

Abstract
AIMS
To test prospectively the predictive value of germinal gene polymorphisms related to fluorouracil (FU) and oxaliplatin (Oxa) pharmacodynamics on toxicity and responsiveness of colorectal cancer (CRC) patients receiving FOLFOX therapy.
METHODS
Advanced CRC patients (n= 117) receiving FOLFOX 7 therapy were enrolled. Gene polymorphisms relevant for FU [thymidylate synthase (TYMS, 28 bp repeats including the G→C mutation + 6 bp deletion in 3'UTR), methylenetetrahydrofolate reductase (MTHFR, 677C→T, 1298A→C), dihydropyrimidine deshydrogenase (IVS14+1G→A) and Oxa: glutathione S-transferase (GST) π (105Ile→Val, 114Ala→Val), excision repair cross-complementing group 1 (ERCC1) (118AAT→AAC), ERCC2 (XPD, 751Lys→Gln) and XRCC1 (399Arg→Gln)] were determined (blood mononuclear cells).
RESULTS
None of the genotypes was predictive of toxicity. Response rate (54.7% complete response + partial response) was related to FU pharmacogenetics, with both 677C→T (P= 0.042) and 1298A→C (P= 0.004) MTHFR genotypes linked to clinical response. Importantly, the score of favourable MTHFR alleles (677T and 1298C) was positively linked to response, with response rates of 37.1, 53.3, 62.5 and 80.0% in patients bearing no, one, two or three favourable alleles, respectively (P= 0.040). Polymorphisms of genes related to Oxa pharmacodynamics showed an influence on progression-free survival, with a better outcome in patients bearing GSTπ 105 Val/Val genotype or XPD 751Lys-containing genotype (P= 0.054).
CONCLUSIONS
These results show that response to FOLFOX therapy in CRC patients may be driven by MTHFR germinal polymorphisms.
Keywords: colon cancer, FOLFOX, gene polymorphism, glutathion S transferase, methylenetetrahydrofolate reductase, pharmacogenetics, XPD
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Numerous clinical studies, including a few prospective ones, have reported conflicting results on the impact of gene polymorphisms related to fluorouracil (FU) and oxaliplatin pharmacodynamics.
WHAT THIS STUDY ADDS
This prospective study is the first to report that clinical response to FOLFOX is significantly related to methylenetetrahydrofolate reductase (MTHFR) gene polymorphisms (677C→T and 1298A→C), with a response rate of 37, 53, 63 and 80% in patients harbouring no, one, two or three favourable MTHFR alleles, respectively.
Only polymorphisms of genes related to oxaliplatin pharmacodynamics (GSTπ 105Ile→Val and XPD 751Ly→Gln) influenced progression-free survival.
These results corroborate the observation that response was related to the cumulative FU dose, whereas progression-free survival was related to the cumulative oxaliplatin dose.
Introduction
Standard chemotherapies in advanced colorectal cancer (CRC) patients have evolved from simple fluorouracil (FU)-based treatment to FU-folinic modulation, FU combination with oxaliplatin (FOLFOX) or irinotecan (FOLFIRI), and finally to associations of FU-containing chemotherapies with biological targeted therapies [1–6]. The wide range of treatment options creates a need for individual predictive factors in order to choose the optimal treatment for a given patient. To this end, tumour molecular markers constitute a valuable approach. Recent data on metastatic CRC patients have clearly demonstrated the predictive value of the absence of K-Ras somatic mutations for selecting patients likely to benefit from the addition of antiepidermal growth factor receptor (EGFR) therapies [7]. However, the benefit of EGFR-targeted therapy in chemotherapeutic care is relatively limited at close to 10% [8]. Moreover, K-Ras status is not relevant when discriminating response to chemotherapy alone [9]. There is still a need for reliable predictive markers aimed at orienting medical treatment in advanced CRC. In this context, two complementary approaches can be considered: tumour markers on the one hand and host-dependent biological factors on the other. Pharmacogenetics belongs to this latter category. Pharmacogenetics, which examines the links between germinal gene polymorphisms and the variability of drug pharmacodynamics, is thus of special interest for anticancer agents. The purpose of the present study was to evaluate prospectively the predictive value of gene polymorphisms potentially related to FU and oxaliplatin pharmacodynamics, taking into account toxicity, response rate and progression-free survival (PFS). The chemotherapeutic protocol was FOLFOX, which is considered a standard in CRC in both advanced disease and the adjuvant setting.
Methods
Patients
One hundred and seventeen patients with advanced CRC were enrolled in this prospective ancillary pharmacogenetic study as part of the multicentre Phase II OPTIMOX 2 trial by the GERCOR group [10]. The study was carried out with ethics committee approval. Patient characteristics are shown in Table 1. All patients received FOLFOX 7 therapy. The main goal of the trial was to assess a new strategy with chemotherapy interruptions in an attempt to improve survival and quality of life. Therefore, patients were randomized in order to receive either six cycles of modified FOLFOX 7 regimen [mFOLFOX 7, 2-h infusion of oxaliplatin 100 mg m−2+ 400 mg m−2 leucovorin (LV) followed by 46-h infusion of FU 3 g m−2, day 1 = day 15] (arm 2, n= 58) or six cycles of mFOLFOX 7 followed by LV–5FU maintenance therapy consisting of a simplified bimonthly regimen from cycle 7 until progression (2-h infusion of LV 400 mg m−2 followed by a bolus of FU 400 mg m−2 and then a 46-h infusion of FU 3 g m−2) (arm 1, n= 59). The number of mFOLFOX 7 cycles was not significantly different between arm 1 and arm 2. The sum of oxaliplatin doses and of FU doses administered during the cycles of FOLFOX 7 therapy were computed. This cumulative oxaliplatin dose and this cumulative FU dose were not significantly different between arm 1 and arm 2.
