while the CYP3A5*1 allele was 26.0%. Of the 104 individuals evaluated in this study, six were homozygous for the wild type (*1/*1, 5.8%), 42 were heterozygous (*1/*3, 40.4%) and 56 were the mutant type (*3/*3, 53.8%).
The two variant alleles for CYP2C19, CYP2C19*2 and CYP2C19*3, were evaluated in the 104 subjects. PCR products from the DNA of individuals with the wild-type allele were cleaved using a restriction enzyme, whereas those from homozygous individuals with the *2/*2 or *3/*3 mutations lacked the Sma I and Bam HI site, respectively. Analysis of the CYP2C19 genotypes exposed six diverse patterns. Specifically, subjects were classified as extensive metabolizers (EMs), intermediate metabolizers (IMs) or PMs based on their level of CYP2C19 activity. Of the 104 subjects included in this study, 52 (50.0%) subjects that were homozygous for the CYP2C19*1/*1 allele were defined as EMs. In addition, 38 (36.5%) subjects that were heterozygous for mutant alleles (CYP2C19*1/*2 and *1/*3) were defined as IMs, while 14 (13.5%) subjects that contained the mutant alleles (CYP2C19*2/*2, *2/*3 and *3/*3) were defined as PMs. The allelic frequencies of CYP2C19*2 and *3 were 22.1% and 9.6%, respectively.
A total of 104 subjects were evaluated for polymorphisms of the ABCB1 exon 12 C1236T, exon 21 G2677A/T (Ala893Ser/Thr) and exon 26 C3435T polymorphisms. When the ABCB1 C1236T polymorphism was evaluated, 28 subjects were found to be homozygous for the wild type (CC, 26.9%), 50 were heterozygous (CT, 48.1%) and 26 were the mutant type (TT, 25.0%). For the G2677A/T polymorphism, GG, GA, GT, AT, TT and AA genotypes were detected in 32 (30.8%), 14 (13.5%), 42 (40.4%), four (3.8%), 12 (11.5%) and 0 (0.0%) individuals, respectively. When the ABCB1 C3435T polymorphism was evaluated, CC, CT and TT genotypes were detected in 50 (48.1%), 42 (40.4%) and 12 (11.5%) individuals, respectively. For all 104 individuals analysed, the allele frequencies of ABCB11236T, 2677T, 2677A and 3435T were 49.0, 33.7, 8.7 and 31.7%, respectively. All genotype frequencies were found to be in Hardy–Weinberg equilibrium (P < 0.05). To ensure the validity of genotyping, all genotype analyses were conducted in triplicate, and the 100% concordance genotyping results were used in the actual genotype run. The genotype characteristics of the 104 subjects that participated in this PK study are summarized in Table 1.
Table 1.
Genotype and allele frequencies for the studied variant genes (n= 104)
| Genotype | No. (frequency, %) | |
|---|---|---|
| CYP3A5 | *1/*1 | 6 (5.8) |
| *1/*3 | 42 (40.4) | |
| *3/*3 | 56 (53.8) | |
| CYP2C19 | *1/*1 | 52 (50.0) |
| *1/*2 or *1/*3 | 38 (36.5) | |
| *2/*2,*3/*3 or *2/*3 | 14 (13.5) | |
| ABCB1 | ||
| Exon 12 (C1236T) | CC | 28 (26.9) |
| CT | 50 (48.1) | |
| TT | 26 (25.0) | |
| Exon 21 (G2677T/A) | GG | 32 (30.8) |
| GT, GA | 56 (53.9) | |
| TT, TA, AA | 16 (15.3) | |
| Exon 26 (C3435T) | CC | 50 (48.1) |
| CT | 42 (40.4) | |
| TT | 12 (11.5) | |
Population pharmacokinetic analysis
The serum concentration profile of the cilostazol vs. time was well described by the two-compartment model with first-order absorption from the central compartment and a lag time. This model is similar to that described in a previous study [29]. In the initial analyses, the two-compartment model decreased the OFV significantly (P < 0.001) when compared with the one- and three-compartment model. The OFV of the two-compartment model decreased significantly in response to the inclusion of a random effect on CL/F, Vc/F, Q/F (P < 0.001) and Vp/F (P < 0.05). However, the effect of including a random effect on KA and ALAG1 was not significant. Residual variability was modelled using the combined additive-proportional error. Inclusion of a lag time in the model led to a significant decrease in the OFV (ΔOFV =−215.061, P < 0.001). This model was selected as the base model.
