

Abstract
The goal of this study was to evaluate a new approach that can be applied for labeling biomolecules with 211At. Many astatine compounds that have been synthesized are unstable in vivo, providing motivation for seeking different 211At labeling strategies. The approach evaluated in this study was to attach astatide anions to soft metal cations, which are also complexed by a bifunctional ligand. Ultimately, this complex could in principle be subsequently conjugated to a biomolecule with the proper selection of ligand functionality. We report here the attachment of 211At− and *I− (*I = 131I or 125I) anions to the soft metal cations Rh(III) and Ir(III), which are complexed by the 1,5,9,13-tetrathiacyclohexadecane-3,11-diol (16aneS4-diol) ligand. Radioactive *I− anions were used for preliminary studies directed at the optimization of reaction conditions and to provide a baseline for comparison of results with 211At. Four complexes Rh[16aneS4-diol]*I/211At and Ir[16aneS4-diol]*I/211 At were synthesized in high yield in a one-step procedure, and the products were characterized mainly by paper electrophoresis and reversed-phase HPLC. The influences of time and temperature of heating and concentrations of metal cations and sulfur ligand 16aneS4-diol, as well as pH on the reaction yields were determined. Yields of about 80% were obtained when the quantities of Rh(III) or Ir(III) cations and 16aneS4-diol ligand in the solutions were 62.5 nmol and 250 nmol, respectively, and the pH ranged 3.0–4.0. Syntheses required heating for 1–1.5 h at 75–80 °C. The influence of microwave heating on the time and completeness of the complexation reaction was evaluated and compared with the conventional method of heating in an oil bath. Microwave synthesis accelerates reactions significantly. With microwave heating, yields of about 75% for Rh[16aneS4-diol]131I and Ir[16aneS4-diol]131I complexes were obtained after only 20 min exposure of the reaction mixtures to microwave radiation. In conclusion, this study has shown that it is possible to attach an astatide anion to soft metal cations in a simple and fast one-step procedure, with high yields. These complexes will be evaluated as reagents for labeling biomolecules.
INTRODUCTION
In the field of targeted radiotherapy, the selection of radionuclide is related to the type of treated disease. Currently, many radionuclides are under intensive investigation for therapeutic applications, particularly the Auger emitter 67Ga (t1/2 = 3.3 d), 111In (t1/2 = 2.8 d), α-particle emitters 211At (t1/2 = 7.2 h), 212Bi (t1/2 = 1 h), 213Bi (t1/2 = 45.6 m), and β−-particle emitters 90Y (t1/2 = 64 h), 188Re (t1/2 = 16.7 h), and 177Lu (t1/2 = 6.7 d) (1). An important factor is the nature of the cytotoxic radionuclide, mainly its physical parameters such as half-life and the nature of its associated emissions. Solid tumors generally have been pursued with β− emitters including 90Y and 131I, because their β− particles have a tissue range of several millimeters. The effective tissue range of β− particles is not optimal for treatment of tumors as small clusters of cells or single cells, and micrometastases, because much of the decay energy is deposited outside the boundary of the tumor. Treatment of these diseases might be more effective with α-emitters, which combine short-range and high linear energy transfer, a combination that results in the high relative biological effect and cytotoxicity (2). Thus, α particles are able to make double-strand breaks in DNA, lesions that have a relatively high probability of leading to a “kill effect” on tumor cells (3). The shorter path length (40–100 μm) of α particles also should help limit radiotoxicity to neighboring normal tissue. For these reasons; considerable effort has been placed recently on the development of the α-emitters 223Ra (t1/2 = 11.4 d), 225Ac (t1/2 = 10 d), generator-obtained 212Bi, 213Bi, as well as the cyclotron-produced 211At. The latter may perhaps be the most promising candidate for targeted radiotherapy, because its 7.2 h half-life allows sufficient time for performing its transportation, synthetic chemistry and multistep labeling, quality control, and clinical application without the problematic characteristic of having relatively long-lived α-particle-emitting daughters. Besides, each event of astatine decay is accompanied by emission of high-energy α particles with average energy of 6.4 MeV, corresponding to a mean range in human tissue of 65 μm (4). Therefore, the ionization occurs within a small volume, and when astatine is localized within a tumor, the surrounding tissue will not suffer from its radiation. Additionally, its electron capture decay branch gives rise to Po K X-rays making 211At easy to follow with standard nuclear detection devices including γ cameras for imaging (5). Dosimetry calculation and preclinical testing of compounds labeled with 211At have indicated a significant therapeutic potential, at least in certain settings, relative to the use of radionuclides that emit particles with low linear energy transfer (6-12).
Because astatine is the heaviest member of the halogen group, it has generally been recognized as a nonmetal. It is also similar to iodine with regard to its biochemical properties. Thus, standard radioiodination protocols had been commonly adapted for preliminary 211At labeling of biomolecules. Unfortunately, proteins labeled by direct electrophilic astatination were unstable due to rapid loss of 211At in both in vitro and in vivo conditions (13). The reason was probably the unspecific binding of astatine to sulfur atoms in peptides instead of attaching to tyrosine residue, like in the case of radioiodine (14). This problem was circumvented later by using two-step procedures, where 211At first was electrophilically incorporated into an aryl compound (e.g., benzoates) and, in the second step, coupled to a protein (15-19). Unfortunately, biomolecules labeled by this method have been found to not always be stable to in vivo deastatination (20). One of the reasons can be the nature of labeled carrier molecule, especially its rate and mode of metabolism (21), but this behavior also probably reflects the weaker aryl carbon–halogen bond for astatine when compared to that with iodine (bond strength for astatine is ~49 kcal/mol and for iodine is ~62 kcal/mol) (22, 23). This fact prompted investigations directed at identifying other labeling approaches that can yield rapid astatination and high stability. For example, the boron–astatine bond should be stronger than the carbon–astatine bond based on the general trend for boron–halogen bond energies (24). Therefore, boron cage moieties have been studied as pendant groups for radiolabeling proteins and peptides with radioiodine (25, 26) and with 211At (27, 28). Studies with nido-carborane derivatives did not provide adequate stability of astatination of these compounds (21), whereas anionic monocarboranes were not reactive enough to be useful for radio-halogenation (29). However, recent experiments with closo-decaborate(2-) derivatives showed good stability and fast labeling of biomolecules with high yield (30), and studies on the optimization of the closo-decaborate(2-) conjugates for protein labeling are underway.