Table 1.
Patient characteristics
| Age | Median, extremes | 67 (31–80) |
| Sex | Men | 65 |
| Women | 52 | |
| Arm | 1 | 59 |
| 2 | 58 | |
| Arm 1 | ||
| Number of cycles* | Median, extremes | 6 (2–8) |
| Total cumulative oxaliplatin dose*† | Median, extremes (mg) | 1000 (320–1200) |
| Total cumulative FU dose*‡ | Median, extremes (g) | 29.8 (9.6–39.2) |
| Arm 2 | ||
| Number of cycles* | Median, extremes | 6 (3–7) |
| Total cumulative oxaliplatin dose*† | Median, extremes (mg) | 983 (600–1200) |
| Total cumulative FU dose*‡ | Median, extremes (g) | 29.5 (13.5–68.4) |
| Previous adjuvant therapy§ | None | 96 |
| Chemotherapy | 14 | |
| Radiotherapy | 13 | |
| WHO performance status | 0 | 60 |
| 1 | 51 | |
| 2 | 6 | |
| Primary localization | Colon | 74 |
| Rectum | 40 | |
| Both | 3 | |
| Metastasis site§ | Liver | 97 |
| Lung | 52 | |
| Peritoneum | 24 | |
| Lymph node | 19 | |
| Bone | 3 | |
| Others | 13 | |
The number of cycles and the cumulative oxaliplatin and FU doses administered during the FOLFOX 7 therapy were not significantly different between arm 1 and arm 2.
Cumulative oxaliplatin dose during initial FOLFOX 7 (cycles 1 to n).
Cumulative FU dose given as continuous infusion during initial FOLFOX 7 therapy (cycles 1 to n).
Sum not equal to 117 due to multiple choice.
Toxicity evaluation
For each toxicity pattern (neutropenia, thrombocytopenia, anaemia, nausea, vomiting, mucositis, diarrhoea, hand-foot syndrome, neurotoxicity and alopecia), the maximum observed toxicity grade was recorded (National Cancer Institute Common Terminology Criteria for Adverse Events grading). For each patient, we considered the maximum observed toxicity grade (whatever the toxic pattern) and the toxicity score (sum of each toxicity pattern grade).
Efficacy evaluation
Objective tumour response was assessed according to Response Evaluation Criteria in Solid Tumors criteria. The best response was analysed, as well as PFS and overall survival (both computed from randomization). At time of analysis, 79 patients had died and median follow-up was 37.8 months (reverse Kaplan–Meier method).
Pharmacogenetic analyses
Constitutional gene polymorphisms were analysed on DNA extracted from a 9 ml blood sample (Paxgene Blood DNA kit; Preanalytics). Germinal polymorphisms of genes relevant for FU, i.e. thymidylate synthase (TYMS), methylenetetrahydrofolate reductase (MTHFR), dihydropyrimidine deshydrogenase (DPYD), and for oxaliplatin, i.e. glutathione S-transferase (GST) π, excision repair cross-complementing group 1 (ERCC1), ERCC2 (XPD) and XRCC1 were analysed as follows:
TYMS: (i) 28 bp repeat polymorphisms (2R or 3R) in the 5'UTR [polymerase chain reaction (PCR)], along with the G→C mutation in the second repeat of the 3R allele [PCR-restriction fragment length polymorphism (RFLP)][11]. The 2R allele presents one E-box binding site for upstream stimulatory factor (USF), whereas the 3R allele presents two E-box binding sites for USF. The presence of a G→C mutation in the 3R allele alters the USF binding, so that the 3RC allele exhibits a single functional E-box. Thus, TYMS genotype was classified as a function of the number of theoretical E-box binding sites likely to bind USF proteins: class 2 (2R2R or 2R3RC or 3RC3RC), class 3 (2R3RG or 3RC3RG), class 4 (3RG3RG). (ii) 6 bp deletion at position 1494 in the 3'UTR (PCR + electrophoresis) [12].
MTHFR: 677C→T and 1298A→C (melting curve analysis) [12].
DPYD: IVS14+1G→A (PCR-RFLP using the Nde I restriction enzyme) [11].
GSTπ (105Ile→Val and 114Ala→Val), ERCC1 (118AAT→AAC), XPD (751Lys→Gln) and XRCC1 (399Arg→Gln) genotypes were determined by PCR-RFLP using restriction enzymes Alw26I, AciI, BsrDI, PstI and MspI, respectively, as previously described [13, 14]. Wild-type (wt) and mutated cell lines were used as controls.
