

Abstract

O-Acetyl oximes serve as effective directing groups for Pd-catalyzed sp2 and sp3 C–H functionalization reactions. The C–H functionalization products can be subsequently transformed into ortho- or β-functionalized ketones, alcohols, amines, and heterocycles.
Over the past decade, palladium-catalyzed ligand-directed C–H functionalization has been extensively exploited to convert unactivated carbon–hydrogen bonds into carbon–heteroatom and carbon–carbon bonds.1 Despite the rapidly growing arsenal of methods in the field, the synthetic utility of these transformations is limited by the requirement that a directing group be built into the substrate. In particular, many effective directing groups are nitrogen-containing heterocycles that are not easily transformed or removed following the C–H functionalization event.1
The ideal directing ligand would be sufficiently robust to tolerate C–H activation/functionalization conditions but then readily converted into diverse functional groups. Instances of such directing groups in Pd-catalyzed reactions are relatively rare. Yu and Shi have independently demonstrated C–H functionalization of triflamide2a- and dimethyl2b-protected benzyl amines followed by nucleophilic displacement or reduction of the amine directing group. Oxazolines have been used for Pd-catalyzed C–I bond forming reactions followed by H2SO4-catalyzed hydrolysis to afford carboxylic acids.3 Carboxylic acids have also been directly employed in Pd-catalyzed C–H arylation and then removed by decarboxylation.4 Finally, amides have proven effective for directing Pd-catalyzed C–C and C–halogen bond formation, and have been subsequently transformed into nitriles5a or carboxylic acids.5b
Ketones are particularly versatile and widely-used synthetic intermediates.6 However, ketones are poor ligands for PdII, and thus are generally ineffective directing groups for Pd-catalyzed C–H functionalization.7,8 We sought to temporarily mask ketones as more coordinating imine or oxime derivatives during C–H functionalization and then subsequently remove this group to reveal the ketone functionality (Scheme 1). This communication describes the development of O-acetyl oximes as versatile and readily transformable directing groups for Pd-catalyzed C–H functionalization.
Scheme 1.

Approach to β-C–H Functionalization of Ketones
Two key challenges exist in the design of a ketone surrogate for Pd-catalyzed C–H functionalization. First, the protecting group must be stable to the catalytic conditions. Second, the group must be readily removed in high yield without affecting the newly installed functional group. Previous studies have shown that oxime ethers such as 1 (eq 1) are effective directing groups for Pd-catalyzed sp2 and sp3 C–H acetoxylation reactions with PhI(OAc)2.9,10 However, removal of the oxime ether protecting group from β-functionalized products like 2 is problematic. Acid-catalyzed hydrolysis11 is sluggish and produces significant quantities of elimination products 3 and 4 (eq. 1).12 The use of super-stoichiometric TiIIICl3 is effective in some cases,13 but this expensive, air sensitive reagent is not practical for general application.
 |
(1) |
Imines would be a versatile alternative to oxime ethers, as they are readily hydrolyzed under mild conditions. Pd-catalyzed imine-directed acetoxylation of an sp2 C–H bond has been reported;9a however, imines have proven too labile for analogous sp3 C–H acetoxylations. For example, reaction of 5 under standard acetoxylation conditions affords the hydrolyzed ketone 6 as the major identifiable product (39% yield, eq 2).
 |
(2) |
 |
(3) |
Simple hydroxyl (OH) oximes are another attractive ketone surrogate. These are readily available, stable, often crystalline starting materials, are known to direct stoichiometric cyclopalladation of both sp2 and sp3 C–H bonds,14 and are much more readily cleaved than their oxime ether counterparts.15 In addition, they are prepared from NH2OH•HCl, which is nearly 100-fold less expensive than NH2OMe•HCl.16 Despite these advantages, oximes are known to undergo rapid oxidative cleavage in the presence of oxidants like PhI(OAc)2.17 For example, subjecting oxime 7 to Pd(OAc)2 and PhI(OAc)2 in AcOH resulted in a nearly instantaneous color change from colorless to blue-green, concomitant with regeneration of the parent ketone 6. However, encouragingly, a similar color change was not observed when the solvent was changed from AcOH to AcOH/Ac2O (1:1). Under these conditions, only traces of ketone 6 (4% by GC) were formed; instead the major product (68% by GC) was the O-acetylated/C–H acetoxylated compound 9 (eq 3).
