

Abstract
AIMS
To assess the two-way pharmacokinetic interaction between maraviroc and raltegravir.
METHODS
In this open-label, multiple-dose, fixed-sequence study, 18 healthy, human immunodeficiency virus (HIV)-seronegative subjects received the following: days 1–3 raltegravir 400 mg q12h, days 4–5 washout, days 6–11 maraviroc 300 mg q12h, and days 12–14 raltegravir 400 mg q12h + maraviroc 300 mg q12h. Serial 12-h blood samples were collected on days 3 (raltegravir), 11 (maraviroc) and 14 (raltegravir + maraviroc). Plasma samples were assayed by validated liquid chromatography tandem mass spectrometry assays. Test/reference ratios and 95% confidence intervals (CIs) were determined for pharmacokinetic parameters.
RESULTS
For maraviroc, the test/reference % ratio (95% CI) for AUCτ was 85.8 (78.7, 93.5), for Cmax was 79.5 (64.8, 97.5) and for Cmin was 90.3 (84.2, 96.9). For raltegravir, the test/reference % ratio (95% CI) for AUCτ was 63.3 (41.0, 97.6), for Cmax was 66.8 (37.1, 120.0) and for Cmin was 72.4 (55.1, 95.2). In all subjects, maraviroc average concentrations (AUCτ divided by 12) were >100 ng ml−1, the threshold value below which there is an increased risk of virological failure. Based on clinical experience for raltegravir, mean Cmin decreases >60% are considered to be clinically relevant for short-term activity; however, in the present study mean changes were only 28% and thus not considered to be of clinical relevance.
CONCLUSIONS
Co-administration of maraviroc and raltegravir decreased systemic exposure of both drugs; however, these are not likely to be clinically relevant. Safety and efficacy studies may help in understanding the role of this combination in the treatment of HIV infection.
Keywords: maraviroc, pharmacokinetics, raltegravir
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Maraviroc is a CCR5 receptor antagonist, while raltegravir is a HIV-1 integrase inhibitor.
Based on the known metabolic pathways (CYP3A4 for maraviroc and UGT1A1 for raltegravir), interaction between the two drugs is unlikely. However, unexpected interactions have been reported for other antiretroviral drugs.
As both these drugs are likely to be used in combination, this study evaluated the pharmacokinetic interaction between them.
WHAT THIS STUDY ADDS
Relative to individual monotherapy, co-administration resulted in a 20% and 33% decrease in mean Cmax, and 14% and 37% decrease in mean AUC of maraviroc and raltegravir, respectively.
Co-administration was generally safe and well tolerated in healthy subjects.
These changes are not likely to be clinically relevant, thus no dose adjustment is necessary.
Introduction
According to the World Health Organization, an estimated 33 million people worldwide are human immunodeficiency virus (HIV) positive, at least 9.7 million of whom are in need of antiretroviral drugs [1]. Standard antiretroviral therapy consists of the use of at least three antiretroviral drugs to suppress maximally the HIV virus and stop the progression of disease [2]. Unfortunately, there is always the potential of drug resistance and thus the need for new antiretroviral drugs with different mechanism of action [3].
One new antiretroviral drug is maraviroc (Selzentry®; Celsentri®), a CCR5 receptor antagonist, recently approved for the combination treatment of HIV-1 infection in treatment-experienced adults infected with CCR5 tropic HIV [4, 5]. Following oral administration, maraviroc is rapidly absorbed, with peak plasma concentration achieved within 0.5–4 h after dosing [6]. Maraviroc is mainly metabolized by CYP3A4 and is also a substrate for P-glycoprotein (P-gp) [6]. Drug interactions have been investigated when maraviroc is co-administered with potent CYP3A4 inhibitors [7] or with potent CYP3A4 inducers [8]. When administered with a potent CYP3A4 inhibitor (with or without a CYP3A4 inducer), the recommended dose of maraviroc is 150 mg twice daily (q12h), whereas the recommended dose is 600 mg q12h when administered with a potent CYP3A4 inducer (without a potent CYP3A4 inhibitor) [4]. Maraviroc did not inhibit the activity of CYP1A2, 2B6, 2C8, 2C9, 2C19 and 3A at clinically relevant concentrations in vitro[4]. The terminal half-life of maraviroc following oral dosing to steady state in healthy subjects was 14–18 h, and steady state was reached within 5 days with q12h dosing [9].
