

Abstract
Hyperlipidemia with accompanying increase in peripheral inflammation is a risk factor for stroke. The effect of excess lipids on stroke-induced injury and the mechanism by which lipid-mediated inflammatory responses contribute to stroke are not known. We investigated these uncertainties by subjecting normal and hyperlipidemic mice to transient middle cerebral artery occlusion, followed by measurement of stroke severity and inflammatory response. Infarct size, swelling, and lipid contents were significantly increased in the high-fat fed ApoE knock-out mice, as was the expression of the inflammatory mediators CD36 and monocyte chemoattractant protein 1 (MCP-1) in the brain and periphery. Furthermore, the hyperlipidemic mice exhibited numerous foam cells, a probable cause of increased swelling and postischemic inflammation, in the peri-infarct area. Genetic deletion of cd36 in the hyperlipidemic condition reduced proinflammatory chemokine/receptor and cytokines (MCP-1, CC chemokine receptor 2, and interleukins 1β and 6), in the brain 6 h after ischemia. The reduced proinflammatory response also resulted in smaller ischemic injury, less swelling, and fewer foam cells at 3 d after ischemia. The results show that hyperlipidemia-induced inflammation is a negative factor for stroke outcomes and indicate that downregulating CD36 may be an effective therapeutic strategy for reducing the impact of stroke in hyperlipidemic subjects.
Keywords: CD36, foam cell formation, inflammation, ischemia, macrophage, stroke
Introduction
Strategies that are effective in reducing stroke severity in animal models have been essentially ineffective in clinical trials. The lack of successful translation from laboratory to the clinic may be the results of prevailing clinical conditions, such as hyperlipidemia, hypertension, and insulin/glucose intolerance, which are correlated with higher incidence of stroke (Strandgaard, 1996; Engström et al., 2002, 2003; Hutter et al., 2004; Kernan and Inzucchi, 2004; Pinto et al., 2004), not being included in animal models of stroke.
Among the risk factors, hyperlipidemia is a risk factor for stroke. Although hypercholesterolemia per se is not a direct predictor of stroke, the condition with high plasma levels of inflammatory-sensitive proteins links to the increased incidence of stroke (Engström et al., 2002). Positive correlations between excess lipid levels and the promotion of inflammation have been documented. Lipid-lowering drugs such as statins have also been shown to antagonize inflammation (Evans et al., 2004; Willerson and Ridker, 2004; Elkind, 2006). A promising model of hyperlipidemic condition is the spontaneously hyperlipidemic ApoE knock-out (KO) mice, in which a high-fat diet (HD) induces inflammation and exacerbates development of severe lesions in arteries (Nakashima et al., 1994).
CD36, also known as a fatty acid translocase, belongs to a class B scavenger receptor and shows high affinity toward lipid-based ligands such as oxidized (oxLDL) or modified (mLDL) low-density lipoprotein and long chain-fatty acids among other ligands, including thrombospondins, fibrillar β-amyloid, and membranes of cells undergoing apoptosis (Abumrad et al., 1993; Savill, 1997; Febbraio et al., 2001; Medeiros et al., 2004). The receptors are expressed in microglia, monocytes/macrophages, microvascular endothelium, cardiac and skeletal muscle, adipocytes, and platelets. The multiple ligands/receptor interaction elicits a wide variety of physiological and pathological processes, including inflammation, lipid metabolism, phagocytosis, angiogenesis, and migration of microglia (Febbraio et al., 2001; Hirano et al., 2003; Stuart et al., 2007).
Several studies implicate a role of CD36 in atherosclerosis and inflammation. Although the effect of CD36 on atherogenesis is debatable (Moore et al., 2005; Moore and Freeman, 2006), uptake of proinflammatory oxLDL or mLDL by macrophage CD36 is a critical step that leads to foam cell formation, atheroma, and a chronic proinflammatory state (Febbraio et al., 2000; Podrez et al., 2002a,b; Rahaman et al., 2006; Guy et al., 2007). In CD36-deficient macrophages, oxLDL-induced proinflammatory gene expression has also been shown to be defective (Janabi et al., 2000). In addition, CD36 mediates innate host response in response to β-amyloid, linking CD36 with the proinflammatory events associated with Alzheimer's disease (El Khoury et al., 2003).