This initial result suggested that the in situ reaction of 7 with Ac2O affords a stable O-acetyl oxime (8) that can direct C–H acetoxylation. Further study showed that this in situ O-acetylation occurred quantitatively upon stirring the oxime starting material in AcOH/Ac2O for 2 h at 25 °C. Subsequent addition of Pd catalyst and oxidant, followed by heating the reaction mixture at 100 °C for 12 h afforded 8 in 70% GC yield (49% isolated).
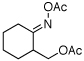
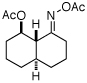
As summarized in Table 1, a number of dialkyl oximes underwent in situ acetylation/β-acetoxylation in modest to good yields (entries 1–6). The observed trends in reactivity and selectivity were similar to those in sp3 C–H functionalization reactions of related oxime ethers.9b For example, oxidation occurred selectively at 1° β-sp3 C–H bonds versus the analogous 2° sites (entries 1–4). Acetoxylation of a 2° C–H bond could be achieved in modest yield in the rigid trans-decalone system (entry 6). The reaction conditions were compatible with a number of functional groups, including alkyl chlorides (entry 4) and protected amines (entry 3); furthermore, remote benzylic C–H bonds were well tolerated under the oxidizing reaction conditions (entries 1, 9, 14).18
Table 1.
O-Acetyl Oxime-Directed Acetoxylation of C–H Bondsa
| entry | starting material | product | yieldb |
|---|---|---|---|
| 1c | 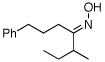 |
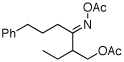 |
49% (70%)d |
| 2c | 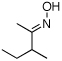 |
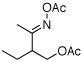 |
61% |
| 3c | 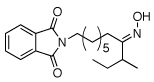 |
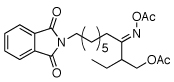 |
65% |
| 4c | 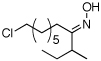 |
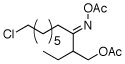 |
33% |
| 5c | 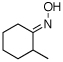 |
 |
66% |
| 6 | 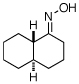 |
 |
41% |
| 7e | 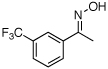 |
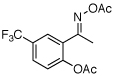 |
61% |
| 8e | 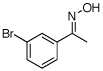 |
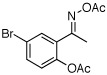 |
86% |
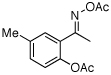
| 9e | 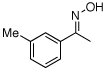 |
 |
72% |
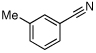
| 10 | 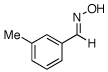 |
 |
77%d |
| 11e | 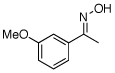 |
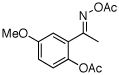 |
77% |
| 12e | 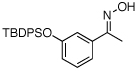 |
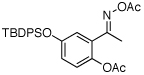 |
79% |
| 13e | 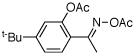 |
80% | |
| 14 | 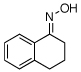 |
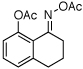 |
55% |
Conditions: 0.12 M in AcOH/Ac2O (1:1), 2 h, 25 °C; then 5 mol % Pd(OAc)2, 1–3 equiv PhI(OAc)2, 80 or 100 °C, 4–12 h.
The remaining mass balance (as determined by GC of the crude reaction mixtures) was generally unreacted O-acetyloxime (analogous to 8 in eq 3).
Starting material and product consisted of a mixture of oxime E/Z stereoisomers.
GC yield.
Product consisted of a mixture of oxime E/Z stereoisomers.