Herein, we have proposed an alternative method of labeling molecules with 211At to solve stability problems of formed conjugates. Instead of using an electrophilic reaction, we decided to attach astatide anions to soft metal cations, which are also complexed by a bifunctional ligand. Ultimately, this complex could in principle be subsequently conjugated to a biomolecule with proper selection of ligand functionality. It can be expected that 211At−, similar to I− anions, should demonstrate soft ligand properties and form strong complexes with soft metal cations, like Hg(II), Pt(II), Rh(III), and Ir(III), according to the hard and soft acids and bases theory (HSAB) (31). On the basis of the generally observed trend that stability constant values for soft metal cation–halogen anion complexes increase in the halogen group, we can expect that a complex with astatide should be stronger than one with iodide. Previously, we reported that 211At− formed strong complexes with Hg(II) cations, much stronger than I− (32). In the present paper, we describe the results of our studies on attaching 211At− to complexes of Rh(III) or Ir(III) with the thioether ligand 1,5,9,13-tetrathiacyclohexadecane-3,11-diol (16aneS4-diol). Rh(III) and Ir(III) cations were chosen because they are moderately soft metal cations, and we hypothesize that they will form strong bonds with the soft astatide anion. In addition, the very high kinetic inertness of low-spin Rh(III) and Ir(III) d6 complexes is particularly well-suited to the formation of a stable conjugate. The 16aneS4-diol ligand was selected for these initial model compound studies based on literature reports that it forms stable complexes with Rh(III) (33). Additionally, this macrocyclic tetra-thioether with diol functionality can be easily modified to create a bifunctional chelate ligand (34).
EXPERIMENTAL PROCEDURES
Reagents and Radioactivity
The macrocyclic crown thioether 1,5,9,13-tetrathiacyclohexadecane-3,11-diol (16aneS4-diol) cis/trans mixture (Figure 1, panel A) was purchased from Aldrich Chemical Co. and used without further purification. Rhodium(III) nitrate hydrate Rh(NO3)3 •3H2O and iridium(III) chloride hydrate IrCl3 •3H2O were obtained from Alfa Aesar, a Johnson Matthey Company. Solvents for HPLC analysis were obtained as HPLC grade and degassed before use by ultrasonification for 15–20 min. All other chemicals were of pure reagent grade and used as received unless otherwise specified. The ion exchange resins Dowex 1 × 8 and Dowex 50W × 8 of different mesh sizes were purchased at Sigma-Aldrich Company. The 131I and 125I in the form of Na*I (*I = 131I or 125I) in 0.1 M NaOH solution were supplied from Perkin-Elmer Life and Analytical Sciences (formerly NEN/Dupont, Billerica, MA) and from the Isotope Production Centre Polatom in Swierk (Poland) as high concentration/high specific activity radioiodide. 211At was produced on the CS-30 cyclotron at the Duke University Medical Center. The targets were prepared from pure metallic bismuth and were irradiated with 28 MeV α particles using the 209Bi(α,2n)211At reaction. Separation of 211At from the bismuth target was performed with a dry distillation method described in ref (35). Finally, the 211At activity was washed out from the cooled trap by MeOH with Na2SO3 (10−4 M) addition to obtain 211At in astatide form. All radioactive materials were handled according to approved protocols at the Institute of Nuclear Chemistry and Technology and at Duke University.
Figure 1.
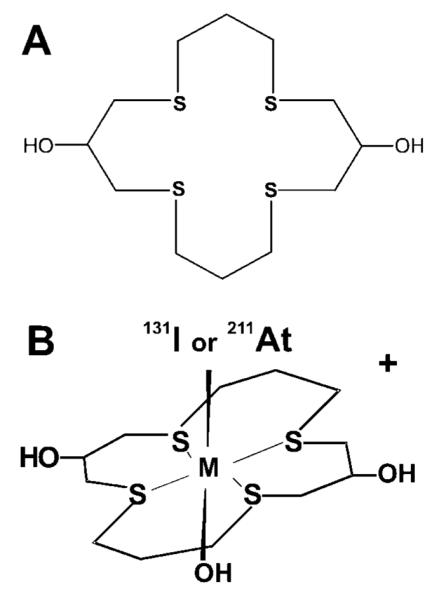
Structure of (A) macrocyclic thioether ligand 1,5,9,13-tetrathiacyclohexadecane-3,11-diol (16aneS4-diol) and (B) cationic complexes trans-M[16aneS4-diol]*I/211At (M = Rh(III) or Ir(III)).
Measurements of 131I, 125I, and 211At at Duke University were accomplished in a dose calibrator on a Capintec CRC-7 Radioisotope (Ramsey, NJ). The 133Xe setting was used with a multiplication factor of 2.3, to count 211At on that instrument. The 131I radioactivity at the Institute of Nuclear Chemistry and Technology was measured in a NaI(Tl) well counter (Polon Alfa, Warszawa).
Preparation of Rh[16aneS4-diol]*I/211At and Ir[16aneS4-diol]*I/211At Complexes and Optimization of the Reaction Conditions
Rh[16aneS4-diol]*I/211At and Ir[16aneS4-diol]*I/211At complexes were prepared by addition 20 μL of *I− or 211At− activity (6.5–70 MBq for *I and 4.5–30 MBq for 211At) to the mixture of 125 μL Rh(III) or Ir(III) and 25 μL 16aneS4-diol in water–ethanol solution. The quantities of metal cations and sulfur ligand were varied during the optimization reactions over the ranges 0.125–125 nmol and 18.75–250 nmol, respectively. After adjusting the pH to 4.0, by dropwise addition of 0.001–0.1 M HNO3 or NaOH, the solutions were heated in sealed Eppendorf tubes or glass vials capped with Teflon-coated septa for about 2 h at 80 °C in an oil bath. Control experiments were also performed in which 125 μL of aqueous Rh(III) or Ir(III) solution was mixed with 25 μL of ethanol without sulfur ligand, and 20 μL of *I− or 211At− solution was added (blank 1). Blank 2 was prepared by mixing 125 μL of water without metal cation and 25 μL of 16aneS4-diol ligand in ethanol and 20 μL of *I− or 211At−. All control experiments were adjusted to pH 4.0 and heated under the same conditions as the main samples for the complexes. Solutions were analyzed mainly by paper electrophoresis and HPLC methods, but also by sorption on ion exchange resins.
The optimization of reaction conditions was focused on increasing the yield of synthesis, ideally, in a reaction time suitable for use with 7.2 h half-life 211At. The influences of time and temperature of heating, concentrations of metal cations, sulfur ligand 16aneS4-diol, and pH of the solutions on the reaction yields were determined. Optimization of each parameter was repeated 2 times for astatine complexes and 3 times for *I complexes in separate experiments. Each set of conditions during 1 experiment was replicated 2–3 times. First, optimization of reaction conditions was performed with *I radionuclides and later repeated with 211At. The reaction volume in all cases was kept at 170 μL.
The formation of complexes was studied by heating solutions for different time periods (30–120 min) and over a wide range of temperatures (40–90 °C). For this experiment, the reaction mixtures contained 125 nmol of metal cation and 250 nmol of sulfur ligand, and the reaction was performed at pH 4.0.