Covariates analysis
A summary of covariate model building steps is shown in Table 2, which represents the steps that resulted in statistical significance in the OFV during the development of the pharmacostatistical model. Screening of the effects of covariates on PK parameters during the GAM analysis suggested that the inclusion of the CYP3A5 and CYP2C19 had an effect on the CL/F (Figure 1). Inclusion of the genotypes of CYP2C19 (CYP2C19*2 and *3) was found to have a significant effect (ΔOFV =−10.318, P= 0.0057, two degrees of freedom) on the CL/F. In addition, the effect of the CYP3A5 genotypes on the CL/F was significant (ΔOFV =−19.171, P < 0.001, two degrees of freedom). Furthermore, a combination of CYP2C19 and CYP3A5 genotypes was found to be associated with a statistically significant difference (P < 0.005) in the CL/F (Table 3). Other covariates including age, body weight, BSA and the ABCB1 genotype did not appear to affect any of the PK parameters. As a result, Model 4 was selected as the final population PK model:
Table 2.
Summary of the covariate model-building steps
| Model 1 | Model 2 | Model 3 | Model 4* | |
|---|---|---|---|---|
| Parameter | Base model as two-compartment with lag time | CYP2C19 as covariate for CL/F | CYP3A5 as covariate for CL/F | CYP2C19 and CYP3A5 as covariate for CL/F |
| OFV† | −4001.51 | −4011.83 | −4020.68 | −4032.29 |
| ΔOFV (P-value‡) | – | −10.32 (<0.01) | −19.17 (<0.001) | −11.61 (<0.005) |
| CL/F (l h−1) (%RSE§) | 7.74 (5.57) | 8.66 (11.2) | 11.0 (12.8) | 12.8 (13.9) |
| Vc/F (l) (%RSE) | 19.7 (9.34) | 19.5 (9.59) | 20.3 (7.98) | 20.5 (8.24) |
| Q/F (l h−1) (%RSE) | 5.74 (13.3) | 5.72 (31.1) | 5.65 (19.3) | 5.64 (13.6) |
| Vp/F (l) (%RSE) | 88.8 (28.5) | 90.9 (82.6) | 75.7 (19.4) | 73.1 (21.9) |
| Ka (h−1) (%RSE) | 0.233 (6.31) | 0.232 (5.99) | 0.242 (5.66) | 0.244 (4.80) |
| TLAG (h) (%RSE) | 0.564 (5.05) | 0.564 (5.35) | 0.565 (4.94) | 0.565 (5.12) |
| CL/F, θIMs (%RSE) | – | −0.191 (28.8) | – | −0.147 (33.7) |
| CL/F, θPMs (%RSE) | – | −0.256 (53.5) | – | −0.272 (34.9) |
| CL/F, θ*1/*3 (%RSE) | – | – | −0.183 (70.0) | −0.223 (48.0) |
| CL/F, θ*3/*3 (%RSE) | – | – | −0.384 (22.0) | −0.407 (20.2) |
| ω2CL/F | 0.116 | 0.100 | 0.0862 | 0.0748 |
| ω2Vc/F | 0.480 | 0.479 | 0.482 | 0.481 |
| ω2Q/F | 0.926 | 0.906 | 1.13 | 1.16 |
| ω2Vp/F | 0.639 | 0.640 | 0.707 | 0.688 |
| ωVp/F, CL/F | 0.102 | 0.0964 | 0.0763 | 0.0685 |
| ωQ/F, Vc/F | −0.298 | −0.291 | −0.350 | −0.355 |
| ωVp/F, Q/F | 0.234 | 0.214 | 0.445 | 0.460 |
| σpro | 0.108 | 0.107 | 0.113 | 0.114 |
| σadd (ug ml−1) | 0.0539 | 0.0540 | 0.0517 | 0.0511 |
Final model.
Objective function value.
Calculated by the log-likelihood ratio test.
Percent relative standard error (SE/estimate × 100).