O-Acetyl ketoximes were also effective directing groups for Pd-catalyzed acetoxylation at sp2 C–H sites. Both electron poor and electron rich aryl rings underwent mono-ortho-oxygenation in high yields (entries 7 and 11). Further, aryl bromides (entry 8) and silyl-protected phenols (entry 12) were compatible with the reaction conditions. One notable limitation of this method is that O-acetyl aldoximes (for example, entry 10) were susceptible to elimination of AcOH to generate nitriles.19
The C–H acetoxylation products in Table 1 were obtained in yields comparable to those previously reported with oxime ether derivatives.9b,d However, the O-acetyl oxime directing group is significantly more readily deprotected. In general, β-hydroxy ketones are accessible via alcoholysis of both acetate groups (to afford β-hydroxy oximes) followed by removal of the oxime functionality. The first step can be accomplished by treatment of starting materials like 9 with K2CO3 in MeOH to afford 10 in 91% isolated yield.
While numerous methods exist for the second step (conversion of an oxime to a ketone), many of our substrates are susceptible to competing formation of side products (e.g., via alcohol oxidation, isoxazoline formation, or elimination). After extensive experimentation, we identified the use of NaHSO3 in EtOH/H2O20 as the most general and high yielding method to transform these oximes into β-hydroxy ketones, while circumventing undesired side reactions and persistent byproducts. Under these conditions, substrate 10 was converted cleanly to 11 in 80% yield. Compound 11 was obtained in pure form by a simple extraction, obviating the need for chromatography.
The two deprotection steps could also be combined to provide an operationally simple, high yielding, one-pot route from O-acetyl oxime C–H oxidation products to β-hydroxy ketones (eq 4). For example, treatment of 9 with K2CO3 in MeOH, followed by addition of NaHSO3 and H2O provided 11 in 80% yield after a simple extractive work-up (eq 4). As shown in Table 2, this one-pot deprotection could also be achieved with other substrates. Under these conditions elimination products were not observed, and isoxazoline formation was limited to ≤5%.
Table 2.
Deprotection to β- and ortho-Hydroxy Ketonesa
| entry | starting material | product | yield |
|---|---|---|---|
| 1 | 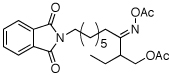 |
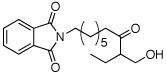 |
80% |
| 2 | 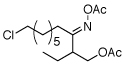 |
 |
56% |
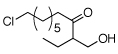
| 3 | 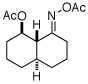 |
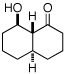 |
83% |
| 4 | 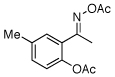 |
 |
89% |
Conditions entries 1–3 (one pot): K2CO3 (3 × 0.15 equiv/2.5 h), MeOH, 25 °C, then 3.5 equiv NaHSO3, H2O, 80 °C, 3 h; Conditions entry 4 (two steps): (i) 0.15 equiv K2CO3, MeOH, 25 °C, 1 h, then (ii) 3.5 equiv NaHSO3, H2O, EtOH, 90 °C, 12 h.
 |
(4) |
An important characteristic of the O-acetyl oxime directing group is that it enables access to a variety of additional structural motifs from a common synthetic intermediate. As exemplified with 12 in Scheme 2, K2CO3-catalyzed methanolysis of the acetyl groups provided oxime 13 in quantitative yield. Product 13 could then be converted to the corresponding acetophenone 14, to oxazoline 15 (via Beckmann rearrangement21 followed by intramolecular condensation), to amino phenol 16 (via reduction),22 and to diol 17 (via oxime hydrolysis followed by reduction).23
Scheme 2.

Diverse Transformations on C–H Functionalization Product 12
Preliminary results indicate that these O-acetyl oximes are also effective directing groups for other Pd-catalyzed C–H functionalization reactions. For example, as shown in Scheme 3, the Pd-catalyzed iodination of 18 and chlorination of 19 proceeded to form aryl halides 20 and 21 in modest to good yields.24 Interestingly, under standard Pd-catalyzed C–H arylation conditions with PhI and AgOAc in TFA,25 19 underwent in situ Beckmann rearrangement/C–H phenylation to afford acetamide 22.