Statistics
The exact P-values for Hardy–Weinberg equilibrium were tested on http://innateimmunity.net/IIPGA2. The influence of each gene polymorphism was evaluated on toxicity, tumour response, PFS and overall survival. For TYMS genotypes, these analyses were conducted in three different ways by considering: (i) the 6-bp deletion (wt/wt vs. wt/del vs. del/del), (ii) the 28-bp repeats (2R2R vs. 2R3R vs. 3R3R), and (iii) the 28-bp repeats including the G→C mutation (class 2 vs. class 3 vs. class 4, as previously defined). Nonparametric tests were performed for comparisons (Mann–Whitney or Kruskal–Wallis). χ2 tests were applied for categorical variables. A logistic model was applied for multivariate analysis of response predictors [1 = complete response (CR) + partial response (PR), 0 = stable disease (SD) + progressive disease (PD)]. Survival curves were plotted according to the Kaplan–Meier method. The influence of the various tested parameters on PFS and overall survival was assessed by means of log rank test, or Cox analysis for continuous variables. Statistics were performed on SPSS software (version 15.0; SPSS Inc., Chicago, IL, USA)).
Results
Description of analysed genotypes
Table 2 depicts the frequency of analysed genotypes. All patients (n= 117) exhibited the wt genotype for the IVS14+1G→A polymorphism of the DPYD gene. With the exception of MTHFR 1298A→C polymorphism, all genotypes agree with those predicted by the Hardy–Weinberg equilibrium. Linkage disequilibriums were observed between TYMS 28-bp repeats and 6-bp deletion (association between 3RG3RG and 6 bp del, P < 0.001), MTHFR 677C→T and 1298A→C (no homozygous patients for both variants, P < 0.001), GSTπ 105Ile→Val and 114Ala→Val (association between 105Val and 114Val, P= 0.003) and between XPD 751Lys/Gln and ERCC1 118AAT→AAC (association between XPD 751Gln and ERCC1 118AAC, P= 0.001).
Table 2.
Genotypes distribution
| Gene | Genotype | n | |
|---|---|---|---|
| TYMS | 28 bp repeats | 2R2R | 28 |
| 2R3R | 53 | ||
| 3R3R | 30 | ||
| Class including G→C | 2 (2R2R or 2R3RC or 3RC3RC) | 66 | |
| 3 (2R3RG or 3RC3RG) | 38 | ||
| 4 (3RG3RG) | 7 | ||
| 6 bp deletion | wt/wt | 61 | |
| wt/del | 43 | ||
| del/del | 12 | ||
| MTHFR | 677C→T | CC | 44 |
| CT | 58 | ||
| TT | 14 | ||
| 1298A→C | AA | 50 | |
| AC | 61 | ||
| CC | 5 | ||
| GSTπ | 105Ile→Val | Ile/Ile | 56 |
| Ile/Val | 49 | ||
| Val/Val | 12 | ||
| 114Ala→Val | Ala/Ala | 104 | |
| Ala/Val | 11 | ||
| Val/Val | 0 | ||
| ERCC1 | 118AAT→AAC | TT | 32 |
| TC | 62 | ||
| CC | 23 | ||
| XPD | 751Lys→Gln | Lys/Lys | 41 |
| Lys/Gln | 58 | ||
| Gln/Gln | 16 | ||
| XRCC1 | 399Arg→Gln | Arg/Arg | 56 |
| Arg/Gln | 52 | ||
| Gln/Gln | 6 |
Impact of gene polymorphisms on toxicity
Tolerance was satisfactory, with grade 3–4 toxicity observed in 13% of patients for neutropenia, 4.3% for thrombocytopenia and nausea, 3.4% for diarrhoea, 2.6% for vomiting, 0.9% for mucositis and no grade 3–4 for anaemia, hand-foot syndrome, alopecia or neurotoxicity. Whatever the toxicity pattern, grade 3–4 was recorded in 22.9% of patients and the toxicity score ranged from 2 to 15 (median 7). Toxicity was not statistically different according to the treatment arm. None of the analysed gene polymorphisms were predictive of toxicity considered either as the maximum observed grade, or as the toxicity score.
Impact of gene polymorphisms on response
Best response was CR in two patients, PR in 62 patients, SD in 45 patients and PD in eight patients, accounting for a total of 54.7% clinical responses (CR+PR). Best response was not statistically different between the two arms (55.9% in arm 1 vs. 53.4% in arm 2, P= 0.79), but was significantly related to the total cumulative FU dose administered during the cycles of FOLFOX 7 therapy (median 27 and 30.4 g in SD+PD and CR+PR, respectively, P= 0.021) and total cumulative oxaliplatin dose (median 960 and 1020 mg in SD+PD and CR+PR, respectively, P= 0.037).
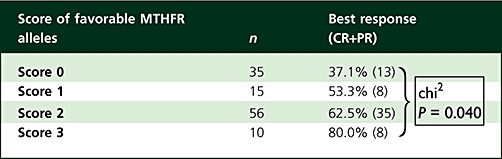
Both the MTHFR 677C→T and 1298A→C genotypes (Figure 1) were linked to clinical response (P= 0.042 and 0.004, respectively), with the rare allele linked to improved response. We defined a score of favourable MTHFR alleles corresponding to the sum of 677T and 1298C alleles. Importantly, tumour response continuously increased with the score of favourable MTHFR alleles (Table 3, P= 0.040). Response rates were 37.1, 53.3, 62.5 and 80.0% in patients bearing no, one, two or three favourable alleles, respectively. None of the other genotypes was linked to tumour response.
Figure 1.
