

Abstract
The total free energy of a molecule includes the classical molecular mechanical energy (which is understood as the free energy in vacuum) and the solvation energy which is caused by the change of the environment of the molecule (solute) from vacuum to solvent. The solvation energy is important to the study of the inter-molecular interactions. In this paper we develop a fast surface-based generalized Born method to compute the electrostatic solvation energy along with the energy derivatives for the solvation forces. The most time-consuming computation is the evaluation of the surface integrals over an algebraic spline molecular surface (ASMS) and the fast computation is achieved by the use of the nonequispaced fast Fourier transform (NFFT) algorithm. The main results of this paper involve (a) an efficient sampling of quadrature points over the molecular surface by using nonlinear patches, (b) fast linear time estimation of energy and inter-molecular forces, (c) error analysis, and (d) efficient implementation combining fast pairwise summation and the continuum integration using nonlinear patches.
Keywords: generalized Born, molecular surface, fast summation, error analysis
1. Introduction
Most of the protein molecules live in the aqueous solvent environment and the stabilities of the molecules depend largely upon their configuration and the solvent type. Since the solvation energy term models the interaction between a molecule and the solvent, the computation of the molecular solvation energy (also known as molecule - solvent interaction energy) is a key issue in molecular dynamics (MD) simulations, as well as in determining the inter-molecular binding affinities “in-vivo” for drug screening. Molecular dynamics simulations where the solvent molecules are explicitly represented at atomic resolution, for example as in the popular package NAMD [1], provide direct information about the important influence of solvation. Moreover, as the total number of atoms of solvent molecules far outnumber the atoms of the solute, a larger fraction of the time is spent on computing the trajectory of the solvent molecules, even though the primary focus of the simulation is the configuration and energetics of the solute molecule. Implicit solvent models, attempt to considerably lower the cost of computation through a continuum representation (mean-field approximation) of the solvent [2]. In the implicit model, the solvation free energy Gsol which is the free energy change to transfer a molecule from vacuum to solvent, consists of three components: the energy to form a cavity in the solvent which is also known as the hydrophobic interactions, the van der Waals interactions between the molecule and the solvent, and the electrostatic potential energy between the molecule and the solvent (also known as polarization energy), Gsol = Gcav + Gvdw + Gpol. Based on the Weeks-Chandler-Andersen (WCA) perturbation theory [3, 4], the non-polar solvation energies are of the form Gcav + Gvdw = G(rep) + G(rep). In [5], G(rep) is described as the weighted sum of the solvent-accessible surface area Ai of the atoms. In [31], a volume term is added: , where p is the solvent pressure parameter and V is the solvent-accessible volume. In [6], the attractive van der Waals dispersion energy , where , ρ0 is the bulk density, is the van der Waals dispersive component of the interaction between atom i in the solute and the volume of solvent at y, θ(y) is a density distribution function for the solvent. Hence the non-polar solvation energies
| (1.1) |
The electrostatic solvation energy is caused by the induced polarization in the solvent when the molecule is dissolved in the solvent, therefore
| (1.2) |
where φreaction = φsolvent − φgas-phase, φ(r) and ρ(r) are the electrostatic potential and the charge density at r, respectively.
The Poisson-Boltzmann (PB) model was developed to compute the electrostatic solvation energy by solving the equation −∇(ε(x)∇φ(x)) = ρ(x) for the electrostatic potential φ. Numerical methods to solve the equation include the finite difference method [7, 8], finite element method [9, 10], and boundary element method [11]. However the PB methods are prohibitive for large molecules such as proteins due to the limited computational resources. As an alternative, (1.2) is approximated by a generalized Born (GB) model which is in the form of discrete sum [12]
| (1.3) |
where , εp and εw are the solute (low) and solvent (high) dielectric constants, qi and Ri are the charge and effective Born radius of atom i, respectively, and ri j is the distance between atoms i and j. The solvation force acting on atom α, which is part of the forces driving dynamics is computed as
| (1.4) |
Because the GB calculation is much faster than solving the PB equation, the GB model is widely used in the MD simulations. Programs which implement the GB methods include CHARMM [13], Amber [14], Tinker [15], and Impact which is now part of Schrodinger, Inc.’s FirstDiscovery program suite. Even though the GB computation is much faster than the PB model, the computation of the Born radius Ri is still slow. During the MD simulation, the Born radii need to be frequently recomputed at different time steps. Because this part of computation is too time-consuming, there are attempts to accelerate the MD simulation by computing the Born radii at a larger time step. For example, in [16] in their test of a 3 ns GB simulation of a 10-base pair DNA duplex, they change the time step of computing the Born radii and long-range electrostatic energy from 1 fs to 2 fs. This reduces the time of carrying out the simulation from 13.84 hours to 7.16 hours. From this example we can see that the calculation of the Born radii takes a large percentage of total computation time in the MD simulation. In the long dynamic runs, this decrease in the frequency of evaluating the effective Born radii are not accurate enough to conserve energy which restricts the MD simulation of the protein folding process to small time scale [17]. Hence it is demanding to calculate the Born radii and the solvation energy accurately and efficiently.
In this paper we develop a method for fast computation of the GB solvation energy, along with the energy derivatives for the solvation forces, based on a discrete and continuum model of the molecules (Figure 1.1). An efficient method of sampling quadrature points on the nonlinear patch is given. We also show that the error of the Born radius calculation is controlled by the size of the triangulation mesh and the regularity of the periodic function used in the fast summation algorithm. The time complexity of the forces computation is reduced from the original O(MN +M2) to nearly linear time O(N +M +n3 logn +M logM), where M is the number of atoms of a molecule, N is the number of integration points that we sample on the surface of the molecule when we compute the Born radius for each atom, and n is a parameter introduced in the fast summation algorithm. The fast summation method shows its advantage when it is applied to the Born radius calculations for macromolecules, where there could be tens of thousands or millions of atoms, and N could be even larger. In the fast summation method, one only need to choose a small n which is much smaller than M and N to get a good approximation, which makes the new fast summation based GB method more efficient.