Raltegravir (Isentress®) was also recently approved for combination treatment of HIV-1 infection in treatment-experienced adult patients who have evidence of viral replication and HIV-1 strains resistant to multiple antiretroviral agents [10, 11]. Raltegravir inhibits the catalytic activity of HIV-1 integrase, an enzyme required for viral replication. Following oral administration, raltegravir is rapidly absorbed, with peak concentrations achieved approximately 3 h after dosing. Steady state is achieved within approximately the first 2 days with q12h dosing. The apparent terminal half-life of raltegravir is approximately 9 h [12]. Raltegravir is eliminated mainly by metabolism via UDP glucuronosyltransferase (UGT1A1) [13]. It is not a substrate of cytochrome P450 enzymes, and does not inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 3A in vitro (IC50 > 100 µm) [14]. Raltegravir does not induce or inhibit CYP3A4 and is also not an inhibitor of UGT1A1 or UGT2B7 (IC50 > 50 µm). However, co-administration with strong inducers of UGT1A1 such as rifampin results in reduced plasma concentrations of raltegravir. Raltegravir does not inhibit P-gp-mediated drug transport [14].
Maraviroc and raltegravir are two antiretrovirals with new mechanisms of action. It is likely that treatment-experienced patients will receive combination treatment that includes these two agents [15]. Hence, this study was undertaken to evaluate systematically the two-way pharmacokinetic interaction between maraviroc and raltegravir in healthy subjects.
Methods
Subjects
Eighteen healthy male and female subjects between the ages of 18 and 55 years were enrolled. To be eligible to participate, subjects had to be in good health as determined by a detailed medical history, full physical examination, 12-lead electrocardiogram, and clinical laboratory tests. Female subjects were required to have a negative serum pregnancy test at screening and prior to commencing the study. The results of a urine drug screen had to be negative. Subjects who were taking or had taken any prescription or over-the-counter drug, vitamins or dietary supplement within 7 days or 5 half-lives, whichever was longer, before the first dose of study treatment were excluded. Subjects were not permitted to consume grapefruit juice within 4 days before dosing or use herbal supplements including St John's Wort within 30 days before dosing.
The study was conducted from February 2008 to March 2008, and subjects' written informed consent was obtained before participation, in conformity with the ethical principles originating in or derived from the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice Guidelines. The study protocol was approved by the Institutional Review Board IntegReview Ltd, Austin, TX, USA.
Study design
This was an open-label, multiple-dose, fixed-sequence study. On days 1–3, subjects received raltegravir 400 mg q12h followed by a washout period on days 4–5. Subjects received maraviroc 300 mg q12h on days 6–11, and the combination of maraviroc 300 mg q12h plus raltegravir 400 mg q12h on days 12–14. Subjects were required to be confined at the Clinical Research Unit for the entire duration of the study. The approved doses of maraviroc (in the absence of a CYP3A4 inducer or inhibitor) and raltegravir were utilized in the study (4, 10). Study drugs were administered on an empty stomach (no food for 1 h before and after dosing). Additionally, on the days of pharmacokinetic evaluations, subjects were required to fast overnight for at least 8 h prior to and for 4 h after dosing.
Pharmacokinetic assessments
Steady-state pharmacokinetics was determined on day 3 (raltegravir), day 11 (maraviroc) and day 14 (maraviroc and raltegravir). Serial blood samples (5–6 ml each) were collected in tubes containing sodium heparin (for maraviroc) or dipotassium ethylenediamine tetraaceticacid (for raltegravir) at the following time points: predose (immediately before the morning dose) and 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 12 h postdose. Blood samples were centrifuged at 1700 g for 10 min at 4°C, and the resulting plasma was stored in appropriately labelled screw-capped polypropylene tubes at −20°C within 1 h of collection.