Figure 1.
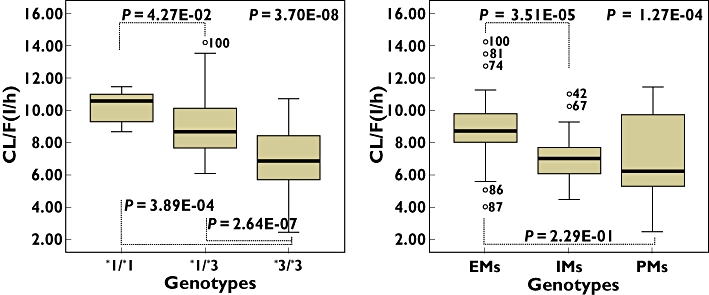
Oral clearance of cilostazol in CYP3A5 (left panel) or CYP2C19 (right panel) genotypes as determined by a Kruskal–Wallis test to compare three genotypes and by a Mann–Whitney test to compare two genotypes
Table 3.
Population pharmacokinetic parameters for cilostazol in 104 subjects and the results of bootstrap validation (final model)
| Final model | Bootstrap replicates | |||
|---|---|---|---|---|
| Parameter | Estimate | CI (95%)* | Median | CI (95%)† |
| TV (CL/F) (l h−1) | 12.8 | 8.2, 17.4 | 12.7 | 8.9, 18.5 |
| TV (Vc/F) (l) | 20.5 | 16.4, 24.6 | 20.6 | 16.6, 25.3 |
| TV (Q/F) (l h−1) | 5.64 | 3.81, 7.47 | 5.88 | 4.34, 7.85 |
| TV (Vp/F) (l) | 73.1 | 32.14, 114 | 75.3 | 42.8, 126 |
| TV (Ka) (h−1) | 0.244 | 0.215, 0.273 | 0.248 | 0.214, 0.285 |
| TV (TLag) (h) | 0.565 | 0.507, 0.623 | 0.566 | 0.480, 0.628 |
| CL/F, θIMs | −0.147 | −0.247, −0.047 | −0.143 | −0.241, −0.016 |
| CL/F, θPMs | −0.272 | −0.460, −0.084 | −0.275 | −0.552, −0.020 |
| CL/F, θ*1/*3 | −0.223 | −0.507, 0.061 | −0.203 | −0.460, −0.070 |
| CL/F, θ*3/*3 | −0.407 | −0.627, −0.187 | −0.388 | −0.592, −0.153 |
| ω2CL/F | 0.075 | 0.039, 0.111 | 0.067 | 0.018, 0.102 |
| ω2Vc/F | 0.481 | 0.327, 0.635 | 0.488 | 0.337, 0.705 |
| ω2Q/F | 1.16 | 0.49, 1.83 | 1.15 | 0.66, 1.87 |
| ω2Vp/F | 0.688 | 0.284, 1.09 | 0.738 | 0.431, 1.43 |
| ωVp/F, CL/F | 0.0685 | 0.0058, 0.131 | 0.367 | 0.019, 0.776 |
| ωQ/F, Vc/F | −0.355 | −0.528, −0.182 | −0.479 | −0.659, −0.267 |
| ωVp/F, Q/F | 0.460 | −0.089, 1.01 | 0.549 | −0.038, 0.851 |
| σ2pro | 0.0131 | 0.0036, 0.0226 | 0.0129 | 0.0040, 0.0337 |
| σ2add | 0.00261 | 0.00121, 0.00401 | 0.00254 | 0.00081, 0.00440 |
Estimate ± 1.96 × (standard error of the estimate).
The 2.5th and 97.5th percentile of the bootstrap parameter estimates. A total of 1000 replicates of bootstrap analysis. θCOVARIATE, influential factor for covariate. *1/*3, CYP3A5*1/*3; *3/*3, CYP3A5*3/*3. ω, interindividual variability; σ, residual variability; CI, confidence interval; IMs, intermediate metabolizers; PMs, poor metabolizers.