Fig. 1.1.

Top left: the discrete van der Waals surface model (436 atoms); top middle: the triangulation of the continuum Gaussian surface model with 6004 triangles; top right: the regularized triangular mesh where the quality of the elements is improved (making each as close as possible to an equilateral triangles); bottom left: the continuum ASMS model generated from the triangular mesh up right; bottom right: the molecular surface rendered according to the interaction with the solvent where red means strong and blue means weak interaction.
The rest of the paper is organized as follows: in Section 2 we explain the geometric model that our energy and force computation are based on; we discuss in detail the energy computation in Section 3 and the force computation in Section 4; some implementation results are shown in Section 5; some details such as the fast summation algorithm and the NFFT algorithm are discussed in the appendix.
2. Geometric model
2.1. Gaussian surface
The electron density and shape are used in a similar sense in the literature with respect to the modeling of molecular surfaces or interfaces between the molecule and its solvent. The electron density of atom i at a point x is represented as a Gaussian function: where xi, ri are the position of the center and radius of the atom k. If we consider the function value of 1, we see that it is satisfied at the surface of the sphere (x: |x − xi| = ri). Using this model, the electron density at x due to a protein with M atoms is just a summation of Gaussians:
| (2.1) |
where β is a parameter used to control the rate of decay of the Gaussian and known as the blobbiness of the Gaussian. In [18] β = −2.3, isovalue = 1 is indicated as a good approximation to the molecular surface.
2.2. Triangular mesh
The triangular mesh of the Gaussian surface is generated by using the dual contouring method [19, 20]. In the dual contouring method a top-down octree is recursively constructed to enforce that each cell has at most one isocontour patch. The edges whose endpoints lie on different side of the isocontour are tagged as sign change edges. In each cube that contains a sign change edge, we compute the intersection points (and their unit normals) of the isocontour and the edges of the cube, denoted as pi and ni, and compute the minimizer point in this cube which minimizes the quadratic error function (QEF) [21]:
Since each sign change edge is shared by either four cubes (uniform grid) or three cubes (adaptive grid), connecting the minimizer points of these neighboring cubes forms a quad or a triangle that approximates the isocontour. We divide the quads into triangles to generate the pure triangular mesh.
2.3. Algebraic spline molecular surface (ASMS)
The triangular mesh is a linear approximation to the Gaussian surface. In our solvation energy computation, we generate another higher order approximation called ASMS model (Figure 2.3(f)) based on the triangular mesh to improve accuracy and efficiency [22]. Starting from the triangular mesh, we first construct a prism scaffold as follows. Let [vivjvk] be a triangle of the mesh where vi, vj, vk are the vertices of the triangle and ni, nj, nk be their unit normals. Define vl(λ) = vl + λnl. Then the prism is define as
Fig. 2.3 .

(a) is the discrete van der Waals model of protein 1BGX with 19,647 atoms; (b) and (c) are the zoom-in views of the the initial triangulation of the continuum surface with 85656 triangles; (d) and (e) are the zoom-in views of the quality improved mesh; (f) is the a continuum ASMS model generated based on the quality improved mesh.
where b1, b2, b3 ∈ [0, 1], b1 + b2 + b3 = 1, and Iijk is a maximal open interval such that (i) 0 ∈ Iijk, (ii) for any λ ∈ Iijk, vi(λ), vj(λ) and vk(λ) are not collinear, and (iii) for any λ ∈ Iijk, ni, nj and nk point to the same side of the plane Pijk(λ):= {p: p = b1vi(λ) + b2vj(λ) + b3vk(λ)} (Figure 2.1).
Fig. 2.1 .

A prism Dijk constructed with a triangle [vivjvk] as a basis.
Next we define a function over the prism Dijk in the cubic Bernstein-Bezier (BB) basis:
| (2.2) |
where . The ASMS denoted as Γ is the zero contour of F. The scheme for defining the coefficients bijk are defined is described in detail in [22]. In short they are defined such that
the vertices of the triangular mesh are points on Γ;
Γ is C1 at the vertices of mesh;
Γ is C1 at the midpoints of the mesh edges.
Later, given the barycentric coordinates of a point (b1, b2, b3) in triangle [vivjvk], we solve the equation F(b1, b2, b3, λ) = 0 for λ by Newton’s method. In this way we can get the corresponding point (x,y,z) on Γ:
| (2.3) |
We have proved in [22] that the ASMS model is C1 everywhere if the normals of the mesh satisfy certain symmetry conditions. The error between the ASMS and the Gaussian surface is bounded and we have shown that the ASMS converges to the Gaussian surface at the rate of O(h3) where h is the maximum edge length of the mesh.
3. Fast solvation energy computation
3.1. Method
Similarly to what is done for other GB models, we use (1.3) as the electrostatic solvation energy function. Before we compute (1.3), we need to first compute the effective Born radius Ri for every atom which reflects the depth a charge buried inside the molecule (Figure: 3.1). An atom buried deep in a molecule has a larger Born radius, whereas an atom near the surface has a smaller radius. Hence surfactant atoms have a stronger impact on the polarization. Given a discrete van der Waals (vdW) atom model, as long as we know Ri for each atom, we can compute (1.3) by using the fast multipole method (FMM) [23] with the time complexity O(M logM). However the Born radii computation is not easy and is very time-consuming. There are various ways of computing the Born radius as summarized in [24]. These methods can be divided into two categories: volume integration based methods and surface integration based methods. In general, the surface integration methods are more efficient than the volume integration methods due to the decreased dimension. So we adopt the surface integration method given in [25] to compute the Born radius:
| (3.1) |
where Γ is the molecule-solvent interface, xi is the center of atom i, and n(r) is the unit normal on the surface at r and we use ASMS as the model of Γ.