Plasma samples were assayed for maraviroc by Tandem Laboratories (West Trenton, NJ, USA) using a validated liquid chromatography tandem mass spectrometry (LC/MS/MS) method. In brief, human plasma (0.050 ml) was mixed with [D5]-maraviroc internal standard in acetonitrile and vortex-mixed. Following centrifugation, an aliquot (0.010–0.020 ml) was injected into the high-performance liquid chromatography system. Chromatographic separation was achieved using a Fluophase PFP column (4.6 × 50 mm, 5 µm; Thermo Electron Corp., Pittsburgh, PA, USA) and a mobile phase consisting of 80 : 20 v : v acetonitrile : 25 mM ammonium acetate in aqueous 0.2% formic acid, at a flow rate of 1 ml min−1. The analytes were detected using an Applied-Biosystem Sciex API 4000 LC/MS/MS system (MDI SCIEX, Concord, ON, Canada) operating in positive TurboIonSpray® mode. The precursor/product ion transitions were m/z 514.1→389.1 for maraviroc, m/z 519.1→394.1 for [D5]-maraviroc. The range of quantification for the maraviroc assay was 0.500–500 ng ml−1. The accuracy (percent difference from nominal) of the quality control samples used during sample analysis ranged from 4.5 to 5.8%, with a precision (as measured by percent relative standard deviation) of ≤6.0%.
Plasma samples for raltegravir were analysed by Bioanalytical Systems, Inc. (McMinnville, OR, USA) using a published validated LC/MS/MS assay [16]. Aliquots (0.200 ml) of plasma containing analyte and internal standard were extracted using liquid/liquid extraction. The range of quantification of the raltegravir assay was 2.00–1000 ng ml−1. The accuracy (percent difference from nominal) of the quality control samples used during sample analysis ranged from 0.8 to 3.2%, with a precision (as measured by percent relative standard deviation) of ≤6.0%.
Pharmacokinetic analyses were carried out using standard noncompartmental methods. Maximum observed plasma concentrations (Cmax), time of first occurrence of Cmax (Tmax) and trough concentrations at 12 h post dose (Cmin) were estimated directly from experimental data. Area under the concentration–time curve over the dosing interval (AUCτ), where τ was 12 h for maraviroc and raltegravir, was estimated using the linear/log trapezoidal method. The average plasma concentration (Cave) was determined by dividing AUCτ by the dosing interval of 12 h.
Safety assessments
Safety was evaluated throughout the study based on adverse event (AE) monitoring, clinical laboratory values, vital sign measurements, electrocardiogram results and physical examination findings.
Statistical analysis
Natural log transformed AUCτ, Cmax and Cmin were analysed using a mixed-effect model with treatment as a fixed effect and subject as a random effect. Estimates of the adjusted mean differences (test/reference) and corresponding 95% confidence intervals (CIs) were obtained from the model. The adjusted mean differences and 95% CIs for the differences were exponentiated to provide estimates of the ratio of adjusted geometric means (test/reference) and 95% CIs for the ratios. Maraviroc or raltegravir administered alone was the reference treatment and maraviroc plus raltegravir was the test treatment. Lack of interaction would be demonstrated if the estimated 95% CI for the ratios for AUCτ and Cmax fell entirely within (80%, 125%). Statistical analyses was conducted using SAS software (Unix SAS Version 8.2, 1999–2001; SAS Institute Inc., Cary, NC, USA).