Scheme 3.

Other Pd-Catalyzed C–H Functionalization Reactions of O-Acetyl Oximes
In conclusion, this letter describes the use of in situ generated O-acetyl oximes as effective directing groups in Pd-catalyzed C–H functionalization reactions. These directing groups are stable under the catalytic reaction conditions but can then be readily manipulated to afford ketones, alcohols, amines, and heterocycles.
Supplementary Material
Acknowledgment
We thank the NIH NIGMS (GM-073836) for support of this research. Additional support from Dupont, AstraZeneca, Merck, Amgen, Abbott, GlaxoSmithKline, Roche, Eli Lilly, and Bristol Myers Squibb is gratefully acknowledged.
Footnotes
Supporting Information Available. Experimental details and spectroscopic data for new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For reviews, see Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. Daugulis O, Zaitsev VG, Shabashov D, Pham QN, Lazareva A. Synlett. 2006:3382. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Li BJ, Yang SD, Shi ZJ. Synlett. 2008:949. Deprez NR, Sanford MS. Inorg. Chem. 2007;46:1924. doi: 10.1021/ic0620337. Chen X, Engle KM, Wang DH, Yu JQ. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273..
- 2.(a) Wang X, Mei TS, Yu JQ. J. Am. Chem. Soc. 2009;131:7520. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]; (b) Cai G, Fu Y, Li Y, Wan X, Shi Z. J. Am. Chem. Soc. 2007;129:7666. doi: 10.1021/ja070588a. [DOI] [PubMed] [Google Scholar]
- 3.Giri R, Wasa M, Breazzano SP, Yu JQ. Org. Lett. 2006;8:5685. doi: 10.1021/ol0618858. [DOI] [PubMed] [Google Scholar]
- 4.Chiong HA, Pham QN, Daugulis O. J. Am. Chem. Soc. 2007;129:9879. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]
- 5.(a) Shabashov D, Maldonado JRM, Daugulis O. J. Org. Chem. 2008;73:7818. doi: 10.1021/jo801300y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wasa M, Engle KM, Yu JQ. J. Am. Chem. Soc. 2009;131:9886. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larock RC. Comprehensive Organic Transformations. 2nd ed. New York: John Wiley & Sons, Inc.; 1999. pp. 1197–1620. [Google Scholar]
- 7.For rare examples of ketones or aldehydes serving as directing groups at Pd, see: Terao Y, Kametani Y, Wakui H, Satoh T, Miura M, Nomura M. Tetrahedron. 2001;57:5967. Gürbüz N, Özdemir I, Çetinkaya B. Tetrahedron Lett. 2005;46:2273..
- 8.For reviews on ketone-directed C–H alkylation and arylation reactions catalyzed by metals other than Pd, see: Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Colby DA, Bergman RG, Ellman JA. [accessed Dec 10, 2009];Chem. Rev. [Online early access]. DOI: 10.1021/cr900005n. Published Online: May 13, 2009. http://pubs.acs.org/doi/pdf/10.1021/cr900005n.
- 9.(a) Dick AR, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:2300. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; (b) Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; (c) Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:3387. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (d) Desai LV, Stowers KJ, Sanford MS. J. Am. Chem. Soc. 2008;130:13285. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For related C–H acetoxylation reactions of other substrates, see: Kalyani D, Sanford MS. Org. Lett. 2005;7:4149. doi: 10.1021/ol051486x. Giri R, Liang J, Lei JQ, Li JJ, Wang DH, Chen X, Naggar IC, Guo C, Foxman BM, Yu JQ. Angew. Chem., Int. Ed. 2005;44:7420. doi: 10.1002/anie.200502767. Wang DH, Hao XS, Wu DF, Yu JQ. Org. Lett. 2006;8:3387. doi: 10.1021/ol061384m. Reddy BVS, Reddy LR, Corey EJ. Org. Lett. 2006;8:3391. doi: 10.1021/ol061389j. Wang GW, Yuan TT, Wu XL. J. Org. Chem. 2008;73:4717. doi: 10.1021/jo8003088. Zhang J, Khaskin E, Anderson NP, Zavalij PY, Vedernikov AN. Chem. Commun. 2008:3625. doi: 10.1039/b803156h. Stowers KJ, Sanford MS. Org. Lett. 2009;11:4584. doi: 10.1021/ol901820w..