Fig. 3.1 .
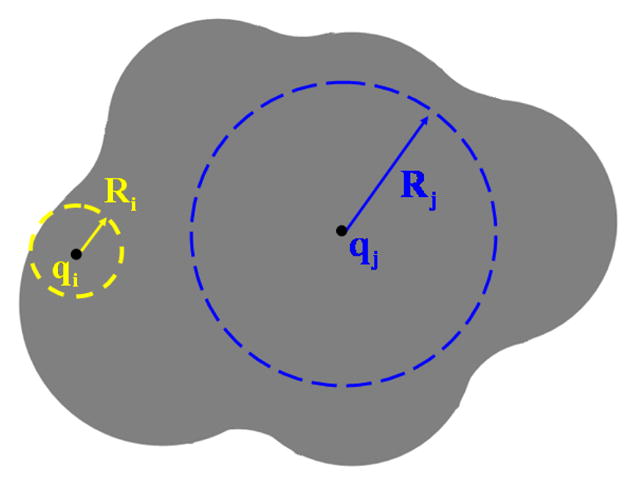
The effective Born radius reflects how deep a charge is buried inside the molecule. The Born radius of an atom is small if the atom is close to the surface of the molecule, otherwise the Born radius is large therefore has weaker interaction with the solvent.
Applying the Gaussian quadrature, We compute (3.1) numerically:
| (3.2) |
where wk and rk are the Gaussian integration weights and nodes on Γ (Figure 3.2). rk are computed by mapping the Gaussian nodes of a master triangle to the algebraic patch via the transformation 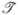 . Let
and
be one of the Gaussian nodes and weights on the master triangle. Then the corresponding node rk and weight wk are
. Let
and
be one of the Gaussian nodes and weights on the master triangle. Then the corresponding node rk and weight wk are 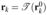 and
and 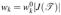 where |J()| is the Jacobian determinant of .
where |J()| is the Jacobian determinant of .
Fig. 3.2 .

Gaussian integration points on the surface of protein (a) 1PPE, (b) 1ANA, (c) 1MAG, and (d) 1CGI l. The surfaces are partitioned into 24244 triangular patches for (a), 28620 triangular patches for (b), 30624 triangular patches for (c), and 29108 triangular patches for (d). There are three Gaussian quadrature nodes per triangle. The nodes are then mapped onto the ASMS to form the red point cloud.
We formalize (3.2) in two steps. First we split it into two parts:
| (3.3) |
Then we split the second summation in (3.3) into three components:
| (3.4) |
The first summation in (3.3) and the three summations in (3.4) without the coefficients in front are of the common form:
| (3.5) |
with the kernel function and the coefficient ck = wkrk · n(rk), , respectively. (3.5) can be efficiently computed by using the fast summation algorithm introduced in [26] with complexity O(M + N + n3 logn), where n is a parameter used in the fast summation algorithm.
3.2. Fast summation
The fast summation algorithm is published in [26]. For convenience, we discuss this algorithm in this section briefly. The fast summation algorithm is often applied to compute the summations of the form
| (3.6) |
where the kernel function g is a fast decaying function. Cutting off the tail of g, one can assume that the support of g is bounded. In our Born radii computation, since the distance between xi and rk is no less than the smallest radius of the atoms, there is no singularity in g. Without loss of generality, we assume . After duplicating g in the other intervals, g can be extended to be a periodic function of period one in ℝ3 and this periodic function can be decomposed into the Fourier series:
| (3.7) |
where I∞:= {(ω1, ω2, ω3) ∈ ℤ3} and gω = ∫Π g(x)e−2πiω·x dx. We approximate (3.7) by a truncated series:
| (3.8) |
where . We compute the Fourier coefficients gω numerically by
| (3.9) |
by using the fast Fourier transform (FFT) algorithm with complexity O(n3 logn).
Plugging (3.8) into (3.6), we get
| (3.10) |
where
| (3.11) |
(3.10) is computed by using the NFFT algorithm with complexity O(n3 logn +M) and (3.11) is computed by the NFFTT algorithm with complexity O(n3 logn +N). Hence the total complexity of computing (3.6) is O(N + M + n3 logn), which is significantly faster than the the trivial O(MN) summation method once the number of terms in the Fourier series n is much smaller than M and N. We explain the NFFT algorithm and the NFFTT algorithm in Appendix A and B, respectively.
3.3. Error analysis
The numerical analysis of the error introduced during the computation of (3.1) can be decomposed as follows: (i) the sum of a quadrature error EQ; (ii) some “fast computation” error in the evaluation of the quadrature itself. The latter error is then decomposed in three terms, which correspond to different steps in the numerical procedure. They are the truncation error EFS when we truncate the Fourier series (3.7) into finite terms, NFFTT errors Eω when we compute the coefficients (3.11), and an NFFT error ENFFT when we finally evaluate (3.10) by the NFFT algorithm.
Let Ii and Ĩi denote the exact integration and the numerical output of (3.1) for atom i, respectively. Then We have
Let . We have
| (3.12) |
Next we will analyze each individual error ||EQ||∞, ||EFS||∞, ||ENFFT||∞, and ||ENFFTT||∞.