The effect of metal cation concentration on complex formation yield was measured in solutions at pH 4.0 containing 250 nmol of sulfur ligand with the quantity of metal cation being varied from 0.125 to 125 nmol. Solutions were heated for 1–1.5 h at 75–80 °C. The influence of 16aneS4-diol concentrations on reaction yield was evaluated by keeping the quantity of metal cation at 62.5 nmol, while varying the quantity of sulfur ligand in the range 18.75–250 nmol. The reactions were carried out at 75–80 °C for 1–1.5 h and at pH 4.0.
The effect of pH on the yield of the radiohalogenation reactions was also determined. For these studies, the reaction mixtures contained 62.5 nmol of metal cation and 250 nmol of 16aneS4-diol. The pH was varied from 2 to 8.5 by addition of 0.001–0.1 M HNO3 or NaOH, and the reaction mixtures were heated for 1–1.5 h at 75–80 °C. Buffers were not used to stabilize the pH, because studies in the literature indicating that several buffers (e.g., phosphate, carbonate) interfered with rhodium complexation (33, 36).
The influence of microwave heating on the yield of Rh[16aneS4-diol]131I and Ir[16aneS4-diol]131I complexes was determined under the best conditions found during the optimization experiments. The quantities of Rh(III) or Ir(III) cations and sulfur ligand in the solutions were kept at 125 nmol and 250 nmol, respectively, and the pH of the reaction mixture was between 3.0 and 4.0. A UniClever II microwave unit (Plazmatronika, Poland) was used for these studies. This device provides control of the processing time, radiation power, temperature, and pressure in a hermetically sealed autoclave. Prepared samples in glass vials were put into the UniClever apparatus, and microwave heating was performed for 5–35 min at 50% of power mode (corresponds to the 300 W absolute power). During this time, the temperature of the samples was maintained between 75 and 80 °C.
Radiochemical Analyses of Synthesized Complexes
The charge of the radioactive products was determined using paper electrophoresis and ion exchange methods. Electromigration was measured utilizing Sigma-Aldrich and Bio-Rad units on glass fiber Whatman Paper Chromedia GF83 (W. & R. Balston, Ltd., England) and cellulose fiber Whatman 3MM (Whatman International Ltd., England) at the potential gradient equal to 10 V/cm for 25–60 min. The electrolyte was 0.02 M phosphate buffer at pH 4.0 and 7.4. An aliquot of the radioactive reaction mixture was spotted on the center of paper strip, and the power supply was switched on. The distribution of radioactivity on the paper strips was measured by cutting the paper into 1 cm pieces and counting them in a NaI(Tl) well counter. Alternatively, a radiochromatographic strip scanner (BioScan System 200 Imaging Scanner) was utilized.
Dowex 1 × 8 and Dowex 50W × 8 ion exchange resins were rinsed with 250–500 mL 1 M NaNO3 and then washed with deionized water. The resins were dried at 60–70 °C and stored in a desiccator. The distribution of radioactive compounds between the aqueous phase, and the ion exchange resin was analyzed by the batch method. Between 50 and 150 mg of ion exchange resin was added to Eppendorf tubes (two replicates for each sample during one experiment) containing 1.5 mL of 0.05 M NaNO3 at pH 4.0 to which 5–20 μL of the radioactive compound was added. Tubes were shaken for 15–20 min, centrifuged, phases separated, and the radioactivity in the resin and solution counted on either a NaI(Tl) well-type counter or an automated gamma counter (1282 LKB Pharmacia, Turku, Finland).
Additionally, radiochemical identification of the reaction products was accomplished using reversed-phase HPLC with a Waters XTerra C18 column (5 μm, 250 × 4.5 mm) on a Beckman System Gold device equipped with both UV and radiometric (Gamma Detector 170) detectors. The gradient elution system utilized mobile phase A (deionized H2O) and mobile phase B (100% acetonitrile) and flow rate of 1 mL/min, starting with 95% A/5% B for 5 min; then, the gradient was increased to 100% B over the next 30 min and then held at 100% B for 5 min, after which gradient parameters returned to the initial conditions during the next 5 min. The HPLC analysis took about 45 min. Both mobile phases contained 0.1% TFA.
RESULTS AND DISCUSSION
Astatine-211 is one of only a few α-emitters that have properties suitable for application to targeted radiotherapy of cancer, especially of micrometastatic disease, cancer resistant to other forms of radiation (e.g., melanoma), and ovarian carcinoma (12, 37). Although a large number of cancer cell selective monoclonal antibodies (mAbs) and other cancer cell targeting agents suitable for targeted radiotherapy have been developed, using them in the treatment of cancer is only possible when the radionuclide is stably coupled with the carrier molecule. In the case of highly toxic α-emitters, only conjugates that are stable toward in vivo release of the α-particle-emitting radionuclide can be used in targeted radiotherapy.
The stability of astatinated biomolecules has been a major impediment to the development of 211At-labeled radiotherapeutics. Direct electrophilic astatination of proteins and peptides was unsuccessful. The yield of the reaction was low, and the labeled molecules were unstable to in vivo deastatination (13). More stable astatinated proteins have been prepared by acylation with a variety of astatobenzoic acid derivatives prepared from trialkylstannyl precursors (15, 16), but the problem with stability appeared when smaller, more rapidly metabolized molecules and mAb fragments (Fab’) were used (20, 38). The conjugates undergo rapidly deastatination, even with molecules for which in vitro studies indicated quite good stability in serum (20). Therefore, without a good chemical method of attaching 211At to these potential cancer targeting agents that will provide high in vivo stability of formed conjugate, the application of this radionuclide in targeted radiotherapy will be limited.
The aim of this work was focused on finding a new stable labeled precursor for use in labeling of biomolecules with 211At. The original idea is to attach astatide anions to soft metal cations, which are complexed by bifunctional ligand bearing a functionality that would facilitate attaching this complex to a biomolecule. We believe that the proposed combination of high kinetic inertness of Rh(III) and Ir(III) compounds with the formation of strong complexes between soft metal cations and soft anions will result in high stability of formed metal–astatide bond.