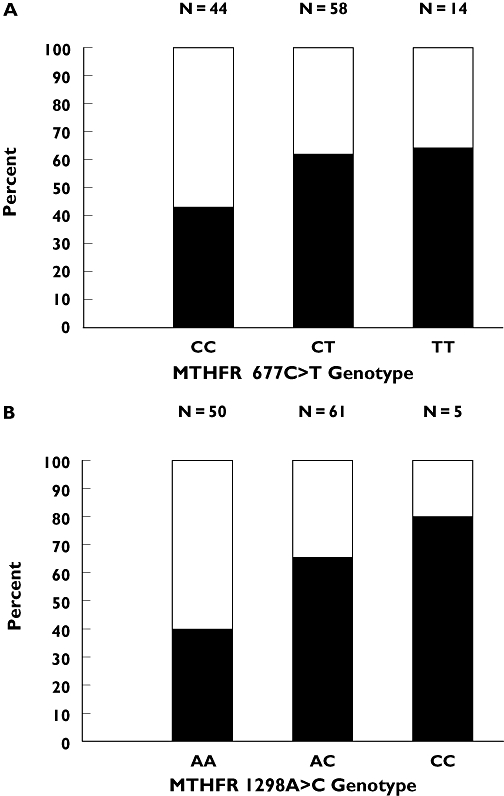
(a) Influence of methylenetetrahydrofolate reductase (MTHFR) 677C→T genotype on objective tumour response. Tumour response was 43.2, 62.1 and 64.3% in CC, CT and TT patients, respectively. χ2 tests: CC vs. CT vs. TT, P= 0.126; CC vs. CT+TT, P= 0.042. (b) Influence of MTHFR 1298A→C genotype on objective tumour response. Tumour response was 40.0, 65.6 and 80.0% in AA, AC and CC patients, respectively. χ2 tests: AA vs. AC vs. CC, P= 0.014; AA vs. AC+CC, P= 0.004. CR+PR ( ); SD+PD (
); SD+PD ( )
)
Table 3.
Influence of MTHFR genotype on tumour response
 |
Finally, a multivariate approach including cumulative FU and oxaliplatin doses revealed that both the score of favourable MTHFR alleles (P= 0.024) and the cumulative FU dose administered during the cycles of FOLFOX 7 therapy (P= 0.017) were significantly related to tumour response.
Impact of gene polymorphisms on survival
At time of analysis, 111 patients had progressed and median PFS was 7.5 months. PFS was influenced by the treatment arm (median 9.1 vs. 6.3 months in arms 1 and 2, respectively, P= 0.015) and by the cumulative oxaliplatin dose (P= 0.031). None of the analysed genotypes significantly influenced PFS, including the score of favourable MTHFR alleles. However, PFS plots according to GSTπ and XPD polymorphisms revealed an improved PFS in patients bearing GSTπ 105 Val/Val genotype or XPD 751 Lys-containing genotype (see Figures S1 and S2). Therefore, we built a score of favourable genotype taking into account both polymorphisms of GSTπ (105Val/Val corresponding to the favourable genotype) and XPD (751Lys/Lys or Lys/Gln corresponding to the favourable genotype). As shown in Figure 2, the score of favourable genotype tends to discriminate PFS with a median of 6.0, 7.6 and 9.8 months in patients bearing no, one or two favourable genotypes, respectively (P= 0.054). The score of favourable genotypes exhibited a similar pattern in both arm 1 and arm 2; however, adjustment of treatment arm did not improve its statistical significance (P= 0.065).
Figure 2.
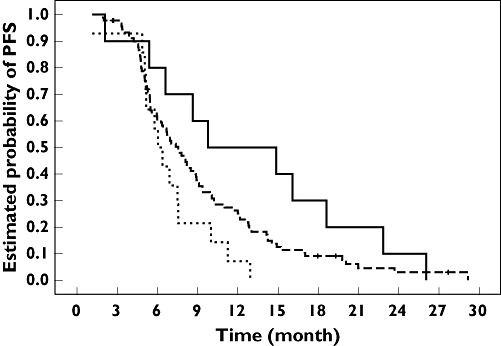
Progression-free survival (PFS) probability according to the score of favourable genotypes, including glutathione S-transferase (GST) π 105Ile→Val and XPD 751Lys→Gln polymorphisms. Favourable genotypes correspond to GSTπ 105 Val/Val, and XPD 751Lys/Lys or 751Lys/Gln. Median PFS was 6.0 months for score 0 (14 patients, 14 events), 7.6 months for score 1 (91 patients, 85 events) and 9.8 months for score 2 (10 patients, 10 events). Log rank test: P= 0.054. Score 0 ( ); Score 1 (
); Score 1 ( ); Score 2 (—)
); Score 2 (—)
At time of analysis, 79 patients had died and median overall survival was 22.8 months. Overall survival was not influenced by the treatment arm, nor by the cumulative oxaliplatin or FU dose. None of the analysed genotypes had a significant impact on overall survival, including the score of favourable MTHFR alleles and the score of favourable GSTπ-XPD alleles.
Discussion
The present pharmacogenetic study was conducted on a prospective cohort of 117 advanced CRC patients, all receiving FOLFOX therapy. This study included two treatment modalities differing according to a chemotherapy interruption strategy (six mFOLFOX 7 cycles ± simplified LV5FU2 regimen). Importantly, the number of mFOLFOX 7 cycles, the cumulative oxaliplatin dose and the cumulative continuous FU dose administered during the cycles of FOLFOX 7 therapy were similar in both treatment arms (Table 1). Also, toxicity and tumour response were not significantly different between treatment arms. The aim of the present study was to examine the role of various germinal polymorphisms on toxicity, response and PFS. The selected genes included both genes relevant for FU (TYMS, MTHFR, DPYD) and oxaliplatin (GSTπ along with DNA repair enzymes ERCC1, XPD and XRCC1).