3.3.1. Quadrature error
Let Γe be one of the algebraic patches on the molecular surface Γ. Suppose Γe is built based on a triangle e:= [vi, vj, vk]. Any point (b1, b2, b3) ∈ e can be mapped to a point r(b1, b2) ∈ Γe. The integration (3.1) over Γe is
| (3.13) |
where Ω0 is the canonical triangle, (b1, b2, b3) is the barycentric coordinates of the points in Ω0 and |J| is the Jacobian. Let f (b1, b2) denote the integrand in (3.13). As we discuss in Appendix C, f (b1, b2) ∈ C∞(Ω0). Suppose we use an s-th order quadrature rule on element e, then
| (3.14) |
We expand f(b1, b2) in a Taylor series around a point :
| (3.15) |
where Ps(b1, b2) is a polynomial of degree s:
| (3.16) |
and the residue Rs is
| (3.17) |
Then the error E becomes
Let Wk = max(|wk|), we get
Within Ω0, and , hence
| (3.18) |
where denotes . By the chain rule,
According to (2.3), we have . Let hmax be the maximum edge length of the triangular mesh, λmax = max{|λ |}, and h = max(hmax, λmax). Then we have . Similarly, we can get the same bound for the derivatives of x, y, z with respect to b1 and b2. Therefore
| (3.19) |
where , and the constant . Noticing that the area of Ω0 is 1/2, we can write
| (3.20) |
Even though a greater number of quadrature nodes correspond to the higher order of accuracy, the increase in complexity is a limiting factor. Meanwhile, since the ASMS error is of the order h3, there is no point in a very accurate approximation of (3.20) to too high an order. As a trade-off, we use a two dimensional 3-point Gaussian quadrature over the triangle Ω0 which is of order 2 [27]. So s = 2 and se = 3. The nodes are ( ) and its permutations. for k = 1,2,3. Then
| (3.21) |
Suppose there are Ne patches on Γ, then |EQ| ≤ 2NeCh3. So we have the same bound
| (3.22) |
3.3.2. Fast summation error
According to the fast summation method described in Section 3.2, the Fourier series is truncated into a finite series
where denotes the truncation error of the Fourier series. Hence
| (3.23) |
where ,
| (3.24) |
with
| (3.25) |
and g being the the kernel function in the fast summation. In the Born radii calculation, . As defined in Section 3.2, Π is bounded and excludes 0. Let ω = (ω1, ω2, ω3). Then we rewrite Σω∈I∞\In|gω| as
| (3.26) |
By successive integration by parts for each dimension, we get
where m = m1 + m2 + m3 and . Therefore
Let μm = ∫Π |Dmg(x)| dx. We obtain . For the other terms in (3.26) we have the same upper bound. If we assume m1, m2, m3 ≥ 2, then
For m1 = m2 = m3, we have
| (3.27) |
Then for (3.24), we have
| (3.28) |
In fact, the right hand side of (3.28) is independent of i. Therefore we get
| (3.29) |
3.3.3. NFFT error
The error analysis of the NFFT algorithm is thoroughly discussed at the end of Appendix A. This error estimation is derived based on the analysis in [28]. In summary, the NFFT error is split into the aliasing error and the truncation error [28]:
The error bounds of and are
| (3.30) |
| (3.31) |
where ξ is a 1-periodic window function defined in Appendix A, Cω (ξ) are the Fourier co-efficients of ξ, and η is a truncated version of ξ. In the fast summation method (3.10), ||Ĝ||1 = Σω∈In|gωaω|, where gω and aω are defined in Section 3.2. Combining (3.30) and (3.31), one obtains
| (3.32) |
In [26], the coefficient C(ξ, m, σ) is given for some special ξ. They are
3.3.4. NFFTT error
As we mentioned in Section 3.2, (3.11) is computed by the NFFTT algorithm and then they are plugged in (3.10) for the following evaluation of the summation. So the NFFT error ENFFTT is
| (3.33) |
where Eω denotes the error of the NFFTT algorithm and gω is the same as is defined in (3.25). Then we have
| (3.34) |
with and .
As we discussed in Appendix B, the NFFTT error Eω is decomposed into the aliasing error ( ) and the truncation error . So
where and . Based on the error bounds derived in Appendix B,
| (3.35) |
and
| (3.36) |
where . Comparing (3.35) with (3.30) and comparing (3.36) with (3.31) yield the error estimation of Eω which is similar to ENFFT:
Hence
| (3.37) |
The inequality (3.37) is independent of i, therefore,
| (3.38) |
4. Fast solvation force computation
The solvation force acting at the center of atom α, which is part of the forces driving dynamics is
| (4.1) |
Partition the solvation energy into polar and non-polar parts:
| (4.2) |
The non-polar force is proportional to the derivatives of the volume and/or the surface area with respect to the atomic coordinates. There has been previous work on analytically computing the derivatives of the area/volume [31, 32, 33]. To compute the polar force, we first define
| (4.3) |
Then
| (4.4) |
Differentiating (4.4) w.r.t. x, one gets
| (4.5) |
where
| (4.6) |
From (4.3), one can easily compute and , which are
is nonzero if i or j = α which will be . In (4.6) the computation of for i = 1,…,M is not trivial. Because Γ depends on the position of the atoms, it is not easy to compute the derivative of Ri directly from (3.1). To solve this problem, we convert the integration domain back to the volume:
| (4.7) |
Then by defining a volumetric density function to distinguish the exterior from the interior of the molecule, we may have an integration domain that is independent of {xi}. One way of defining the volumetric function is given in [34] where they first define a density function for each of the atoms
and then define the volumetric function by following the inclusion-exclusion principle
| (4.8) |
There are some nice properties of this model. For example, the exterior region of the molecule is well characterized by ρ = 0 and two atoms i and j are disconnected if for any r ∈ ℝ3, χi(r)χ j(r) = 0. The drawback of this model is that function χ is not smooth, which makes it inapplicable to the derivative computation. Therefore we smoothen χ by introducing a cubic spline near the atom boundary:
| (4.9) |
with x =|| r − xi||. The region defined by ρi ≠ 0 is regarded as the interior of atom i and this region converges to the van der Waals volume of the atom as w goes to 0. In the SES model, two atoms are considered to be completely separated if the distance between the centers is greater than the sum of the radii plus the probe diameter. Otherwise they can be connected by the reentrant surface of the rolling probe. By setting w = 1.4 Å, atoms i and j are disconnected in the same sense as in the SES model iff ρi(r)ρj(r) = 0, for any r ∈ ℝ3. In addition to this modification, we neglect the cases that more than four atoms overlap simultaneously. Therefore the molecular volumetric density function becomes
| (4.10) |
We define the complementary function ρ̄ = 1 − ρ. It is easy to show that within the VWS of the molecule, ρ̄ is always 0, beyond the SAS, ρ̄ is always 1, in between, 0 < ρ̄ < 1. Then (4.7) can be rewritten as
| (4.11) |
Differentiating both sides of (4.11), one gets
| (4.12) |
So
| (4.13) |
For the first integral in (4.13),
where j, k, l are the atoms overlapping with atom α, g = 1 − Σj ρj + Σj<k ρjρk − Σj<k<l ρjρkρl, and
with x = ||r − xα||. Noticing that only if aα < |r − xα| < aα + w, the first integral in (4.13) is simplified as
| (4.14) |
The integration domain of (4.14) is a regular spherical shell of the width w around atom α (Figure 4.1(a)). We switch to the spherical coordinate system:
where (r, θ, φ) ∈ [0, w] × [0, 2π] × [0, π]. We sample r, θ, φ by using the 2-point Gaussian quadrature nodes in each dimension. For all the atoms in the molecule, they share the same set of sampling points (r, θ, φ).