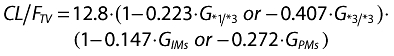 |
where 12.8 l h−1 is the estimated oral clearance for individuals with the combined CYP3A5*1/*1 and CYP2C19 EM genotypes, −0.223 or −0.407 is the fractional change in the oral clearance for the hetero or mutant-type group of CYP3A5, −0.147 or −0.272 is the fractional change for IMs or PMs of CYP2C19, and GTYPE is the index variable for genotypes.
The population parameter values and inter- and intraindividual variability estimated by the final model are shown in Table 2. The PK parameters were generally well estimated, with the relative standard error (%RSE) for estimation being around 5.12–21.89% of the estimated population parameter mean values. The %RSE of the interindividual variability of parameter estimates was low (15.62–26.89%). The values of η shrinkage were relatively small (all <0.28) in the final model. The residual error (reflecting intrasubject variability, measurement error and model misspecification) was low, being 11.4% for the proportional error and 0.0511 µg ml−1 for the additive error. The basic goodness-of-fit plots are shown in Figure 2. The diagnostic plots of the final population PK model revealed no systematic bias. There was good agreement between the individual predicted and observed values. The weighted predictions for the final population PK model were generally distributed around zero and were relatively symmetric. The population and individual post hoc predictions were distributed around the line of identity.
Figure 2.
Goodness of fit plots for the final model
Model evaluation
The predicted concentrations for 1000 simulated datasets by the final model (Model 4) for single oral doses of cilostazol 50 and 100 mg are depicted in Figure 3. In spite of a little underestimation of the observed Cmax for the 50-mg dose, the majority of the observed cilostazol plasma concentrations were within the range of the lower (5%) and upper (95%) percentiles of the simulated concentrations.
Figure 3.
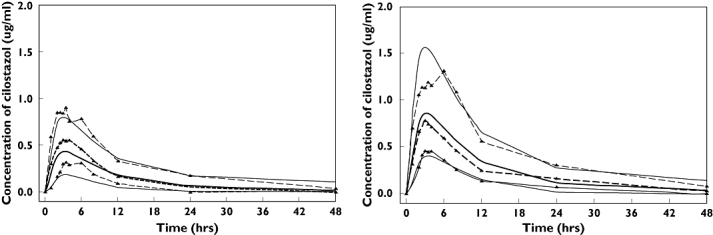
Visual predictive check of the final pharmacokinetic model between 0 and 48 h after a single oral administration of 50 (left panel) and 100 mg (right panel) cilostazol. A total of 1000 datasets were simulated using the final pharmacokinetic parameter estimates. Depicted are the observations (dashed lines with symbols) and the predictions (solid lines); median is thick lines, 90% prediction intervals are thin lines
When bootstrap analysis was conducted, the population estimates of the final model were in the 2.5–97.5 percentiles of the bootstrap replicates (Table 3). It demonstrates that the population estimates of the final model were not statistically different from the parameters estimated during the bootstrap process.
Discussion
The present study provides a population PK analysis of a single oral dose of 50 or 100 mg of cilostazol in healthy adult volunteers. The population PK of cilostazol was well fitted by a two-compartment model with a first-order absorption and lag time. The PK parameters of cilostazol showed considerable interindividual variability. One of the common causes of individual variations in drug response can be genetic polymorphisms of drug-metabolizing enzymes.
The major pharmacologically active metabolites of cilostazol are dehydrocilostazol (OPC-13015) and monohydroxycilostazol (OPC-13213). Dehydrocilostazol is primarily metabolized by CYP3A4, whereas monohydroxycilostazol is primarily metabolized by CYP2C19 and CYP3A5 [30]. Therefore, this study was conducted to evaluate the relationship between genetic polymorphisms in CYP3A4*1B, CYP3A5*3, CYP2C19*2 and *3 alleles and cilostazol PK in healthy Korean volunteers. In addition, we examined the effects of the major ABCB1 polymorphisms on cilostazol PK.
We selected and genotyped the CYP3A4*1B allele because it is known to be associated with enhanced CYP3A4 expression in vitro[31]. However, additional genotyping of 226 Korean subjects did not reveal any individuals carrying the CYP3A4*1B allele. It has been reported that genetic polymorphisms in CYP3A4 are rare in Asians [12]. Taken together, these findings indicate that the CYP3A4*1B allele is not the cause of variability in cilostazol disposition in Koreans.