Tolerance was good, with 19.3% of patients exhibiting grade 3 toxicity and 3.7% grade 4 toxicity. None of the genes related to oxaliplatin pharmacodynamics was linked to toxicity. Also, TYMS and MTHFR polymorphisms were not predictive of toxicity, and all patients exhibited the common IVS14+1G variant for DPYD gene. The absence of impact of MTHFR polymorphism on toxicity concords with recent results obtained on >600 cancer patients receiving FU monotherapy [15].
A clinical response was observed in 54.7% of patients. Among all analysed polymorphisms, only MTHFR gene polymorphisms were related to tumour response (Figure 1, Table 3). The gene coding for TYMS is carried by chromosome 18p, frequently prone to loss of heterozygosity in CRC, and Uchida et al.[16] have clearly reported that TYMS germinal polymorphism does not faithfully reflect tumoral polymorphism. The fact that TYMS polymorphisms were analysed on blood mononuclear cells may thus explain the lack of association with tumour response. The MTHFR enzyme is located at a major folate metabolic cross-roads, irreversibly converting 5–10 methylenetetrahydrofolate (CH2FH4) into 5-methyltetrahydrofolate (CH3FH4). FU acts mainly via fluorodeoxyuridine monophosphate (FdUMP), which inhibits TYMS and subsequent DNA synthesis through the formation of an inactive ternary complex between TYMS, FdUMP and the methyl donor CH2FH4. Experimental [17] and clinical [18] studies have shown that optimal FU cytotoxicity requires elevated CH2FH4 tumoral concentrations. Accordingly, clinical studies have demonstrated higher efficacy when FU is associated with folinic acid, a precursor of CH2FH4 [19, 20]. The MTHFR gene is subject to several polymorphisms, of which the 677C→T (Ala to Val at codon 222) [21] and 1298A→C (Glu to Ala at codon 428) [22] single nucleotide polymorphisms are the two most commonly linked with altered enzyme activity and increased homocysteine levels [23]. Even though the impact of MTFHR genotype on tumoral CH2FH4 concentrations has not been clearly established, deficient MTHFR genotypes may theoretically favour an increase in intracellular CH2FH4 concentrations. It can thus be hypothesized that tumors exhibiting deficient MTHFR variants (677T or 1298C) may be more sensitive to FU cytotoxicity than tumours bearing the common MTHFR variants (677C or 1298A).
The impact of MTHFR polymorphisms on FU efficacy has been previously reported in vitro and in vivo. An experimental study on 19 human cancer cell lines of various origins has reported a greater FU efficacy in cell lines homozygous for the 1298C variant compared with cells homozygous for the 1298A variant [12]. Also, Sohn et al.[24] demonstrated, on human cancer cell lines transfected with 677C or 677T MTHFR cDNA, significantly higher sensitivity to FU in the 677T cell lines relative to the 677C cell lines. The impact of MTHFR gene polymorphisms on treatment outcome in the clinical setting is more varied. Cohen et al.[25] were the first to describe a link between the 677C→T MTHFR polymorphism and tumour response to FU-based chemotherapy. In this study conducted on 43 metastatic CRC patients, all five 677TT patients responded to treatment, whereas response rate was approximately 50% in 677CC patients [25]. In a retrospective study from our group including 98 CRC patients with liver metastases receiving FUFOL, responsiveness was significantly linked to 677C→T genotype, with an increased response rate in 677TT tumours relative to 677CC (odds ratio = 1.88) [26]. In contrast, a study by Marcuello et al.[27] failed to show a link between MTHFR polymorphisms and clinical response in 94 metastatic CRC patients receiving FU associated with irinotecan or oxaliplatin. Also, Suh et al.[28] reported that MTHFR 677C→T polymorphism was not a significant predictor of response in 54 patients receiving FOLFOX treatment. More recently, Ruzzo and coworkers [29] have reported the absence of influence of 677C→T and 1298A→C MTHFR genotypes on objective response in 166 advanced CRC patients receiving first-line FOLFOX chemotherapy. Of note, the FOLFOX regimens differed among these studies according to the FU and oxaliplatin dose intensity. This could explain some of the discrepancies. In contrast, the present study clearly shows that both 677C→T and 1298A→C polymorphisms were significantly linked to FOLFOX responsiveness (Figure 1). Moreover, response rate continuously increased with the score of favourable MTHFR allele (Table 3), with only 37% of response in patients without any favourable allele, up to 80% in patients bearing three favourable alleles (i.e. homozygous for one variant and heterozygous for the second). None of the patients were homozygous for both 677T and 1298C. The divergence between present results and those of Ruzzo [29], both obtained on relatively large prospective cohorts of patients receiving FOLFOX therapy, is difficult to explain. Numerous studies on the role of MTHFR 677C→T and 1298C→A polymorphisms in CRC risk have clearly shown that physio-pathological consequences of a deficient MTHFR genotype are closely dependent on the folate status intake. It can thus be hypothesized that the influence of MTHFR polymorphisms on FU-based responsiveness in the Ruzzo study may have been blurred by a wide interpatient variability in folate status. Unfortunately, the Ruzzo study, like ours, did not provide information on the folate dietary status.