Fig. 4.1 .

When computing the derivatives of the Born radii , the quadrature points of the first integral are points within a spherical shell around atom α, as shown in (a), whereas the second integral is necessary when i ≡ α and the quadrature points are points on the surface, as shown in (b). The dark region represents the molecule, the light grey region is the shell of width w around atom α.
The second integral in (4.13) is nonzero if i ≡ α. In that case
| (4.15) |
We compute each component of (4.15) individually and convert to the surface integration (Figure: 4.1(b)) by the divergence theorem:
| (4.16) |
| (4.17) |
| (4.18) |
where the quadrature weights and points (wk, rk) and the unit normals ( ) are the same as those used in Section 3. We compute (4.16), (4.17), and (4.18) by directly applying the fast summation method with the coefficients , respectively. Since the same algorithm is used in the Born radius derivative calculation, the error analysis is similar to the error analysis of the Born radius calculation except that a quadrature error of the integration over the shell region needs to be added.
To compute the force acting on each of the M atoms, we need to compute (4.15) for i = 1, …, M. By using the fast summation algorithm, the computational complexity of this part is O(N + M + n3 log n), the same as the energy computation. To compute (4.14), since the shell integration domain is narrow, only a small number of atoms have non-zero densities in this region, therefore the complexity of computing (4.14) for a fixed α for i = 1, …, M is O(M). Moreover, since the integrand in (4.14) is very small if atom i and atom α are far apart, we use a cut-off distance d0 in our computation and compute (4.14) only if d(i, α) ≤ d0. Therefore the overall time complexity of computing (4.13) is O(N +M +n3 log n).
5. Results
We compare the polarization energy computed based on the fast summation algorithm and the trivial summation in Table 5.1 for four proteins (PDB ID: 1CGI_l, 1BGX, 1DE4, 1N2C). An ASMS model is constructed for each protein with Ne number of patches. A three-point Gaussian quadrature is used on each algebraic patch. We also compare the overall computation time of the two methods. As we see from the table, for the small proteins (e.g. 1CGI_l), the fast summation method is slower than the trivial summation. However as the protein size gets larger (e.g. 1BGX, 1DE4, 1N2C), the fast summation is apparently faster than the trivial summation without losing too much accuracy. The relative error ε between the fast summation and the trivial summation is small. As for the trade-off between efficiency and accuracy, since in the current research of the MD simulation efficiency is more concerned, the fast-summation-based GB is superior to the trivial GB method.
Table 5.1.
Comparison of the electrostatic solvation energy Gpol (kcal/mol) and computation time (second) of the fast summation method (A) and the trivial summation method (B). M is the number of atoms. Ne is the number of patches, N is the number of integration points. n, σ, and m are parameters in the fast summation method. ε is the relative percentage error .
| Protein ID | 1CGI_l | 1BGX | 1DE4 | 1N2C | |
|---|---|---|---|---|---|
| M | 852 | 19,647 | 26,003 | 39,946 | |
| Ne | 29,108 | 112,636 | 105,288 | 83,528 | |
| N | 116,432 | 450,544 | 421,152 | 334,112 | |
| n | 100 | 100 | 100 | 100 | |
| σ | 2 | 2 | 2 | 2 | |
| m | 4 | 4 | 4 | 4 | |
| A | Gpol | −1380.988 | −19734.848 | −25754.552 | −41408.959 |
| timing | 86 | 358 | 863 | 631 | |
| B | Gpol | −1343.150 | −19297.528 | −25388.455 | −40675.383 |
| timing | 49 | 4327 | 5368 | 9925 | |
| ε | 2.8% | 2.3% | 1.4% | 1.8% | |
In Figure 5.1 we compare Gpol computed by the fast summation based GB and the trivial summation method along with their computation time for proteins of various sizes. For all these proteins, we generate the ASMS of the same number of patches (in our test we use 20,000 patches for each protein). We choose the fixed parameters n = 30, m = 4, and σ = 2 for all the proteins. We observe that the Gpol computed by the fastsum GB is close to that computed by the trivial GB methods and the error gets larger as the molecule gets bigger. Even though the error analysis in Section 3.3 does not show that the error depends on the size of the molecule, the analysis is based on the assumption that the kernel function is defined on the domain . To ensure that xi − rk, i = 1, …, M, k = 1, …, N are all within this range, we scale the molecule. The larger the molecule, the larger the scaling factor. Later on when we scale back to the original coordinates by multiplying the scaling factor, the error gets amplified. As we expect, computation time of the fastsum GB increases as M becomes large but is much faster than the traditional GB method.
Fig. 5.1 .

In (a) we compare Gpol computed by the fastsum GB and the non-fastsum GB for various proteins containing different number of atoms. In (b) we compare the computation time of the two methods.