With a sample size of at least 18 subjects, this study had at least 90% overall power to demonstrate a lack of interaction of the test to reference treatments for maraviroc pharmacokinetic parameters (i.e. equivalence in AUCτ and Cmax). For maraviroc, sample size calculations were based on the intrasubject percent coefficient of variation (%CV) of 0.1082 and 0.1853 for log AUC and log Cmax, respectively, which were obtained from the average of three previous maraviroc studies in healthy subjects (Pfizer data on file). The sample size estimate was also based on the assumption that the true ratio of test to reference treatments for AUCτ and Cmax for maraviroc was 1.0. For raltegravir, sample size calculations were based on intrasubject %CV of 0.290 and 0.414 for log AUC and log Cmax, respectively, as obtained from the average of two drug–drug interaction studies (atazanavir/ritonavir, 400 mg q12h; and tenofovir) [10]. A sample size of 18 subjects provided 90% CI for the difference between treatments of raltegravir (± 0.205 and 0.293) on the natural log scale for AUCτ and Cmax, respectively, with 90% coverage probability.
Results
Study population
A total of 18 subjects (16 male and two female) were enrolled into the study and received treatment. Seventeen subjects completed the study as planned. One subject discontinued on day 10 during treatment with maraviroc for personal reasons. For pharmacokinetics, 17 and 18 subjects were included for maraviroc and raltegravir analyses, respectively. All subjects were included in the safety analyses. The mean age was 36.3 years (range 25–54) and mean body mass index was 26.2 kg m−2 (range 19.9–30.4). Six subjects (33.3%) were White, nine (50.0%) Black and three (16.7%) were other/multiracial.
Effect of raltegravir on maraviroc pharmacokinetics
The concentration–time profiles for maraviroc administered alone (day 11) and when co-administered with raltegravir (day 14) are shown in Figure 1. Administration of raltegravir slightly decreased the mean concentrations of maraviroc. Geometric mean maraviroc Cmax and AUCτ following co-administration with raltegravir were decreased by approximately 21% (785–624 ng ml−1) and 14% (2853–2447 ng h−1 ml−1), respectively (Table 1). There was also a 10% decrease in mean trough values (51.6–46.6 ng ml−1) due to co-administration with raltegravir. Median Tmax values were similar for both treatments (Table 1). The lower 95% CI for AUCτ and Cmax fell outside the lower boundary of 80%, 125%; however, for Ctrough both 95% CIs were entirely contained within this range. Figure 2 shows the individual subject change in average plasma concentration of maraviroc when co-administered with raltegravir. The majority of subjects (13 of 17) showed a decrease in average concentrations.
Figure 1.
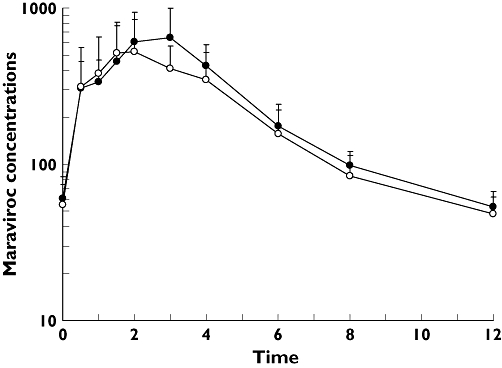
Mean (SD) maraviroc plasma concentration (ng ml−1) vs. time (h) profiles on day 11 (maraviroc) and day 14 (maraviroc + raltegravir). Maraviroc (n = 17) (–•–); Maraviroc + Raltegravir (n = 17) (–○–)
Table 1.
Geometric mean (%CV) pharmacokinetic parameters and statistical analyses for maraviroc
| Parameters (units) | Maraviroc alone | Maraviroc + raltegravir | % Ratio (95% CI)* |
|---|---|---|---|
| AUCτ (ng h−1 ml−1) | 2853 (28%) | 2447 (31%) | 85.8 (78.7, 93.5) |
| Cmax (ng ml−1) | 785 (41%) | 624 (47%) | 79.5 (64.8, 97.5) |
| Cmin (ng ml−1) | 51.6 (26%) | 46.6 (28%) | 90.3 (84.2, 96.9) |
| Tmax (h)† | 2.0 (0.5–4.0) | 2.0 (0.5–4.0) | NA‡ |
Ratio based on adjusted geometric means for combination treatment vs. single agent obtained from anova model.
Median (range).