We previously reported in a previous study from our group [26] on 98 metastatic CRCs that MTHFR-deficient patients (1298CC genotype) had the shorter specific survival, suggesting a prognostic value of MTHFR polymorphism, probably independent of FU-based therapy. Also, a recent study by Zhang [30] reported a sex-specific influence of MTHFR 1298A→C polymorphism on overall survival in metastatic CRC women, with greater overall survival in MTHFR nondeficient women (1298AA genotype). Present data do not confirm the impact of MTHFR polymorphism on overall survival. A subgroup analysis in women did not reveal any prognostic value of 1298A→C genotype (49 events/52 women, data not shown). None of the analysed genotypes had a significant impact on overall survival.
Of note, PFS was related only to genes involved in oxaliplatin pharmacodynamics, with a tendency for a better outcome in patients bearing GSTπ 105 Val/Val genotype or XPD 751Lys-containing genotype (Figure 2). GSTπ is a phase II metabolic enzyme that inactivates platinum derivatives by adding a glutathione to its electrophile group. It has been shown that lymphocytic activity of GSTπ was significantly reduced in GSTπ 105 Val/Val patients compared with GSTπ 105 Ile/Ile patients [31]. Accordingly, Stoehlmacher et al.[32] reported in CRC patients receiving FOLFOX therapy that individuals bearing the GSTπ 105 Val/Val genotype had a better PFS and overall survival than patients bearing the GSTπ 105 Ile allele. XPD is a DNA repair enzyme of the ERCC2 group. The functional impact of XPD 751 Lys→Gln at the protein level is not clearly established. However, numerous clinical studies on CRC patients receiving oxaliplatin have reported a significantly improved PFS and/or survival in 751 Lys/Lys patients [29, 32–34]. The presently reported influence of GSTπ and XPD polymorphisms agrees well with the literature data.
Of particular interest is the comparison between the influence of cumulative drug doses and of pharmacogenetics on clinical end-points. On one hand, tumour response was significantly associated with elevated cumulative FU doses administered during the cycles of FOLFOX 7 therapy, suggesting that FU plays a preponderant role in tumour response. This observation closely concords with the pharmacogenetic results showing a major role of MTHFR polymorphisms in tumour response. Interestingly, the score of favourable MTHFR alleles and the cumulative FU dose were independent significant predictors of response. On the other hand, as previously reported [35], PFS was related to the cumulative oxaliplatin dose (and not to the cumulative FU dose) and the impact of the oxaliplatin dose was sustained by the influence of GSTπ and XPD polymorphisms on PFS. Even though PFS was linked to tumour response (data not shown), the influence of MTHFR genotypes on tumour response does not translate into an impact on PFS. Conversely, GSTπ and XPD genotypes do not influence tumour response while having an impact on PFS. These observations suggest that PFS is controlled by additional factors, including factors related to oxaliplatin pharmacodynamics.
The clinical usefulness of a routine pharmacogenetic approach to treatment adjustment is not yet established. Analysis of somatic K-Ras mutation is now applied in routine practice to identify CRC patients who will benefit from anti-EGFR therapy. However, more predictive factors are needed in this context since fluoropyrimidines still constitute the core of the treatment. Present data establish the role of MTHFR germinal polymorphism as a potential strong predictor of response to FOLFOX therapy and show for the first time that the response rate to FOLFOX increases continuously with the number of favourable MTHFR alleles. If confirmed in further studies, a possible application of this result may be to propose an alternative regimen containing no FU in the 30% of patients without a favourable MTHFR allele (i.e. score 0). MTHFR polymorphisms may provide a useful marker that presents the advantage of large-scale feasibility of routine analysis based on an easy-to-perform blood sample, combined with relatively low cost.
Competing interests
T.A. received a speaker's honorarium from Sanofi Aventis.
These results were presented in part at the 2008 annual meeting of the American Society of Clinical Oncology (ASCO). We thank the GERCOR group for their helpful collaboration regarding sample collection and data management, and the French Research Ministry (Programme Hospitalier de Recherche Clinique) for financial support.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Progression-free survival (PFS) probability according to the glutathione S-transferase (GST) π 105Ile→Val genotype. Median PFS was 7.36 months for Ile/Ile (56 patients, 53 events), 7.56 months for Ile/Val (49 patients, 46 events) and 9.76 months for Val/Val (12 patients, 12 events). Log rank test: P = 0.20 (P = 0.22 after adjustment on treatment arms)
Figure S2 Progression-free survival (PFS) probability according to the XPD 751Lys→Gln genotype. Median PFS was 6.38 months for Gln/Gln (16 patients, 16 events), 8.01 months for Lys/Gln (58 patients, 55 events) and 6.41 months for Lys/Lys (41 patients, 38 events). Log rank test: P = 0.33 (P = 0.29 after adjustment on treatment arms)