In Figure 5.2, we compare Gpol computed by the fast summation based GB versus the trivial summation method and the computation time for a test protein 1JPS where we generate the ASMS with different numbers of patches. We use the same values for the parameters n, m, and σ as in the previous test. As shown in the figure, as the triangular mesh becomes denser, the fast summation result converges rapidly to the result of the trivial method but takes less computation time.
Fig. 5.2 .

For protein 1JPS, in (a) we compare Gpol computed by the fastsum GB and the non-fastsum GB with various number of surface elements. In (b) we compare the computation time of the two methods.
For the test proteins 1ANA, 1MAG, 1PPE_l, 1CGI_l, we compute the solvation force , for α = 1, …, M. We show the timing results in Table 5.2. In general, if an atom has a strong solvation force, this atom is in favor of being polarized, and hence is an active atom. On the contrary, if an atom has a weak solvation force, it is more likely to be an inactive atom. For every test protein, after we compute the solvation force for each atom, we sort the forces based on their magnitude and choose the top most active atoms and the top inactive atoms. As shown in Figure 5.3, the top 5% of the most active atoms are rendered in red and the bottom 5% of the atoms are rendered in blue. This provides a convenient and cheaper way, alternative to the experimental method, to help the biologists quickly find an active site of a protein.
Table 5.2.
Force calculation timing: M is the number of atoms, N is the number of triangles in the surface triangular mesh, t1 is the time (in seconds) for computing (4.15) for i = 1, …, M and t2 is the time for computing the rest of the terms in (4.6) for i, j, α = 1,…, M. Ttotal is the overall timing.
| Protein ID | M | N | t1 (s) | t2 (s) | Ttotal (s) |
|---|---|---|---|---|---|
| 1ANA | 249 | 6,676 | 66.05 | 0.14 | 66.19 |
| 1MAG | 544 | 7,328 | 69.58 | 0.23 | 69.81 |
| 1PPE_l | 436 | 5,548 | 59.55 | 0.56 | 60.11 |
| 1CGI_l | 852 | 6,792 | 68.71 | 3.27 | 71.98 |
Fig. 5.3 .

Atoms that have the greatest electrostatic solvation force (top 5%) are colored in red; atoms that have the weakest electrostatic solvation force (bottom 5%) are colored in blue.
6. Conclusion
We introduce a fast summation based algorithm to calculate the effective Born radii and their derivatives in the generalized Born model of implicit solvation. The algorithm relies on a variation of the formulation for the Born radii and an additional analytical volumetric density function for the derivatives. For a system of M atoms and N sampling points on the molecular surface, the trivial way of computing the Born radii requires O(MN) arithmetic operations, whereas with the aids of the Fourier expansion of the kernel functions of the Born radii (and their derivatives) and the NFFT algorithm which essentially approximates the complex exponentials in the NDFT by the DFT of a fast decaying smooth window function, the Born radii as well as their derivatives can be obtained at cost of (M +N +n3 log n) where n is the number of frequencies in the Fourier expansion. We show that the error of the algorithm decreases as the mesh gets denser, or as any of the parameters σ, m, n increase. Other than the Born model developed with a Coulomb field approximation, there has been other models for the Born radii evaluation, for example the Kirkwood-Grycuk model [35] where . This model is recently applied to the GBr6NL model which approximates the solvation energy of the nonlinear Poisson-Boltzmann equation [36]. It is interesting to note that we can utilize a similar quadrature point generation via ASMS and the fast summation algorithm to speed up this GBr6NL computation. In fact, by the divergence theorem, and the rest follows similar to the methods in this paper.
Fig. 2.2 .

The control coefficients of the cubic Bernstein-Bezier basis of function F
Acknowledgments
This research was supported in part by NSF grant CNS-0540033 and NIH contracts R01-EB00487, R01-GM074258, R01-GM07308. We thank the reviewers, as well as Dr. Rezaul Chowdhury for all the excellent suggestions that have resulted in a considerably improved paper. We also wish to thank several members of our CVC group for developing and maintaining TexMol, our molecular modeling and visualization software tool, which was used in conjunction with our nFFTGB implementation, to produce all the pictures in our paper (http://cvcweb.ices.utexas.edu/software/).
Appendix A. NFFT
The NFFT [28] is an algorithm for fast computation of multivariate discrete Fourier transforms for nonequispaced data in spacial domain (NDFT1). The NDFT1 problem is to evaluate the trigonometric polynomials
| (A.1) |
where . Without loss of generality, we assume . Instead of computing the summations in (A.1) directly, one can approximate G by a function s(x) which is a linear combination of the shifted 1-periodic kernel function ξ:
| (A.2) |
where and . We have σ > 1 because of the error estimation discussed in Section 3.3.3.
The kernel function ξ is defined as
Good candidates for ξ0 include Gaussian, B-spline, sinc, and Kaiser-Bessel functions. Expand the periodic kernel function ξ by its Fourier series
| (A.3) |
with the Fourier coefficients
Cut off the higher frequencies in (A.3), one can get
| (A4.) |
| (A.5) |
with the coefficients
| (A.6) |
By defining
| (A.7) |
one can immediately get
| (A.8) |
The next problem is to compute gl. From (A.6), one can compute the coefficients gl which are also coefficients in (A.8) by the discrete Fourier transform
| (A.9) |
with complexity O(n3 log n) by the FFT algorithm.
Since the function ξ drops very fast, one can further reduce the computation complexity of (A.8) by cutting off the tail of ξ. Define a function η0:
Construct the one-periodic function η the same way as ξ is constructed:
Replacing ξ with η in (A.8), we obtain that
| (A.10) |
where Iσn,m(xj) = {(l1, l2, l3): σnxj,i − m ≤ li ≤ σnxj,i + m, i = 1, 2, 3}. There are at most (2m + 1)3 nonzero terms in (A.10). Therefore the complexity of evaluating (A.10) for j = 1, …, M is O(m3M). Adding the complexity of computing the coefficients gl, the overall complexity of NFFT algorithm is O(n3 log n + m3M).