NA, not applicable.
Figure 2.
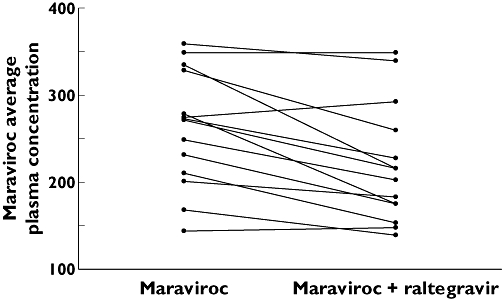
Average maraviroc plasma concentration (ng ml−1) on day 11 (maraviroc) and on day 14 (maraviroc + raltegravir)
Effect of maraviroc on raltegravir pharmacokinetics
The concentration–time profiles for raltegravir administered alone (day 3) and when co-administered with maraviroc (day 14) are shown in Figure 3. Administration of maraviroc decreased the mean steady-state concentration of raltegravir on day 14 relative to raltegravir alone on day 3. The decrease in mean raltegravir AUCτ, Cmax and Cmin following co-administration with maraviroc was approximately 37% (7356.0–4653.0 ng h−1 ml−1), 33% (2116.0–1413.0 ng ml−1) and 28% (66.2–48.0 ng ml−1), respectively (Table 2). The ratio (95% CI) of test/reference treatments was 63.3% (41.0, 97.6) for AUCτ, 66.8% (37.1, 120.0) for Cmax, and 72.4% (55.1, 95.2) for Cmin (Table 2). The point estimate for all three parameters was <80%. The median Tmax value was reduced by 1.5 h when raltegravir was given with maraviroc compared with raltegravir alone (Table 2). The individual subject trough concentrations of raltegravir for the two treatments are shown in Figure 4. There appeared to be wide variability in the change in trough concentrations across subjects. The majority of subjects (15 of 17) showed a decrease in raltegravir trough concentrations upon co-administration, whereas trough concentrations increased in two subjects.
Figure 3.
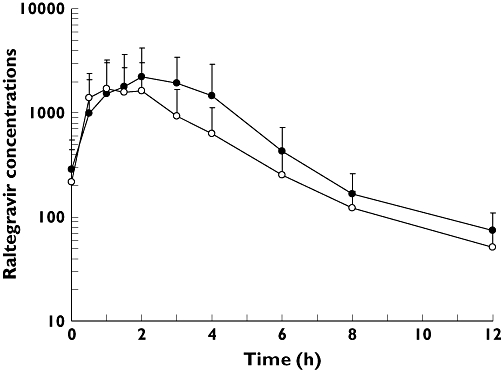
Mean (SD) raltegravir plasma concentration (ng ml−1) vs. time (h) profiles on day 3 (raltegravir) and day 14 (maraviroc + raltegravir). Raltegravir (n = 17) (–•–); Maraviroc + Raltegravir (n = 17) (–○–)
Table 2.
Geometric mean (%CV) pharmacokinetic parameters and statistical analyses for raltegravir
| Parameters (units) | Raltegravir alone | Maraviroc + raltegravir | % Ratio (95% CI)* |
|---|---|---|---|
| AUCτ (ng h−1 ml−1) | 7356 (58) | 4819 (63) | 63.3 (41.0, 97.6) |
| Cmax (ng ml−1) | 2116 (67) | 1486 (72) | 66.8 (37.1, 120.0) |
| Cmin (ng ml−1) | 66.2 (49) | 47.0 (44) | 72.4 (55.1, 95.2) |
| Tmax (h)† | 2.5 (1.0–6.0) | 1.0 (0.0–2.0) | NA‡ |
Ratio based on adjusted geometric means for combination treatment vs. single agent obtained from anova model.
Median (range).
NA, not applicable.
Figure 4.