Results from literature studies show that the 16aneS4-diol ligand reacts with RhCl3 to form a cationic Rh[16aneS4-diol]Cl2 complex with a trans-Rh(III)Cl2 core. In this complex, the sulfur donor atoms of the macrocyclic ligand occupy four equatorial positions, whereas the Cl− anions are in the axial positions (33). Therefore, we hypothesized that analogous complexes could be formed in which one axial position is occupied by either the *I− or 211At− anion. It should be noted that in our studies we used Rh(NO3)3 for synthesis instead of RhCl3, as was used previously (33). Therefore, due to the weak complexing properties of NO3− anions, we believe that the second axial position will be occupied by a OH− group (Figure 1, panel B). We prepared the well-defined macroscopic nonradioactive Rh[16aneS4-diol]Cl2 complex according to a literature procedure (33). Following the same scheme, the Rh[16aneS4-diol]I complex with cold iodine was prepared. The electrophoretic migration of the yellow Rh[16aneS4-diol]Cl2 complex was compared with the orange Rh[16aneS4-diol]I complex and radioactive Rh[16aneS4-diol]131I. In all cases, the migration toward cathode was identical. Also, cospotting Rh[16aneS4-diol]131I with nonradioactive Rh[16aneS4-diol]Cl2 and Rh-[16aneS4-diol]I gave identical electrophoretic behavior. The properties of three complexes were also studied by reverse-phase HPLC using the gradient conditions mentioned above. The UV detection was performed at 275 nm for nonradioactive Rh[16aneS4-diol]Cl2 and Rh[16aneS4-diol]I complexes, and the radioactive Rh[16aneS4-diol]131I complex was detected by the radiometric detector. The retention times for both nonradioactive Rh[16aneS4-diol]Cl2 and Rh[16aneS4-diol]I complexes were identical and comparable to the radioactive Rh[16aneS4-diol]131I, with peaks appearing between 15 and 17 min. From these results, we assumed that the structures of cold Rh[16aneS4-diol]I and radioactive Rh[16aneS4-diol]131I were identical to the structure of Rh[16aneS4-diol]Cl2 presented in ref (33).
Paper electrophoresis and ion exchange studies confirmed the predicted cationic character of the synthesized complexes. For example, the distribution of *I radioactivity on the paper strip for Rh[16aneS4-diol]*I complex and control samples is presented in Figure 2. It is shown that, in the heated solution contained Rh(III) cations, sulfur ligand 16aneS4-diol and *I−, a cationic complex was formed, because most of the radioactivity (80−85%) migrated toward the cathode. During the electrophoretic studies, we also noticed that the percentage of radioactivity distributed on both fiber strips was similar; however, the migration of the complex on cellulose fiber was slower than on the glass one at the same applied voltage. In the case of Whatman GF83, migration of the cationic complex was 4.5–5.0 cm toward the cathode during 25 min, whereas for Whatman 3MM, fiber migration was 3.7–4.0 cm toward the cathode during 1 h. The last result is comparable to the migration reported for the cationic 105Rh[16aneS4-diol]Cl complex also on the cellulose fiber (Whatman #1) (34). The differences in migration range and time are probably due to different components and structures of both fibers. Nevertheless, the migration time did not influence the final results of the overall percentage distribution of radioactivity on the fibers, but for the electrophoretic studies, glass fiber was mainly used, because of the shorter analysis time, an advantage that was of particular importance for the characterization of the astatine complexes. The pH of the sodium phosphate buffer (4.0 or 7.4) used as the electrolyte did not influence the migration range and time for all four complexes, which were found to remain positively charged at the higher pH.
Figure 2.
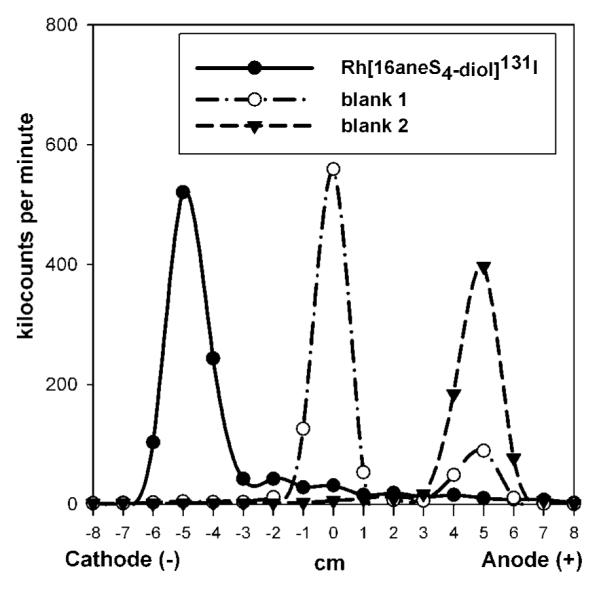
Electrophoretic analysis of heated mixtures: (Rh[16aneS4-diol]*I) contained Rh(III) cations, 16aneS4-diol ligand, and *I−; blank 1 contained Rh(III) and *I− without sulfur ligand; blank 2 contained sulfur ligand and *I− without metal cations. Paper electrophoresis was performed on glass fiber Whatman Paper Chromedia GF83 using phosphate buffer (pH 4.0 or 7.4) at 10 V/cm for 25 min.
Confirmation that the synthesized complex Rh[16aneS4-diol]*I has cationic character was also obtained from an experiment determining its adsorption on a cation exchange resin. In the case of the complex, most of the *I radioactivity was adsorbed on the cationic resin, with only 15–20% remaining in solution; this was nearly completely adsorbed on the anion exchange resin. This suggests that 15–20% of free iodide remains uncomplexed in solution at the end of the synthesis reaction.
Studies on control samples indicated that, in the heated solution of 16aneS4-diol with *I− but without the metal cation (blank 2), all the radioactivity migrated toward the anode, consistent with uncomplexed iodide (Figure 2). This indicates that there are no interactions of the halogen anion with 16aneS4-diol ligand in the absence of the metal. In the case of blank 1, where iodide was heated with Rh(III) cations in the absence of the 16aneS4-diol ligand, most of the radioactivity stayed at the origin and only 10–35% moved toward the anode like iodide (Figure 2). Radioactivity from this compound also was not adsorbed on Dowex 50W × 8, but stayed in solution, even after a few hours of shaking. Only 10–35% was adsorbed on Dowex 1 × 8, suggesting the presence of uncomplexed iodide. We speculate that, in blank 1, the neutral species Rh(OH)2*I was formed.
The results mentioned above were confirmed by reversed-phase HPLC as shown in Figure 3. If iodide was heated with the macrocyclic thioether ligand without metal cations (blank 2), only one peak was visible on the radiochromatogram (Figure 3, panel A) with a retention time of around 3.9 min. corresponding to an uncomplexed iodide standard. In the case of blank 1, where iodide was heated with Rh(III) cations in the absence of 16aneS4-diol, two peaks were found with the retention times 3.9 and 5.4 min, corresponding to free iodide and probably neutral Rh(OH)2*I, respectively (Figure 3, panel B). The synthesized cationic complex Rh[16aneS4-diol]*I appeared at 15–17 min when using the previously described gradient (Figure 3, panel C).
Figure 3.

HPLC radiochromatograms of (A) control sample “blank 2”; (B) control sample “blank 1”; (C) sample containing cationic complex Rh[16aneS4-diol]*I.
Although not shown, the results obtained by paper electrophoresis and reversed-phase HPLC methods for the other cationic complexes that were synthesized, Rh[16aneS4-diol]211At and Ir[16aneS4-diol]*I/211At, were quite similar to those described above for Rh[16aneS4-diol]*I. Therefore, only the results for Rh[16aneS4-diol]*I are shown as examples.