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
REFERENCES
- 1.De Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, Papamichael D, Le Bail N, Louvet C, Hendler D, De Braud F, Wilson C, Morvan F, Bonetti A. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–47. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 2.Douillard JY, Cunningham D, Roth AD, Navarro M, James RD, Karasek P, Jandik P, Iveson T, Carmichael J, Alakl M, Gruia G, Awad L, Rougier P. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355:1041–7. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 3.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 5.Tournigand C, André T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, De Gramont A. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–37. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 6.Van Cutsem E, Nowacki M, Lang I, Cascinu S, Shchepotin I, Maurel J, Rougier P, Cunningham D, Nippgen J, Köhne C. Randomized phase III study of irinotecan and 5FU/FA with or without cetuximab in the first-line treatment of patients with metastatic colorectal cancer: The CRYSTAL trial. J Clin Oncol. 2007;25:164s. abst 4000. [Google Scholar]
- 7.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 8.Moosmann N, Heinemann V. Cetuximab plus XELIRI or XELOX for first-line therapy of metastatic colorectal cancer. Clin Colorectal Cancer. 2008;7:110–7. doi: 10.3816/CCC.2008.n.015. [DOI] [PubMed] [Google Scholar]
- 9.Etienne-Grimaldi MC, Formento JL, Francoual M, François E, Formento P, Renée N, Laurent-Puig P, Chazal M, Benchimol D, Delpero JR, Letoublon C, Pezet D, Seitz JF, Milano G. K-Ras mutations and treatment outcome in colorectal cancer patients receiving exclusive fluoropyrimidine therapy. Clin Cancer Res. 2008;14:4830–5. doi: 10.1158/1078-0432.CCR-07-4906. [DOI] [PubMed] [Google Scholar]
- 10.Chibaudel B, Maindrault-Goebel F, Lledo G, Mineur L, André T, Bennamoun M, Mabro M, Artru P, Carola E, Flesch M, Dupuis O, Colin P, Larsen AK, Afchain P, Tournigand C, Louvet C, de Gramont A. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol. 2009 doi: 10.1200/JCO.2009.23.4344. [online] doi: 10.1200/JCO.2009.23.4344. [DOI] [PubMed] [Google Scholar]
- 11.Largillier R, Etienne-Grimaldi MC, Formento JL, Ciccolini J, Nebbia JF, Ginot A, Francoual M, Renée N, Ferrero JM, Foa C, Namer M, Lacarelle B, Milano G. Pharmacogenetics of capecitabine in advanced breast cancer patients. Clin Cancer Res. 2006;12:5496–502. doi: 10.1158/1078-0432.CCR-06-0320. [DOI] [PubMed] [Google Scholar]
- 12.Etienne MC, Ilc K, Formento JL, Laurent-Puig P, Formento P, Chéradame S, Fischel JL, Milano G. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms: relationships with 5-fluorouracil sensitivity. Br J Cancer. 2004;90:526–34. doi: 10.1038/sj.bjc.6601523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishii T, Matsuse T, Teramoto S, Matsui H, Miyao M, Hosoi T, Takahashi H, Fukuchi Y, Ouchi Y. Glutathione S-transferase P1 (GSTP1) polymorphism in patients with chronic obstructive pulmonary disease. Thorax. 1999;54:693–6. doi: 10.1136/thx.54.8.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, O'Day SJ, Yang D, Boasberg P, Milford R, Kristedja T, Groshen S, Weber J. Impact of gene polymorphisms on clinical outcome for stage IV melanoma patients treated with biochemotherapy: an exploratory study. Clin Cancer Res. 2005;11:1237–46. [PubMed] [Google Scholar]
- 15.Schwab M, Zanger UM, Marx C, Schaeffeler E, Klein K, Dippon J, Kerb R, Blievernicht J, Fischer J, Hofmann U, Bokemeyer C, Eichelbaum M German 5-FU Toxicity Study Group. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26:2131–8. doi: 10.1200/JCO.2006.10.4182. [DOI] [PubMed] [Google Scholar]
- 16.Uchida K, Hayashi K, Kawakami K, Schneider S, Yochim JM, Kuramochi H, Takasaki K, Danenberg KD, Danenberg PV. Loss of heterozygosity at the thymidylate synthase (TS) locus on chromosome 18 affects tumor response and survival in individuals heterozygous for a 28-bp polymorphism in the TS gene. Clin Cancer Res. 2004;10:433–9. doi: 10.1158/1078-0432.ccr-0200-03. [DOI] [PubMed] [Google Scholar]
- 17.Rustum YM, Trave F, Zakrzewski SF, Petrelli N, Herrera L, Mittelman A, Arbuck SG, Creaven PJ. Biochemical and pharmacologic basis for potentiation of 5-fluorouracil action by leucovorin. NCI Monogr. 1987;5:165–70. [PubMed] [Google Scholar]
- 18.Chéradame S, Etienne MC, Formento P, Schneider M, Dassonville O, Demard F, Milano G. Tumoral-reduced folates and clinical resistance to fluorouracil-based treatment in head and neck cancer patients. J Clin Oncol. 1997;15:2604–10. doi: 10.1200/JCO.1997.15.7.2604. [DOI] [PubMed] [Google Scholar]
- 19.Petrelli N, Herrera L, Rustum Y, Burke P, Creaven P, Stulc J, Emrich LJ, Mittelman A. A prospective randomized trial of 5-fluorouracil vs. 5-flurouracil and high-dose leucovorin vs. 5-fluorouracil and methotrexate in previously untreated patients with advanced colorectal carcinoma. J Clin Oncol. 1987;5:1559–65. doi: 10.1200/JCO.1987.5.10.1559. [DOI] [PubMed] [Google Scholar]