The effects of CYP3A5 polymorphisms on the CL/F of cilostazol were significant, and the estimated oral clearance was higher in subjects with the CYP3A5*1/*1 allele than in those with the CYP3A5*3/*3 allele. According to the results of a previous study [13], the presence of the CYP3A5*3 allele in intron 3 results in a truncated protein with loss of CYP3A5 expression. Furthermore, that study found that individuals with the CYP3A5*1 allele had threefold higher total CYP3A protein levels than individuals with the CYP3A5*3/*3 allele. In addition, the variant alleles CYP3A5*3/*3 were related to low clearance values. According to Model 3 in Table 2, the CL/F in CYP3A5*1/*1, *1/*3 and *3/*3 was 11.0, 9.0 and 6.8 l h−1, respectively. When CYP2C19 was evaluated, statistically significant differences in the CL/F were observed among the three groups (EMs, IMs and PMs). According to Model 2 in Table 2, the CL/F in CYP2C19 EMs, IMs and PMs was 8.7, 7.0 and 6.4 l h−1, respectively.
The combination of genetic polymorphisms of CYP3A5 and CYP2C19 was also found to be associated with statistically significant differences in the CL/F. Specifically, both the CYP3A5 and CYP2C19 genotypes were found to affect the CL/F, which indicates that cilostazol is a strong substrate of both CYP3A5 and CYP2C19. After including these genotypes in the model, the interindividual variability of CL/F decreased from 34.1% in the base model to 27.3% in the final model. These findings suggest that both CYP3A5 and CYP2C19 polymorphisms may be useful clinical covariates for individualizing the dosage of cilostazol. With the exception of the CL/F (27.3%) in the final model, the interindividual variability associated with the PK parameters was generally high, especially for the intercompartmental clearance and the peripheral volume of distribution (108% for Q/F and 82.9% for V3/F); however, the %RSE of the interindividual variability of parameter estimates was low (15.62–26.89%). Goodness-of-fit plots indicated that the PK data were well fitted. In addition, the VPC and bootstrap analysis confirmed the predictive ability, model stability and precision of the parameter estimates. According to the final model's equation, the estimated CL/F of CYP2C19 EMs with CYP3A5*1/*1 is about two times larger than that of CYP2C19 PMs with CYP3A5*3/*3. CL/F is in inverse proportion to the concentration of drug. Woo et al. have reported that the antiplatelet and cardiovascular effects of cilostazol depend on the concentration of drug [29]. Therefore, the difference in CL/F of cilostazol among different genotypes could significantly influence pharmacodynamic effects such as antiplatelet aggregation. This suggests that it would be useful for the tailored medicine of cilostazol to analyse an individual's genotypes of CYP3A5 and CYP2C19. Moreover, these results mean that the changes of PK parameters due to genotype and genotype interactions could be estimated by NONMEM method.
In this study, we also investigated the association of P-gp with the cilostazol PK. Allelic variations in exons 12 (C1236T), 21 (G2677T/A) and 26 (C3435T) of the ABCB1 gene have been found to be linked and it has been reported that the C3435T mutation is associated with an altered disposition of P-gp substrates such as fexofenadin and digoxin [32, 33]. However, no significant differences were observed in the cilostazol PK parameters among the ABCB1 genotype groups in the present study. These findings are in agreement with those of previously conducted studies in which P-gp was found to have no significant impact on the intestinal absorption of cilostazol [34]. In addition, other covariates such as age, body weight and BSA also did not induce any obvious effects on the PK parameters. This may have occurred because the study was conducted in healthy adult subjects. Studies of a more diverse patient population may allow a more meaningful examination of covariate influences on PK parameters.
These modelling processes were optimized processes to the data. There was a wave pattern in the CWRES plot of goodness-of-fit and a little underestimation of the 50-mg dose in VPC. For more accurate modelling, it might be necessary to add other covariates such as liver and kidney function.
In conclusion, the results of the present study indicate that CYP3A5 and CYP2C19 polymorphisms explain the substantial interindividual variability in the PK of cilostazol, but the ABCB1 product P-gp does not appear to be associated with the disposition of cilostazol.
Competing interests
None to declare.