The synthesis of inert low-spin d6 metal complexes often requires heating at high temperature and sometimes for extended time periods (39). Our studies also showed that temperature and time of heating are important parameters for high-yield synthesis of the desired complexes. The yield of Rh[16aneS4-diol]*I synthesis was below 20% at 40 °C, even after 2 h of heating (Figure 4, panel A). Raising the temperature from 40 to 90 °C resulted in a sharply increased rate of reaction. The yield of Rh[16aneS4-diol]*I complex formation was around 55–60% after 30 min of heating at 80–90 °C, and it was still increasing during the next 1.5 h, approaching a maximum value of about 90% at 2 h. The temperature-dependence studies for the Ir[16aneS4-diol]*I complex were performed at 40, 60, and 80 °C (Figure 4, panel B). Generally, the obtained results were very similar to those for the Rh[16aneS4-diol]*I complex. The yield of reaction was low at 40 °C and increased up to 87% after heating for 2 h at 80 °C. In the case of Rh[16aneS4-diol]211At and Ir[16aneS4-diol]211At, the effect of heating time on yield was evaluated only at 75–80 °C (Figure 4, panel C). The maximum reaction yield for both astatine complexes was about 80%, and could almost be achieved after 1.5 h of heating.
Figure 4.

Influence of temperature and time of heating on the synthesis yield of complexes: (A) Rh[16aneS4-diol]*I; (B) Ir[16aneS4-diol]*I; (C) M[16aneS4-diol]211At (M = Rh(III) or Ir(III)).
Microwave-assisted synthesis is commonly used in organic chemistry because it enhances chemical yields by reducing reaction times without causing major degradation or introducing undesired reactions. There are also few articles about preparation of transition metals coordination complexes using microwave techniques (40, 41), including studies on Rh(III) complexes with bipyridine, cyclopentadiene, and pyridylazoresorcinol group ligands (39, 42). This technique has been also applied to labeling biomolecules with short-lived radionuclides, which require procedures that afford the labeled product in a short reaction time. Therefore, it is used in the synthesis with 18F (43, 44), but also with other radiohalogens such as 123I (45). Microwave heating may significantly accelerate the synthesis, especially of substitutionally inert second- and third-row transition metal coordination compounds (40). For example, the microwave synthesis of cis-[Rh(bpy)2I2][PF6] (bpy = bipyridine) from RhCl3 reduced the reaction time from 65 min to 12 min (39). In our studies, after 10 min of microwave heating, the yields for Rh[16aneS4-diol]131I and Ir[16aneS4-diol]131I complexes were 51% and 38%, respectively (Figure 5). During the next 10 min, the yields increased up to 75% and 73%, the level which was almost reached in conventional heating in the oil bath for 1.0 h at 75–80 °C. Finally, the yields obtained for Rh[16aneS4-diol]131I and Ir[16aneS4-diol]131I after 35 min of microwave heating were 90% and 83%, respectively. The temperatures of all samples exposed to microwave radiation were kept in the range 75–80 °C by using only 50% of maximum during microwave irradiation.
Figure 5.
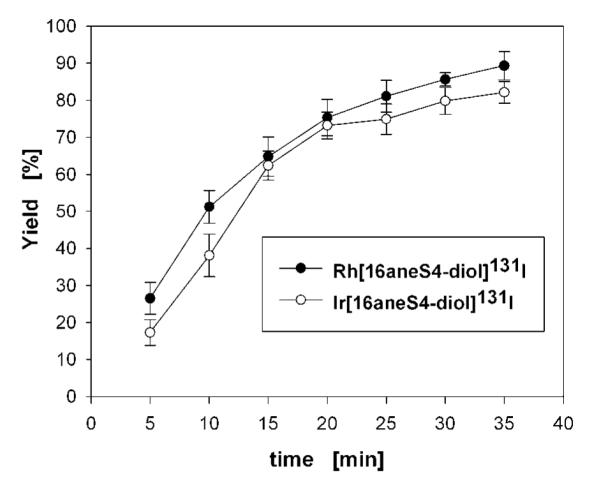
Yield of Rh[16aneS4-diol]131I and Ir[16aneS4-diol]131I complexes versus time of microwave exposition.
Good synthesis yields of about 80% were obtained for Rh[16aneS4-diol]*I/211At and Ir[16aneS4-diol]*I/211At complexes from solutions containing 62.5 nmol of Rh(III) or Ir(III) cations, 250 nmol of 16aneS4-diol ligand, and carrier-free radiohalogen. Decreasing the amount of metals cation in the reaction mixture resulted in a significant decrease in yield for all four complexes (Table 1). High complex formation yields were obtained even when the complexation reaction was performed under a 1:1 stoichiometry ratio of 16aneS4-diol to Rh(III) or Ir(III) at pH 4.0 (nM = 62.5 nmol). The yield was reduced when the ratio was less than 1:1 (Table 2).
Table 1.
Effect of Rh(III) and Ir(III) Concentration on the M[16aneS4-diol]X (M = Rh(III) or Ir(III) and X = *I or 211At) Complexes Yield at pH of ~4.0
| yield ± SD [%] |
||||
|---|---|---|---|---|
| quantity of metal nM (nmol) |
Rh[16aneS4- diol]*I |
Ir[16aneS4- diol]*I |
Rh[16aneS4- diol]211At |
Ir[16aneS4- diol]211At |
| 0.125 | 2.7 ± 1.5 | 1.8 ± 0.6 | 3.3 ± 2.6 | 4.6 ± 2.4 |
| 1.25 | 5.1 ± 2.0 | 7.4 ± 3.2 | 4.3 ± 2.4 | 5.5 ± 3.7 |
| 6.25 | 22.6 ± 5.6 | 19.3 ± 5.6 | 11.3 ± 5.5 | 9.4 ± 8.1 |
| 12.5 | 48.4 ± 3.9 | 50.5 ± 3.9 | 37.0 ± 6.0 | 33.1 ± 6.0 |
| 62.5 | 82.5 ± 4.8 | 79.2 ± 3.3 | 77.2 ± 8.2 | 73.3 ± 6.2 |
| 125.0 | 88.4 ± 3.3 | 85.8 ± 3.6 | 82.8 ± 4.6 | 81.3 ± 7.3 |
Table 2.