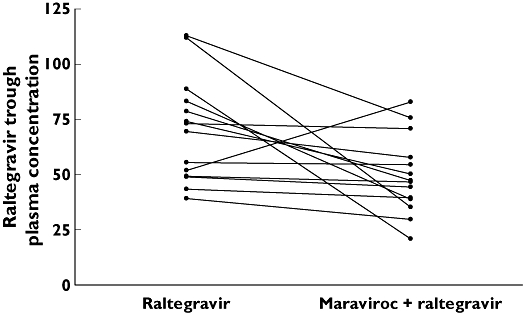
Raltegravir trough plasma concentration (ng ml−1) on day 3 (raltegravir) and day 14 maraviroc + raltegravir)
Safety
Treatment-related AEs were experienced by three subjects (16.7%) during treatment with raltegravir, four subjects (22.2%) during treatment with maraviroc and four subjects (23.5%) during treatment with maraviroc plus raltegravir. All of these events were mild in severity. Headache and somnolence were reported by two subjects each during the study. Other AEs reported by one subject each included constipation, dyspepsia, back pain, folliculitis, and peripheral neuropathy. In the one subject who experienced peripheral neuropathy, this event began about 2.3 h after dosing on study day 1 during raltegravir treatment and resolved on study day 17 after completion of all study treatments. There were no clinically significant changes in safety laboratory test results, vital signs, ECG or physical examination findings during the study.
Discussion
The purpose of this study was to evaluate the two-way pharmacokinetic drug interaction and tolerability of concomitant administration of maraviroc and raltegravir. The results showed that, relative to monotherapy, co-administration of maraviroc and raltegravir was associated with reductions in the systemic exposure of both drugs that are unlikely to be clinically relevant. These results are intriguing because a pharmacokinetic interaction was not anticipated based on the known metabolic profiles of maraviroc and raltegravir. Maraviroc is mainly metabolized by CYP3A4 while raltegravir is eliminated by UGT1A1. Neither drug induces or inhibits cytochrome P450 enzymes. Both drugs are substrates for P-gp. Examination of the mean plasma concentration–time profiles (Figures 1 and 3) showed that the terminal elimination phase was not noticeably affected for either drug due to concomitant administration, suggesting that the interaction does not appear to be related to systemic clearance of either drug. Instead, the reduced peak concentrations of both drugs point towards changes in pre-systemic elimination associated with absorption and/or first-pass metabolism as a potential mechanism of interaction; however, the exact mechanism of interaction cannot be deduced from the present study.
The effect of maraviroc exposure and various prognostic factors on efficacy end-points such as virological failure has been examined using data from two Phase 3 studies in treatment-experienced patients, namely MOTIVATE 1 and 2 [17]. In these analyses, baseline CD4+ cell count was the most important prognostic factor, followed by systemic exposure to maraviroc, in determining viral response (success/failure) at 24 weeks. The exposure–response analyses suggested that treatment-experienced patients who achieved a maraviroc average concentration of at least 100 ng ml−1 had >80% probability of success (defined as achieving <50 copies ml−1). In a similar analysis in treatment-naive patients enrolled in the MERIT study, the probability of success dropped sharply at maraviroc average concentration <75 ng ml−1[18]. In the current study, all subjects had a maraviroc average concentration >100 ng ml−1 when maraviroc was administered alone or co-administered with raltegravir. Overall, it appears that reductions in maraviroc exposure observed due to co-administration with raltegravir are unlikely to affect the antiretroviral activity of maraviroc. Notably, in a combination study with raltegravir, maraviroc and etravirine in heavily pretreated HIV-infected patients harbouring R5 co-receptor-using virus, 90% of patients had undetectable viral load (<50 copies ml−1) at week 48 [15].
The pharmacokinetics of raltegravir is highly variable. In the pivotal Phase 3 studies in treatment-experienced patients BENCHMRK 1 and 2, the coefficient of variation for inter- and intrasubject variability for raltegravir trough concentrations was 212% and 122%, respectively [10]. This variability has limited the development of a robust population pharmacokinetic model, and hence trough concentrations have been evaluated for correlation to clinical outcomes in Phase 2 and 3 studies in treatment-experienced patients [19]. The pharmacokinetics of raltegravir appear to have much less impact on viral load outcome compared with other variables (e.g. baseline viral RNA and other background therapy) and no threshold for trough concentration of raltegravir could be identified [19]. In the absence of definitive correlation between trough concentrations and clinical efficacy, the lower bounds in the clinical programme have been used to define the fold reduction in trough concentrations that are likely to be associated with an increased risk of virological failure [14].