REFERENCES
- 1.Dawson DL, Cutler BS, Meissner MH, Strandness DE., Jr. Cilostazol has beneficial effects in treatment of intermittent claudication: results from a multicenter, randomized, prospective, double-blind trial. Circulation. 1998;98:678–86. doi: 10.1161/01.cir.98.7.678. [DOI] [PubMed] [Google Scholar]
- 2.Shinoda-Tagawa T, Yamasaki Y, Yoshida S, Kajimoto Y, Tsujino T, Hakui N, Matsumoto M, Hori M. A phosphodiesterase inhibitor, cilostazol, prevents the onset of silent brain infarction in Japanese subjects with Type II diabetes. Diabetologia. 2002;45:188–94. doi: 10.1007/s00125-001-0740-2. [DOI] [PubMed] [Google Scholar]
- 3.Choi JM, Shin HK, Kim KY, Lee JH, Hong KW. Neuroprotective effect of cilostazol against focal cerebral ischemia via antiapoptotic action in rats. J Pharmacol Exp Ther. 2002;300:787–93. doi: 10.1124/jpet.300.3.787. [DOI] [PubMed] [Google Scholar]
- 4.Bramer SL, Forbes WP, Mallikaarjun S. Cilostazol pharmacokinetics after single and multiple oral doses in healthy males and patients with intermittent claudication resulting from peripheral arterial disease. Clin Pharmacokinet. 1999;37(Suppl 2):1–11. doi: 10.2165/00003088-199937002-00001. [DOI] [PubMed] [Google Scholar]
- 5.Suri A, Forbes WP, Bramer SL. Pharmacokinetics of multiple-dose oral cilostazol in middle-age and elderly men and women. J Clin Pharmacol. 1998;38:144–50. doi: 10.1002/j.1552-4604.1998.tb04403.x. [DOI] [PubMed] [Google Scholar]
- 6.Akiyama H, Kudo S, Shimizu T. The metabolism of a new antithrombotic and vasodilating agent, cilostazol, in rat, dog and man. Arzneimittelforschung. 1985;35:1133–40. [PubMed] [Google Scholar]
- 7.Abbas R, Chow CP, Browder NJ, Thacker D, Bramer SL, Fu CJ, Forbes W, Odomi M, Flockhart DA. In vitro metabolism and interaction of cilostazol with human hepatic cytochrome P450 isoforms. Hum Exp Toxicol. 2000;19:178–84. doi: 10.1191/096032700678827717. [DOI] [PubMed] [Google Scholar]
- 8.Yoo HD, Park SA, Cho HY, Lee YB. Influence of CYP3A and CYP2C19 genetic polymorphisms on the pharmacokinetics of cilostazol in healthy subjects. Clin Pharmacol Ther. 2009;86:281–4. doi: 10.1038/clpt.2009.90. [DOI] [PubMed] [Google Scholar]
- 9.Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Domanski TL, Finta C, Halpert JR, Zaphiropoulos PG. cDNA cloning and initial characterization of CYP3A43, a novel human cytochrome P450. Mol Pharmacol. 2001;59:386–92. doi: 10.1124/mol.59.2.386. [DOI] [PubMed] [Google Scholar]
- 11.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–23. [PubMed] [Google Scholar]
- 12.Chowbay B, Cumaraswamy S, Cheung YB, Zhou Q, Lee EJ. Genetic polymorphisms in MDR1 and CYP3A4 genes in Asians and the influence of MDR1 haplotypes on cyclosporin disposition in heart transplant recipients. Pharmacogenetics. 2003;13:89–95. doi: 10.1097/00008571-200302000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27:383–91. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 14.Balram C, Zhou Q, Cheung YB, Lee EJ. CYP3A5*3 and *6 single nucleotide polymorphisms in three distinct Asian populations. Eur J Clin Pharmacol. 2003;59:123–6. doi: 10.1007/s00228-003-0594-2. [DOI] [PubMed] [Google Scholar]
- 15.Lee SJ, Usmani KA, Chanas B, Ghanayem B, Xi T, Hodgson E, Mohrenweiser HW, Goldstein JA. Genetic findings and functional studies of human CYP3A5 single nucleotide polymorphisms in different ethnic groups. Pharmacogenetics. 2003;13:461–72. doi: 10.1097/00008571-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 16.de Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem. 1994;269:15419–22. [PubMed] [Google Scholar]