Remark
If we reorganize the above equations, it is not hard to see that, in fact, (A.1) is approximately computed by the expression
| (A.11) |
From a linear algebra point of view, equation (A.11) can be written as the product of a matrix and a vector. For example, for a one dimensional NFFT, (A.11) is equivalent to
| (A.12) |
with vectors
Ξ is a sparse matrix
F is the classical Fourier matrix
and D is an n × n diagonal matrix with the iith element being . For a multi-dimensional NFFT, it is the same as the 1D case as long as one orders the indices of the multi-dimension into one dimension.
As discussed in [28], in the first approximation (A.8), we see that s is equal to G after its higher frequencies in the Fourier series are cut off. Hence the error introduced in (A.8) which is known as the aliasing error is
| (A.13) |
Note that from (A.6), we have the condition G̃ω+iσn = G̃ω, for i ∈ ℤ3 and ω ∈ Iσn. By the definition (A.7), one obtains
| (A.14) |
Let . Then
| (A.15) |
In the second approximation (A.10), since ξ is replaced by η, the so caused error, known as the truncation error, is
| (A.16) |
Thus
| (A.17) |
Appendix B. NFFTT
The NFFTT algorithm deals with the fast computation of multivariate discrete Fourier transforms for nonequispaced data in frequency domain (NDFT2):
| (B.1) |
Define a function
| (B.2) |
where ξ is defined as same as in Appendix A. The Fourier series of A(x) is:
| (B.3) |
On the other hand,
| (B.4) |
Hence we get the relationship of Fourier coefficients of A and ξ:
| (B.5) |
Comparing (B.5) with (B.1) one obtains
| (B.6) |
It remains to compute Cω (A). By definition,
| (B.7) |
Discretizing the integration in (B.7) by the left rectangular rule leads to
| (B.8) |
Replacing ξ with η yields
| (B.9) |
where
| (B.10) |
To compute ĝl, if one scans the rk list, then for each rk there are at most (2m + 1)3 grid points (l) that contribute nonzero η. Hence, the complexity of computing ĝl is O(m3N). After computing ĝl one can easily evaluate (B.9) by the FFT algorithm at the complexity of O(n3 log n). Lastly the complexity of computing (B.6) is O(n3). So the overall complexity of the NFFTT algorithm is O(m3N +n3 log n).
Remark
Similar to the NFFT algorithm, we may write the one-line formula for computing (B.1) by the NFFTT:
| (B.11) |
which in one dimension is equivalent to the linear system:
| (B.12) |
with vectors
Matrix Ξ is similar to that defined in Appendix A
F* is the conjugate transpose of the Fourier matrix F, and D is the same as that defined in Appendix A. From the matrix expression, we see why the algorithm is called the “transpose” of NFFT.
Let Eω designate the error of a(ω). Eω can also be split into the aliasing error introduced in (B.8) and the truncation error introduced in (B.9), , for ω ∈ Iσn. By taking the Fourier expansion of ξ, we get form (B.8), so
Since
we have,
| (B.13) |
By (B.1),
| (B.14) |
Define . Then we have
| (B.15) |
In (B.9), the truncation error
| (B.16) |
which has the bound
| (B.17) |
Appendix C. Continuity of f
As defined in Section 3.3.1,
| (C.1) |
where r ≠ xi and n = ∇F with F given in (2.2). r(b1, b2, λ) is simply defined in (2.3). In this appendix, we mainly discuss the continuity of n. As derived in [22],
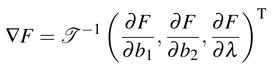 |
(C.2) |
where
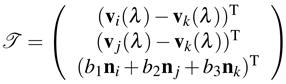 |
is a nonsingular matrix. Hence n is well defined. Consider ( ):
Let . We have
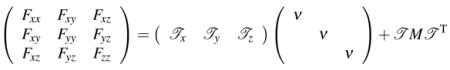 |
(C.3) |
where 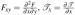 , and
, and
| (C.4) |
To show is differentiable, we take the first row of and compute its derivative with respect to x, i.e. (
) as an example. We write (2.3) in the form of
| (C.5) |
Taking the second derivatives of both sides of (C.5) with respect to x, we get
| (C.6) |
| (C.7) |
| (C.8) |
where
So we get
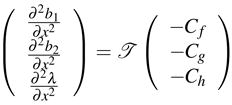 |
(C.9) |
Using the same method, we can get the other rows of 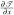 , matrices
, matrices 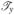 and
and 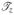 by changing Cf, Cg, Ch in (C.9). Therefore is differentiable. Similarly, we can compute the higher order derivatives of and prove that ∈ C∞, thus prove F ∈ C∞ (Ω0), where Ω0 defined in Section 3.3.1 is the canonical triangle. Therefore, as defined in (C.1), f ∈ C∞(Ω0).
by changing Cf, Cg, Ch in (C.9). Therefore is differentiable. Similarly, we can compute the higher order derivatives of and prove that ∈ C∞, thus prove F ∈ C∞ (Ω0), where Ω0 defined in Section 3.3.1 is the canonical triangle. Therefore, as defined in (C.1), f ∈ C∞(Ω0).
Footnotes
This research was supported in part by NSF grant: CNS-0540033, and in part by NIH grants: P20-RR020647, R01-GM074258, R01-GM073087, and R01-EB004873.
Contributor Information
CHANDRAJIT BAJAJ, Email: bajaj@ices.utexas.edu.
WENQI ZHAO, Email: wzhao@ices.utexas.edu.