- 11.For example, see: Shipe WD, Sorensen EJ. Org. Lett. 2002;4:2063. doi: 10.1021/ol0259342..
- 12.Conversion of oxime ethers to ketones has been reported using Amberlyst 15 at temperatures ranging from 25 °C to 110 °C. See: Sakamoto T, Kikugawa Y. Synthesis. 1993:563. Mears RJ, Sailes HE, Watts JP, Whiting A. J. Chem. Soc., Perkin Trans. 1. 2000:3250.. In our hands, 2 and derivatives were stable to the reported conditions at room temperature. At elevated temperatures (80 °C), 2 reacted to form a complex mixture of products that did not include the expected β-acetoxy or β-hydroxy ketone.
- 13.(a) Desai LV. Ph.D. Thesis. University of Michigan; 2008. Apr, Palladium-Catalyzed Functionalization of C–H Bonds and Alkenes. [Google Scholar]; (b) Corey EJ, Niimura K, Konishi Y, Hashimoto S, Hamada Y. Tetrahedron Lett. 1986;27:2199. [Google Scholar]
- 14.For selected examples, see: Baldwin JE, Jones RH, Nájera C, Yus M. Tetrahedron. 1985;41:699. Baldwin JE, Nájera C, Yus M. J. Chem. Soc., Chem. Commun. 1985:126. Carr K, Saxton HM, Sutherland JK. J. Chem. Soc., Perkin Trans. 1. 1988:1599..
- 15.Corsaro A, Chiacchio U, Pistarà V. Synthesis. 2001:1903. [Google Scholar]
- 16.Cost calculated on a molar basis using: Aldrich Catalog Handbook of Fine Chemicals. Milwaukee, WI: Aldrich Chemical; 2009. [Google Scholar]
- 17.Moriarty RM, Prakash O, Vavilikolanu PR. Synth. Commun. 1986;16:1247. [Google Scholar]
- 18.The acetoxylated products were typically isolated as mixtures of E/Z oxime stereoisomers, which rapidly interconvert under the catalytic reaction conditions.
- 19.For an example under similar conditions, see: Waring P. Aust. J. Chem. 1988;41:667..
- 20.Pines SH, Chermerda JM, Kozlowski MA. J. Org. Chem. 1966;31:3446. [Google Scholar]
- 21.Xiao LF, Xia CG, Chen J. Tetrahedron Lett. 2007;48:7218. [Google Scholar]
- 22.Chung JU, Kim SY, Lim JO, Choi HK, Kang SU, Yoon HS, Ryu H, Kang DW, Lee J, Kang B, Choi S, Toth A, Pearce LV, Pavlyukovets VA, Lundberg DJ, Blumberg PM. Bioorg. Med. Chem. 2007;15:6043. doi: 10.1016/j.bmc.2007.06.041. [DOI] [PubMed] [Google Scholar]
- 23.Prein M, Maurer M, Peters EM, Peters K, von Schnering HG, Adam W. Chem. Eur. J. 1995;1:89. [Google Scholar]
- 24.(a) Kalyani D, Dick AR, Anani WQ, Sanford MS. Org. Lett. 2006;8:2523. doi: 10.1021/ol060747f. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D, Dick AR, Anani WQ, Sanford MS. Tetrahedron. 2006;62:11483. doi: 10.1021/ol060747f. [DOI] [PubMed] [Google Scholar]
- 25.Daugulis O, Zaitsev VG. Angew. Chem. Int. Ed. 2005;44:4046. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.