Overall, the clinical experience with raltegravir across dose-ranging Phase 2 studies and pivotal Phase 3 studies suggests that an average reduction in trough concentrations of up to 60% of raltegravir is not likely to affect virological success [14]; the mean change noted in the present study was 28%. Of the subjects with reduced trough concentrations of raltegravir, 13/15 had <60% decrease, while two subjects had greater decreases (76% and 69%, respectively), highlighting the variability in raltegravir pharmacokinetics. Interestingly, these two subjects had the highest raltegravir concentrations on day 3 (raltegravir alone). Alternatively, using AUC as a measure of exposure, the ratio (95% CI) of test/reference treatments for raltegravir AUCτ was 66.8% (37.1, 120.0). Changes of a similar magnitude were noted in interaction studies with efavirenz and tiprinavir/ritonavir; the ratio (90% CI) of the AUC for test/reference treatments for these interaction was 64% (52, 80) and 76% (49, 119), respectively. No dose adjustment of raltegravir is necessary when co-administering with efavirenz or tiprinavir/ritonavir [14]. Overall, it appears that the reduction in raltegravir exposures due to co-administration with maraviroc observed in this study is not likely to be clinically meaningful.
In summary, co-administration of maraviroc and raltegravir in healthy HIV-seronegative subjects resulted in reductions in the systemic exposure of both drugs. These changes are unlikely to be clinically relevant and hence no dose adjustment of maraviroc or raltegravir is necessary. Safety and efficacy data from studies in patients receiving regimens that include maraviroc and raltegravir will be helpful in further understanding the role of this combination in the treatment of HIV-1 infection.
Competing interests
None to declare.
The study was sponsored by Pfizer Global Research and Development, and was funded by Pfizer Inc. E.A., J.F., P.C., R.T. and B.D. are employees of Pfizer Inc. and hold stock in the company. P.G. was an employee of Pfizer Inc. at the time this research was performed and holds stock in the company. The authors thank Manoli Vourvahis, Pharm D (Pfizer Inc.) for his valuable contributions during the study.
REFERENCES
- 1.World Heath Organization. 2007 AIDS Epidemic Update. Available at http://data.unaids.org/pub/EPISlides/2007/2007_epiupdate_en.pdf (last accessed 2 October 2009.
- 2.Wilson LE, Gallant JE. HIV/AIDS: the management of treatment-experienced HIV-infected patients: new drugs and drug combinations. Clin Infect Dis. 2009;48:214–21. doi: 10.1086/595701. [DOI] [PubMed] [Google Scholar]
- 3.Chen TK, Aldrovandi GM. Review of HIV antiretroviral drug resistance. Pediatr Infect Dis J. 2008;27:749–52. doi: 10.1097/INF.0b013e3181846e2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marviroc (Selzentry®). United States Prescribing Information. New York: Pfizer Inc.; 2008. [Google Scholar]
- 5.MacArthur RD, Novak RM. Reviews of anti-infective agents: Maraviroc: the first of a new class of antiretroviral agents. Clin Infect Dis. 2008;47:236–41. doi: 10.1086/589289. [DOI] [PubMed] [Google Scholar]