References
- 1.Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roux B, Simonson T. Implicit solvent models. Biophys Chem. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 3.Weeks J, Chandler D, Andersen H. Role of repulsive forces in determining the equilibrium structure of simple liquids. J Chemical Physics. 1971;54:5237–5247. [Google Scholar]
- 4.Chandler D, Weeks J, Andersen H. Van der Waals picture of liquids, solids, and phase transformations. Science. 1983;220:787–794. doi: 10.1126/science.220.4599.787. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberg D, Mclachlan AD. Solvation energy in protein folding and binding. Nature (London) 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 6.Wagoner JA, Baker NA. Assessing implicit models for nonpolar mean solvation forces: The importance of dispersion and volume terms. 2006;103:8331–8336. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharp K. Incorporating solvent and ion screening into molecular dynamics using the finite-difference Poisson-Boltzmann method. J Comput Chem. 1991;12:454–468. [Google Scholar]
- 8.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 9.Holst M, Baker N, Wang F. Adaptive multilevel finite element solution of the Poisson-Boltzmann equation I. Algorithms and examples. J Comput Chem. 2000;21:1319–1342. [Google Scholar]
- 10.Baker N, Holst M, Wang F. Adaptive multilevel finite element solution of the Poisson-Boltzmann equation II. Refinement at solvent-accessible surfaces in biomolecular systems. J Comput Chem. 2000;21:1343–1352. [Google Scholar]
- 11.Lu B, Zhang D, McCammon JA. Computation of electrostatic forces between solvated molecules determined by the Poisson-Boltzmann equation using a boundary element method. J Chemical Physics. 2005;122:214102–214109. doi: 10.1063/1.1924448. [DOI] [PubMed] [Google Scholar]
- 12.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 13.MacKerel AD, Jr, Brooks CL, III, Nilsson L, Roux B, Won Y, Karplus M. CHARMM: The Energy Function and Its Parameterization with an Overview of the Program, volume 1 of The Encyclopedia of Computational Chemistry. John Wiley & Sons; Chichester: 1998. pp. 271–277. [Google Scholar]
- 14.Case D, Cheatham T, III, Darden T, Gohlke H, Luo R, Merz K, Jr, Onufriev A, Simmerling C, Wang B, Woods R. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren P, Ponder JW. Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B. 2003;107:5933–5947. [Google Scholar]
- 16.Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers. 2001;56:275–291. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 17.Shih Amy Y, Denisov Ilia G, Phillips James C, Sligar Stephen G, Schulten Klaus. Molecular dynamics simulations of discoidal bilayers assembled from truncated human lipoproteins. Biophys J. 2005;88:548–556. doi: 10.1529/biophysj.104.046896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ritchie David W. Evaluation of protein docking predictions using hex 3.1 in capri rounds 1 and 2. Proteins: Structure, Function, and Genetics. 2003 July;52(1):98–106. doi: 10.1002/prot.10379. [DOI] [PubMed] [Google Scholar]
- 19.Ju T, Losasso F, Schaefer S, Warren J. Dual contouring of hermite data. Proceedings of ACM SIG-GRAPH. 2002:339–346. [Google Scholar]
- 20.Zhang Y, Xu G, Bajaj C. Quality meshing of implicit solvation models of biomolecular structures. Computer Aided Geometric Design. doi: 10.1016/j.cagd.2006.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garland M, Heckbert P. Simplifying surfaces with color and texture using quadric error metrics. IEEE Visualization. 1998:263–270. [Google Scholar]
- 22.Zhao W, Xu G, Bajaj C. An algebraic spline model of molecular surfaces. ACM Symp Sol Phys Model. 2007:297–302. doi: 10.1109/TCBB.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greengard L, Rokhlin V. A fast algorithm for particle simulations. J Chemical Physics. 1987;73:325–348. [Google Scholar]
- 24.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks C., III Performance comparison of generalized Born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh A, Rapp CS, Friesner RA. Generalized Born model based on a surface integral formulation. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 26.Potts D, Steidl G. Fast summation at nonequispaced knots by NFFTs. SIAM J Sci Comput. 2003;24:2013–2037. [Google Scholar]
- 27.Dunavant D. High degree efficient symmetrical Gaussian quadrature rules for the triangle. International Journal of Numerical Methods in Engineering. 1985;21:1129–1148. [Google Scholar]
- 28.Potts D, Steidl G, Tasche M. Modern Samplling Theory: mathematics and Applications. Birkhauser; 2001. Fast Fourier transforms for nonequispaced data: A tutorial; pp. 247–270. [Google Scholar]
- 29.Beylkin G. On the fast Fourier transform of functions with singularities. Appl Comput Harmon Anal. 1995;2:363C–381. [Google Scholar]
- 30.Jackson JI. Selection of a convolution function for Fourier inversion using gridding. IEEE Trans Med Imag. 1991;10:473–C478. doi: 10.1109/42.97598. [DOI] [PubMed] [Google Scholar]
- 31.Edelsbrunner H, Koehl P. The weighted-volume derivative of a space-filling diagram. 2003;100:2203–2208. doi: 10.1073/pnas.0537830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Im W, Lee MS, Brooks C., III Generalized Born model with a simple smoothing function. J Comput Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 33.Bryant R, Edelsbrunner H, Koehl P, Levitt M. The area derivative of a space-filling diagram. Discrete Comput Geom. 2004;32:293–308. [Google Scholar]
- 34.Grant JA, Pickup BT. A Gaussian description of molecular shape. J Phys Chem. 1995;99:3503–3510. [Google Scholar]
- 35.Grycuk T. Deficiency of the Coulomb-field approximation in the generalized Born model: An improved formula for Born radii evaluation. J Chemical Physics. 2003;119:4817–4827. [Google Scholar]
- 36.Tjong H, Zhou H. GBr6NL: A generalized Born method for accurately reproducing solvation energy of the nonlinear Poisson-Boltzmann equation. J Chemical Physics. 2007;126:195102–195106. doi: 10.1063/1.2735322. [DOI] [PubMed] [Google Scholar]