- 17.De Morais SM, Wilkinson GR, Blaisdell J, Meyer UA, Nakamura K, Goldstein JA. Identification of a new genetic defect responsible for the polymorphism of (S)-mephenytoin metabolism in Japanese. Mol Pharmacol. 1994;46:594–8. [PubMed] [Google Scholar]
- 18.Goldstein JA, Ishizaki T, Chiba K, de Morais SM, Bell D, Krahn PM, Evans DA. Frequencies of the defective CYP2C19 alleles responsible for the mephenytoin poor metabolizer phenotype in various Oriental, Caucasian, Saudi Arabian and American black populations. Pharmacogenetics. 1997;7:59–64. doi: 10.1097/00008571-199702000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Benet LZ, Cummins CL. The drug efflux-metabolism alliance: biochemical aspects. Adv Drug Deliv Rev. 2001;50(Suppl 1):S3–11. doi: 10.1016/s0169-409x(01)00178-8. [DOI] [PubMed] [Google Scholar]
- 20.van Schaik RHN, van der Heiden IP, van den Anker JN, Lindemans J. CYP3A5 variant allele frequencies in Dutch Caucasians. Clin Chem. 2002;48:1668–71. [PubMed] [Google Scholar]
- 21.Fiegenbaum M, da Silveira FR, Van der Sand CR, Van der Sand LC, Ferreira ME, Pires RC, Hutz MH. The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin Pharmacol Ther. 2005;78:551–8. doi: 10.1016/j.clpt.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 22.Cascorbi I, Gerloff T, Johne A, Meisel C, Hoffmeyer S, Schwab M, Schaeffeler E, Eichelbaum M, Brinkmann U, Roots I. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–74. doi: 10.1067/mcp.2001.114164. [DOI] [PubMed] [Google Scholar]
- 23.Beal S, Sheiner L. NONMEM User's Guide, Part I. San Francisco, CA: University of California at San Francisco; 1992. [Google Scholar]
- 24.Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic–pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511–28. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 25.Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Meth Programs Biomed. 1999;58:51–64. doi: 10.1016/s0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 26.Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15:1463–8. doi: 10.1023/a:1011970125687. [DOI] [PubMed] [Google Scholar]
- 27.Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24:2187. doi: 10.1007/s11095-007-9361-x. [DOI] [PubMed] [Google Scholar]
- 28.Holford N. Wings for NONMEM. Available at http://wfnsourceforgenet (last accessed December 2008.
- 29.Woo SK, Kang WK, Kwon KI. Pharmacokinetic and pharmacodynamic modeling of the antiplatelet and cardiovascular effects of cilostazol in healthy humans. Clin Pharmacol Ther. 2002;71:246–52. doi: 10.1067/mcp.2002.122474. [DOI] [PubMed] [Google Scholar]
- 30.Hiratsuka M, Hinai Y, Sasaki T, Konno Y, Imagawa K, Ishikawa M, Mizugaki M. Characterization of human cytochrome p450 enzymes involved in the metabolism of cilostazol. Drug Metab Dispos. 2007;35:1730–2. doi: 10.1124/dmd.107.016758. [DOI] [PubMed] [Google Scholar]
- 31.Amirimani B, Ning B, Deitz AC, Weber BL, Kadlubar FF, Rebbeck TR. Increased transcriptional activity of the CYP3A4*1B promoter variant. Environ Mol Mutagen. 2003;42:299–305. doi: 10.1002/em.10199. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97:3473–8. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim RB, Leake BF, Choo EF, Dresser GK, Kubba SV, Schwarz UI, Taylor A, Xie HG, McKinsey J, Zhou S, Lan LB, Schuetz JD, Schuetz EG, Wilkinson GR. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70:189–99. doi: 10.1067/mcp.2001.117412. [DOI] [PubMed] [Google Scholar]
- 34.Toyobuku H, Tamai I, Ueno K, Tsuji A. Limited influence of P-glycoprotein on small-intestinal absorption of cilostazol, a high absorptive permeability drug. J Pharm Sci. 2003;92:2249–59. doi: 10.1002/jps.10490. [DOI] [PubMed] [Google Scholar]