Effect of 16aneS4-diol Concentration on the M[16aneS4-diol]X (M =Rh(III) or Ir(III) and X = *I or 211At) Complexes Yield at pH of ~4.0
| yield ± SD [%] |
||||
|---|---|---|---|---|
| quantity of ligand nS4 (nmol) |
Rh[16aneS4- diol]*I |
Ir[16aneS4- diol]*I |
Rh[16aneS4- diol]211At |
Ir[16aneS4- diol]211At |
| 18.75 | 2.1 ± 1.3 | 2.4 ± 1.8 | 3.1 ± 2.2 | 3.5 ± 2.6 |
| 25.0 | 7.4 ± 2.8 | 18.2 ± 1.3 | 9.5 ± 0.8 | 8.6 ± 4.5 |
| 62.5 | 43.8 ± 7.1 | 38.4 ± 1.3 | 48.7 ± 5.7 | 26.4 ± 7.6 |
| 125.0 | 73.8 ± 6.9 | 70.5 ± 3.1 | 69.6 ± 6.4 | 65.2 ± 8.7 |
| 187.5 | 79.4 ± 2.0 | 77.6 ± 2.8 | 76.5 ± 3.3 | 71.3 ± 6.5 |
| 250.0 | 82.1 ± 3.2 | 80.2 ± 2.5 | 78.3 ± 4.2 | 74.5 ± 2.8 |
The pH was an important parameter, which influenced the formation of all four complexes. The Rh[16aneS4-diol]*I/211At and Ir[16aneS4-diol]*I/211At yields were the highest in the range 3.0–4.0 and dramatically decreased with pH higher than 5.0, declining to 4.5 ± 0.4% and 12.4 ± 6.7% for *I and 211At, respectively at pH 8.5 (Table 3). The reduction in complex formation probably was due to hydrolysis of the metal cations. During pH studies, we noticed that the decrease in yield for Rh[16aneS4-diol]*I/211At was greater than for Ir[16aneS4-diol]*I/211At. This is consistent with the hypothesis that this behavior reflects hydrolysis of the metal cation, because Ir(III) as a larger cation, which is below Rh(III) in the Periodic Table, should have a smaller affinity to hydrolysis reaction than Rh(III).
Table 3.
Yield of the M[16aneS4-diol]X (M = Rh(III) or Ir(III) and X = *I or 211At) Complexes Synthesis vs pH of Solution
| yield ± SD [%] |
||||
|---|---|---|---|---|
| pH | Rh[16aneS4- diol]*I |
Ir[16aneS4- diol]*I |
Rh[16aneS4- diol]211At |
Ir[16aneS4- diol]211At |
| 2.0 | 80.6 ± 3.2 | 74.7 ± 2.4 | 72.2 ± 8.1 | 68.9 ± 6.1 |
| 3.0 | 83.3 ± 2.9 | 76.5 ± 1.7 | 75.3 ± 4.5 | 74.8 ± 3.2 |
| 4.0 | 84.2 ± 4.3 | 80.2 ± 2.9 | 80.4 ± 4.2 | 75.1 ± 6.4 |
| 5.0 | 55.1 ± 9.4 | 65.7 ± 3.1 | 68.1 ± 3.6 | 69.0 ± 6.0 |
| 6.0 | 32.6 ± 8.2 | 62.4 ± 2.5 | 43.3 ± 3.4 | 58.4 ± 5.3 |
| 7.5 | 14.0 ± 7.7 | 27.3 ± 6.7 | 24.7 ± 5.2 | 35.0 ± 1.7 |
| 8.5 | 4.5 ± 0.4 | - | 12.4 ± 6.7 | - |
CONCLUSIONS
Our results demonstrate the feasibility of attaching 211At− anions under mild conditions to moderately soft Rh(III) or Ir(III) cations complexed by 16aneS4-diol to form kinetically inert positively charged Rh[16aneS4-diol]211At and Ir[16aneS4-diol]211At complexes. The macrocyclic ligand 16aneS4-diol with diol functionality was chosen for model studies, because it forms stable complex with Rh(III) and can be easily modified to provide a bifunctional chelate ligand. For further labeling studies with biomolecules, we will use a bifunctional sulfur ligand. A macrocyclic sulfur ligand with an additional carboxylic group as a functionality for attachment will probably be the best for conjugation with biomolecules. This type of ligand has been described previously. Li et al. (34) and Goswami et al. (36, 46) utilized rhodium complexes with different bifunctional sulfur ligands and reported that the carboxylic group did not coordinate with the rhodium core in their complexes, even after heating at high temperatures for a long time. Therefore, we also do not expect the competition reaction and replacement of astatide or iodide in our complexes by the carboxylic group. However, this must be verified experimentally. The complexes labeled with 211At, after appropriate purification, could be used in the future as the precursors for labeling of biomolecules such as monoclonal antibodies. Because the formation of Rh[16aneS4-diol]211At and Ir[16aneS4-diol]211At or complexes with similar bifunctional sulfur ligands requires heating at high temperatures, we anticipate the need for a two-step procedure for labeling biomolecules. First, the appropriate astatine complex should be synthesized and, after purification, conjugated at room temperature with the biomolecule via an appropriate functional group (e.g., carboxylic group mentioned above).
Studies with *I− anions were performed mainly for optimization of reaction conditions. In addition, results obtained during synthesis of Rh[16aneS4-diol]*I and Ir[16aneS4-diol]*I complexes, enable us to compare them with the results for 211At complexes and make final conclusions. Generally, results obtained during optimization processes for all four complexes were comparable. However, we noticed that complexes with the Rh(III) cation core were formed with a few percent higher yield in comparison to complexes with the Ir(III) cation core. This situation was changed only when the effects of reaction pH were evaluated. Also, the yield for complexation with *I radionuclides was a few percent higher than for 211At. Finally, we showed that microwave heating could significantly accelerate the reactions. Using microwaves, instead of conventional heating, reduced the time of reaction from 1–1.5 h to about 20–35 min with the approximate yield of 80%.
Preliminary stability studies of Rh[16aneS4-diol]*I and Ir[16aneS4-diol]*I complexes in PBS and human serum have been performed and gave promising results. Both iodine complexes were stable in PBS and human serum over 51 h incubation at 37 °C. The stability of Rh[16aneS4-diol]211At and Ir[16aneS4-diol]211At complexes was checked only in PBS solution. We did not observe any loss of astatine from the complexes after 6 h (almost one-half-life) incubation at 37 °C. Extended studies on the evaluation of the in vitro and in vivo stability of Rh[16aneS4-diol]*I/211At and Ir[16aneS4-diol]*I/211At complexes will be described in a subsequent publication.
ACKNOWLEDGMENT
This work was done under the N204 081 31/1939 and N204 143 32/3547 grants of Polish Ministry of Science and Higher Education. Part of the work was carried out in the frame of Marie Curie Action for the Transfer of Knowledge, contract no. MTKD-CT-2004-509224 with the European Commission and part was funded by Grant CA42324 from the National Institutes of Health of the United States. We gratefully acknowledge the Polish-US Fulbright Commission for enabling Marek Pruszyński to spend 5 months working at Duke University Medical Center, Durham, NC.