- 20.Poon MA, O'Connell MJ, Moertel CG, Wieand HS, Cullinan SA, Everson LK, Krook JE, Mailliard JA, Laurie JA, Tschetter LK, Wiesenfeld M. Biochemical modulation of fluorouracil: evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;7:1407–18. doi: 10.1200/JCO.1989.7.10.1407. [DOI] [PubMed] [Google Scholar]
- 21.Rozen R. Molecular genetics of methylenetetrahydrofolate reductase deficiency. J Inherti Metab Dis. 1996;19:589–94. doi: 10.1007/BF01799831. [DOI] [PubMed] [Google Scholar]
- 22.Weisberg I, Tran P, Christensen B, Sibani S, Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab. 1998;64:169–72. doi: 10.1006/mgme.1998.2714. [DOI] [PubMed] [Google Scholar]
- 23.DeVos L, Chanson A, Liu Z, Ciappio ED, Parnell LD, Mason JB, Tucker KL, Crott JW. Associations between single nucleotide polymorphisms in folate uptake and metabolizing genes with blood folates, homocysteine, and DNA uracil concentrations. Am J Clin Nut. 2008;88:1149–58. doi: 10.1093/ajcn/88.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sohn KJ, Croxford R, Yates Z, Lucock M, Kim YN. Effect of the methylenetetrahydrofolate reductase C677T polymorphism on chemosensitivity of colon and breast cancer cells to 5-fluorouracil and methotrexate. J Natl Cancer Inst. 2004;96:134–44. doi: 10.1093/jnci/djh015. [DOI] [PubMed] [Google Scholar]
- 25.Cohen V, Panet-Raymond V, Sabbaghian N, Morin I, Batist G, Rozen R. Methylenetetrahydrofolate reductase polymorphism in advanced colorectal cancer: a novel genomic predictor of clinical response to fluoropyrimidine-based chemotherapy. Clin Cancer Res. 2003;9:1611–5. [PubMed] [Google Scholar]
- 26.Etienne MC, Formento JL, Chazal M, Francoual M, Magné N, Formento P, Bourgeon A, Seitz JF, Delpero JR, Letoublon C, Pezet D, Milano G. Methylenetetrahydrofolate reductase gene polymorphisms and response to fluorouracil-based treatment in advanced colorectal cancer patients. Pharmacogenetics. 2004;14:785–92. doi: 10.1097/00008571-200412000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Marcuello E, Altes A, Menoyo A, Rio ED, Baiget M. Methylenetetrahydrofolate reductase gene polymorphisms: genomic predictors of clinical response to fluoropyrimidine-based chemotherapy? Cancer Chemother Pharmacol. 2006;57:835–40. doi: 10.1007/s00280-005-0089-1. [DOI] [PubMed] [Google Scholar]
- 28.Suh KW, Kim JH, Kim Y, Kim YB, Lee C, Choi S. Which gene is a dominant predictor of response during FOLFOX chemotherapy for the treatment of metastatic colorectal cancer, the MTHFR or XRCC1 gene? Ann Surg Oncol. 2006;13:1379–85. doi: 10.1245/s10434-006-9112-y. [DOI] [PubMed] [Google Scholar]
- 29.Ruzzo A, Graziano F, Loupakis F, Rulli E, Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P, Bisonni R, Masi G, Schiavon G, Giordani P, Giustini L, Falcone A, Tonini G, Silva R, Mattioli R, Floriani I, Magnani M. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. J Clin Oncol. 2007;25:1247–54. doi: 10.1200/JCO.2006.08.1844. [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Press OA, Haiman CA, Yang DY, Gordon MA, Fazzone W, El-Khoueiry A, Iqbal S, Sherrod AE, Lurje G, Lenz HJ. Association of methylenetetrahydrofolate reductase gene polymorphisms and sex-specific survival in patients with metastatic colon cancer. J Clin Oncol. 2007;25:3726–31. doi: 10.1200/JCO.2007.11.4710. [DOI] [PubMed] [Google Scholar]
- 31.Dusinská M, Ficek A, Horská A, Raslová K, Petrovská H, Vallová B, Drlicková M, Wood SG, Stupáková A, Gasparovic J, Bobek P, Nagyová A, Kováciková Z, Blazícek P, Liegebel U, Collins AR. Glutathione S-transferase polymorphisms influence the level of oxidative DNA damage and anti-oxidant protection in humans. Mutat Res. 2001;482:47–55. doi: 10.1016/s0027-5107(01)00209-3. [DOI] [PubMed] [Google Scholar]
- 32.Stoehlmacher J, Park DJ, Zhang W, Yang D, Groshen S, Zahedy S, Lenz HJ. A multivariate analysis of genomic polymorphisms: prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br J Cancer. 2004;91:344–54. doi: 10.1038/sj.bjc.6601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park DJ, Stoehlmacher J, Zhang W, Tsao-Wei DD, Groshen S, Lenz HJ. A Xeroderma pigmentosum group D gene polymorphism predicts clinical outcome to platinum-based chemotherapy in patients with advanced colorectal cancer. Cancer Res. 2001;61:8654–8. [PubMed] [Google Scholar]
- 34.Paré L, Marcuello E, Altés A, Del Río E, Sedano L, Salazar J, Cortés A, Barnadas A, Baiget M. Pharmacogenetic prediction of clinical outcome in advanced colorectal cancer patients receiving oxaliplatin/5-fluorouracil as first-line chemotherapy. Br J Cancer. 2008;99:1050–5. doi: 10.1038/sj.bjc.6604671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maindrault-Goebel F, De Gramont A, Louvet C, André T, Carola E, Gilles V, Lotz JP, Tournigand C, Mabro M, Molitor JL, Artru P, Izrael V, Krulik M. Evaluation of dose-intensity in bimonthly leucovorin and 5 fluorouracil continuous infusion regimens (FOLFOX) in pretreated metastatic colorectal cancer. Ann Oncol. 2000;11:1–7. doi: 10.1023/a:1026520812351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.