- 6.Abel S, Russell D, Whitlock LA, Ridgway CE, Nedderman AN, Walker DK. Assessment of the absorption, metabolism and absolute bioavailability of maraviroc in healthy male subjects. Br J Clin Pharmacol. 2008;65(Suppl. 1):60–7. doi: 10.1111/j.1365-2125.2008.03137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abel S, Russell D, Taylor-Worth RJ, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl. 1):27–37. doi: 10.1111/j.1365-2125.2008.03133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abel S, Jenkins TM, Whitlock LA, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inducers with and without CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl. 1):38–46. doi: 10.1111/j.1365-2125.2008.03134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel S, van der Ryst E, Rosario MC, Ridgway CE, Medhurst CG, Taylor-Worth RJ, Muirhead GJ. Assessment of the pharmacokinetics, safety and tolerability of maraviroc, a novel CCR5 antagonist, in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl. 1):5–18. doi: 10.1111/j.1365-2125.2008.03130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raltegravir (Isentress®). United States Prescribing Information. Whitehouse Station, NJ: Merck & Co. Inc.; 2007. [Google Scholar]
- 11.Cocohoba J, Dong BJ. Raltegravir: the first HIV integrase inhibitor. Clin Ther. 2008;30:1747–65. doi: 10.1016/j.clinthera.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Iwamoto M, Wenning LA, Petry AS, Laethem M, De Smet M, Kost JT, Merschman SA, Strohmaier KM, Ramael S, Lasseter KC, Stone JA, Gottesdiener KM, Wagner JA. Safety, tolerability, and pharmacokinetics of raltegravir after single and multiple doses in healthy subjects. Clin Pharmacol Ther. 2008;83:293–9. doi: 10.1038/sj.clpt.6100281. [DOI] [PubMed] [Google Scholar]
- 13.Kassahun K, McIntosh I, Cui D, Hreniuk D, Merschman S, Lasseter K, Azrolan N, Iwamoto M, Wagner JA, Wenning LA. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab Dispos. 2007;35:1657–63. doi: 10.1124/dmd.107.016196. [DOI] [PubMed] [Google Scholar]
- 14.Merck. ISENTRESS™ (raltegravir) 400 mg For Treatment of HIV. 2007. NDA 22-145 Briefing Document. Presented as part of the FDA Antiviral Drugs Advisory Committee meeting, held September 5, 2007. Available at http://www.fda.gov/ohrms/dockets/ac/07/briefing/2007-4314b1-01-Merck.pdf (last accessed 6 March 2009.
- 15.Nozza S, Visco F, Soria A, Galli L, Salpietro S, Gianotti N, Carini E, Bigoloni A, Fusetti G, Tambussi G, Lazzarin A, Castagna A. Excellent short-term CD4 recovery with a PI- and NRTI-sparing regimen in triple-class failure HIV-infected patients: raltegravir, maraviroc, etravirine. 9th International Congress on Drug Therapy in HIV Infection. Glasgow, UK, 9–13 November 2008. Abstract P045.
- 16.Merschman SA, Vallano PT, Wenning LA, Matuszewski BK, Woolf EJ. Determination of the HIV integrase inhibitor, MK-0518 (raltegravir), in human plasma using 96-well liquid–liquid extraction and HPLC-MS/MS. J Chromatogr. 2007;857:15–24. doi: 10.1016/j.jchromb.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 17.McFadyen L, Jacqmin P, Wade JR, Weatherley B, Chan PLS. Maraviroc (MVC) exposure–efficacy relationship HIV-1-infected patients. 11th European AIDS Conference / EACS. Madrid, Spain, 24–27 October 2007. Poster P4.1/06.
- 18.McFadyen L, Jacqmin P, Wade JR, Weatherley B. Maraviroc exposure-efficacy (<50 copies/mL) analysis in HIV-1-infected treatment-naïve subjects – ITT Population (MERIT Study) XVII International AIDS Conference, Mexico City, Mexico, 3–8 August 2008. Poster TUPE0053.
- 19.Wenning L, Nguyen BY, Sun X, Hwang E, Chen Y, Teppler H, Harvey C, Rhodes R, Ryan D, Azrolan N, Stone J. Pharmacokinetic/pharmacodynamic (PK/PD) analyses of raltegravir in phase II and III studies in treatment-experienced HIV patients. 9th International Workshop on Clinical Pharmacology of HIV Therapy. New Orleans, USA, 7–9 April 2008.