LITERATURE CITED
- (1).Zalutsky MR. Radionuclide therapy. In: Rösch F, editor. Radiochemistry and Radiopharmaceutical Chemistry in Life Sciences. Kluwer Academic Publishers; Dordrecht: 2003. pp. 315–348. [Google Scholar]
- (2).Hall EJ. Radiobiology for the Radiologist. Lippincott; Philadelphia: 1988. pp. 161–177. [Google Scholar]
- (3).Ritter MA, Cleaver JW, Tobias CA. High-LET radiations induce a large proportion of non-rejoining DNA breaks. Nature. 1977;266:653–655. doi: 10.1038/266653a0. [DOI] [PubMed] [Google Scholar]
- (4).Lambrecht RM, Mirzadeh S. Cyclotron isotopes and radiopharmaceuticals-XXXV. Astatine-211. Int. J. Appl. Radiat. Isot. 1985;36:443–450. [Google Scholar]
- (5).Turkington TG, Zalutsky MR, Jaszczak RJ, Garg PK, Vaidyanathan G, Coleman RE. Measuring astatine-211 distributions with SPECT. Phys. Med. Biol. 1993;38:1121–1130. doi: 10.1088/0031-9155/38/8/010. [DOI] [PubMed] [Google Scholar]
- (6).Vergote I, Larsen RH, De Vos L, Nesland JM, Bruland Ø, Bjørgum J, Alstad J, Tropé C, Nustad K. Therapeutic efficiacy of the α-emitter 211At bound on microspheres compared with 90Y and 32P colloids in a murine intraperitoneal tumour model. Gynecol. Oncol. 1992;47:366–372. doi: 10.1016/0090-8258(92)90141-5. [DOI] [PubMed] [Google Scholar]
- (7).Strickland DK, Vaidyanathan G, Zalutsky MR. Cytotoxicity of α-particle emitting m-[211At]astatobenzylguanidine on human neuroblastoma cells. Cancer Res. 1994;54:5414–5419. [PubMed] [Google Scholar]
- (8).Larsen RH, Vaidyanathan G, Zalutsky MR. Cytotoxicity of α-particle emitting 5-[211At]astato-2′-deoxyuridine in human cancer cells. Int. J. Radiat. Biol. 1997;72:79–90. doi: 10.1080/095530097143563. [DOI] [PubMed] [Google Scholar]
- (9).Larsen RH, Murud KM, Akabani G, Hoff P, Bruland Ø, Zalutsky MR. At-211 and I-131-labeled bisphosphonates with high in vivo stability and bone accumulation. J. Nucl. Med. 1999;40:1197–1203. [PubMed] [Google Scholar]
- (10).Andersson H, Lindegren S, Back T, Jacobsson L, Leser G, Horvath G. Radioimmunotherapy of nude mice with intraperitoneally growing ovarian cancer xenograft utilizing 211At-labelled monoclonal antibody MOv18. Anticancer Res. 2000;20:459–462. [PubMed] [Google Scholar]
- (11).Humm JL, Roeske JC, Fisher DR, Chen GTY. Microdosimetric concepts in radioimmunotherapy. Med. Phys. 1993;20:535–541. doi: 10.1118/1.597049. [DOI] [PubMed] [Google Scholar]
- (12).Zalutsky MR, Bigner DD. Radioimmunotherapy with alpha-particle emitting radioimmunoconjugates. Acta Oncol. 1996;35:373–379. doi: 10.3109/02841869609101654. [DOI] [PubMed] [Google Scholar]
- (13).Vaughan ATM, Fremlin JH. Preparation of astatine labeled protein using an electrophilic reaction. Int. J. Nucl. Med. Biol. 1978;5:229–230. doi: 10.1016/0047-0740(78)90145-6. [DOI] [PubMed] [Google Scholar]
- (14).Visser GWM, Diemer EL, Kaspersen FM. The nature of the astatine-protein bond. Int. J. Appl. Radiat. Isot. 1981;32:905–912. doi: 10.1016/0020-708x(81)90078-8. [DOI] [PubMed] [Google Scholar]
- (15).Zalutsky MR, Stabin MG, Larsen RH, Bigner DD. Tissue distribution and radiation dosimetry of astatine-211-labeled chimeric 81C6, an α-particle-emitting immunoconjugate. Nucl. Med. Biol. 1997;24:255–261. doi: 10.1016/s0969-8051(97)00060-7. [DOI] [PubMed] [Google Scholar]
- (16).Yordanov AT, Garmestani K, Zhang M, Zhang Z, Yao Z, Phillips KE, Herring B, Horak E, Beitzel MP, Schwarz UP, Gansow OA, Plascjak PS, Eckelman WC, Waldmann TA, Brechbiel MW. Preparation and in vivo evaluation of linkers for 211At labeling of humanized anti-Tac. Nucl. Med. Biol. 2001;28:845–856. doi: 10.1016/s0969-8051(01)00257-8. [DOI] [PubMed] [Google Scholar]
- (17).Friedman AM, Zalutsky MR, Wung W, Buckingham F, Harper PV, Scherr GH, Wainer B, Hunter RL, Appleman EH, Rothberg RM, Fitch FW, Stuart FP, Simonian SJ. Preparation and biologically stable and immunologically competent astatinated protein. Int. J. Nucl. Med. Biol. 1977;4:219–224. doi: 10.1016/0047-0740(77)90146-2. [DOI] [PubMed] [Google Scholar]
- (18).Vaughan ATM. Labelling of proteins with 211At using an Acylation Reaction. Int. J. Appl. Radiat. Isot. 1979;30:576–577. [Google Scholar]
- (19).Zalutsky MR, Narula AS. Astatination of proteins using an N-succinimidyl tri-n-butylstannyl benzoate intermediate. Appl. Radiat. Isot. 1988;39:227–232. doi: 10.1016/0883-2889(88)90176-1. [DOI] [PubMed] [Google Scholar]
- (20).Hadley SW, Wilbur DS, Gray MA, Atcher RW. Astatine-211 labeling of an antimelanoma antibody and its Fab fragment using N-succinimidyl p-[211At]astatobenzoate: comparisons in vivo with the p-[125I]iodobenzoyl conjugate. Bioconjugate Chem. 1991;2:171–179. doi: 10.1021/bc00009a006. [DOI] [PubMed] [Google Scholar]
- (21).Wilbur DS, Chyan MK, Hamlin DK, Kegley BB, Risler R, Pathare PM, Quinn J, Vessella RL, Foulon C, Zalutsky MR, Wedge TJ, Hawthorne MF. Reagents for astatination of biomolecules: comparison of the in vivo distribution and stability of some radioiodinated/astatinated benzamidyl and nido-carboranyl compounds. Bioconjugate Chem. 2004;15:203–223. doi: 10.1021/bc034175k. [DOI] [PubMed] [Google Scholar]
- (22).Berei K, Vasaros L. Organic Chemistry of Astatine. 1981. Hungarian Academy of Sciences ReportKFKI-1981–10. [Google Scholar]
- (23).Garg PK, John CS, Zalutsky MR. Preparation and preliminary evaluation of 4-[211At]astato-N-piperidinoethyl benzamide. Nucl. Med. Biol. 1995;22:467–473. doi: 10.1016/0969-8051(94)00134-6. [DOI] [PubMed] [Google Scholar]
- (24).Kerr JA. Strengths of Chemical Bonds. In: Lide DR, editor. CRC Handbook of Chemistry and Physics. CRC Press; Boca Raton: 1993. pp. 9-123–9-145. [Google Scholar]
- (25).Wilbur DS, Hamlin DK, Srivastava RR. Radioiodinations of nido-carboranes. potential hydrophilic pendant groups for radiohalogenation of biological molecules. J. Labelled Compd. Radiopharm. 1994;35:199–201. [Google Scholar]
- (26).Wilbur DS, Hamlin DK, Buhler KR, Srivastava RR, Suray JE, Daniel J, Vessella RL. Comparison of in vivo distribution of simple radioiodinated borane and carborane cage molecules. Quat. J. Nucl. Med. 1995;39:51–52. [Google Scholar]
- (27).Orlova A, Lebeda O, Tolmachev V, Sjöberg S, Carlsson J, Lundqvist H. Closo-dodecaborate(2-) anion as a prosthetic group for attachment of astatine to proteins. Aspects of the labelling chemistry with Chloramine-T. J. Labelled Compd. Radiopharm. 2000;43:251–260. [Google Scholar]
- (28).Sjöström A, Tolmachev V, Lebeda O, Koziorowski J, Carlsson J, Lundqvist H. Direct astatination of a tumour-binding protein, human epidermal growth factor, using nido-carborane as a prosthetic group. J. Radioanal. Nucl. Chem. 2003;256:191–197. [Google Scholar]
- (29).Wilbur DS, Hamlin DK, Srivastava RR, Chyan MK. Synthesis, radioiodination, and biodistribution of some nido- and closo-monocarbon carborane derivatives. Nucl. Med. Biol. 2004;31:523–530. doi: 10.1016/j.nucmedbio.2003.11.003. [DOI] [PubMed] [Google Scholar]
- (30).Wilbur DS, Chyan MK, Hamlin DK, Vessella RL, Wedge TJ, Hawthorne MF. Reagents for astatination of biomolecules. 2. Conjugation of anionic boron cage pendant groups to a protein provides a method for direct labeling that is stable to in vivo deastatination. Bioconjugate Chem. 2007;18:1226–1240. doi: 10.1021/bc060345s. [DOI] [PubMed] [Google Scholar]
- (31).Pearson RG. Hard and soft acids and bases. J. Am. Chem. Soc. 1963;85:3533–3543. [Google Scholar]
- (32).Pruszyński M, Bilewicz A, Was B, Petelenz B. Formation and stability of astatide-mercury complexes. J. Radioanal. Nucl. Chem. 2006;268:91–94. [Google Scholar]
- (33).Venkatesh M, Goswami N, Volkert WA, Schlemper EO, Ketring AR, Barnes CL, Jurisson S. An Rh-105 complex of tetrathiacyclohexadecane diol with potential for formulating bifunctional chelates. Nucl. Med. Biol. 1996;23:33–40. doi: 10.1016/0969-8051(95)02012-8. [DOI] [PubMed] [Google Scholar]
- (34).Li N, Struttman M, Higginbotham C, Grall AJ, Skerlj JF, Vollano JF, Bridger SA, Ochrymowycz LA, Ketring AR, Abrams MJ, Volkert WA. Biodistribution of model 105Rh-labeled tetradentate thiamacrocycles in rats. Nucl. Med. Biol. 1997;24:85–92. doi: 10.1016/s0969-8051(96)00177-1. [DOI] [PubMed] [Google Scholar]
- (35).Koziorowski J, Lebeda O, Weinreich R. A cryotrap as flow reactor for synthesis of 211At labeled compounds. Appl. Radiat. Isot. 1999;50:527–529. [Google Scholar]
- (36).Goswami N, Higginbotham C, Volkert W, Alberto R, Nef W, Jurisson S. Rhodium-105 tetrathioether complexes: radiochemistry and initial biological evaluation. Nucl. Med. Biol. 1999;26:951–957. doi: 10.1016/s0969-8051(99)00070-0. [DOI] [PubMed] [Google Scholar]
- (37).Wilbur DS. Potential use of alpha emitting radionuclides in the treatment of cancer. Antibody, Immunoconjugates, Radiopharm. 1991;4:85–97. [Google Scholar]
- (38).Garg PK, John CS, Zalutsky MR. Preparation and preliminary evaluation of 4-[211At]astato-N-piperidinoethyl benzamide. Nucl. Med. Biol. 1995;22:467–473. doi: 10.1016/0969-8051(94)00134-6. [DOI] [PubMed] [Google Scholar]
- (39).Amarante D, Cherian C, Emmel C, Chen HY, Dayal S, Koshy M, Megehee EG. Improved synthetic routes to rhodium bipyridine complexes: comparison of microwave vs. conventional synthesis. Inorg. Chim. Acta. 2005;358:2231–2238. [Google Scholar]
- (40).Baghurst DR, Cooper SR, Greene DL, Mingos DMP, Reynolds SM. Application of microwave dielectric loss heating effects for the rapid and convenient synthesis of coordination compounds. Polyhedron. 1990;9:893–895. [Google Scholar]
- (41).Rau S, Schäfer B, Grüßing A, Schebesta S, Lamm K, Vieth J, Görls H, Walther D, Rudolph M, Grummt UW, Birkner E. Efficient synthesis of ruthenium complexes of the type (R-bpy)2RuCl2 and [(R-bpy)2Ru(L-L)]Cl2 by microwave-activated reactions (R: H, Me, tert-But) (L-L: substituted bibenzimidazoles, bipyrimidine, and phenanthroline) Inorg. Chim. Acta. 2004;357:4496–4503. [Google Scholar]
- (42).Dedkov, Yu M, Korsakova NV, Radugina OG. Effect of microwave radiation on the complexation of rhodium(III) and iridium(IV) with reagents of the pyridylazoresorcinol group. J. Anal. Chem. 2000;55:1128–1131. [Google Scholar]
- (43).Hwang DR, Moerlein SM, Welch MJ. Microwave-facilitated synthesis of [18F]-Spiperone. J. Labelled Compd. Radiopharm. 1989;26:391. [Google Scholar]
- (44).Lemaire C, Cantineau R, Christiaens L, Guillaume M. Radiofluorination of Altanserine a potential serotonin receptor binding radiopharmaceutical for positron emission tomography. J. Labelled Compd. Radiopharm. 1989;36:336. [Google Scholar]
- (45).Kumar P, Wiebe LI, Asikoglu M, Tandon M, McEwan AJB. Microwave-assisted (radio)halogenation of nitroimidazole-based hypoxia markers. Appl. Radiat. Isot. 2002;57:697–703. doi: 10.1016/s0969-8043(02)00185-9. [DOI] [PubMed] [Google Scholar]
- (46).Goswami N, Alberto R, Barnes CL, Jurisson S. Rhodium(III) complexes with acyclic tetrathioether ligands. Effects of back-bone chain length on the conformation of the Rh(III) complexes. Inorg. Chem. 1997;35:7546–7555. [Google Scholar]