Ischemia reperfusion elicits a series of inflammatory events that subsequently attract peripheral immune cells into ischemic territory (Iadecola and Alexander, 2001; Iadecola et al., 2004; Huang et al., 2006). Previously, CD36 has been shown to function as an inflammatory mediator in ischemic brain injury (Cho et al., 2005). Furthermore, attenuating CD36 pathways by a novel cell-permeable antioxidant peptide ameliorates ischemia-induced injury (Cho et al., 2007). These observations and a close correlation between hyperlipidemia and inflammation led us to the hypothesis that CD36 could mediate postischemic inflammation in hyperlipidemia. By coupling hyperlipidemia in an experimental mouse stroke model, the present study investigates whether hyperlipidemia exacerbates ischemic outcomes. In addition, we addressed whether any exacerbation of stroke outcomes was linked to CD36, by using cd36-deficient hyperlipidemic mice. We report that hyperlipidemia exacerbates ischemia-induced injury by promoting inflammation in both the CNS and the periphery and that CD36 mediates the exacerbation.
Materials and Methods
Animals and diet.
The use of animals and procedures performed were approved by the Institutional Animal Care and Use Committee of Weill Medical College of Cornell University. Experiments were performed in wild-type (WT), ApoE KO, and ApoE/CD36 double knock-out (DKO) mice. These mice were generated at Weill Cornell Medical College by Dr. Maria Febbraio and housed at the Burke Medical Research Institute. These mice were backcrossed six times into the C57BL/6 strain, and the procedures for breeding and genotyping were described previously (Febbraio et al., 1999, 2000). Male mice were weaned at 4 weeks of age and fed normal chow (4.5% fat, 0% cholesterol; W. F. Fisher & Son, Somerville, NJ) for 2 weeks. After a 2 week acclimation period, some mice were continued on the normal diet (ND) or were switched to an HD (catalog #88137; Harlan Teklad, Madison, WI) for 8 weeks. The HD contains 21% fat by weight, adjusted calories (42% from fat) and 0.15% (w/w) cholesterol. Previously, WT, ApoE KO, and DKO mice that were fed the same HD showed higher levels of total cholesterol, free fatty acids, and triglycerol in plasma (Febbraio et al., 2000, 2004).
Measurement of plasma cholesterol levels.
At the end of 7 weeks of feeding, tail bloods from overnight fasted mice were collected in citrated buffer [25 mm citric acid, 75 mm sodium citrate, 136 mm glucose, and citrate buffer/blood (1:8)]. The diluents were centrifuged at 6000 rpm for 5 min at room temperature. The plasma was kept at −80°C until the measurement of cholesterol level using a cholesterol measuring kit (BioVision, Mountain View, CA).
Transient middle cerebral artery occlusion.
Procedures for middle cerebral artery occlusion (MCAO) were described previously (Cho et al., 2005, 2007). Briefly, mice were anesthetized with a mixture of isoflurane/oxygen/nitrogen. A fiber optic probe was glued to the parietal bone (2 mm posterior and 5 mm lateral to the bregma) and connected to a laser-Doppler flowmetry (Periflux System 5010; Perimed, Järfälla, Sweden) for continuous monitoring of cerebral blood flow (CBF) in the center of the ischemic territory. For MCAO, a 6-0 Teflon-coated black monofilament surgical suture (Doccol, Redland, CA) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the cerebral arterial circle to obstruct the origin of the MCA. The filament was left in place for 30 min and then withdrawn. This procedure typically reduces CBF >80% during MCAO and restores CBF >80% by 10 min of reperfusion and leads to reproducible infarcts involving both the cerebral cortex and the striatum.
Because genetic background and diet intervention may induce vascular changes that contribute to the exacerbation, we examined fatty streaks in internal, external, and common carotid arteries in a preliminary study. ApoE KO mice with 12 weeks of HD exhibited numerous fatty streaks in the arteries and a high mortality after ischemia. To circumvent the problem, we shortened the HD for 8 weeks. The modification did not cause gross fatty streaks. To optimize the acute ischemic condition for ApoE KO mice fed an HD for 8 weeks, we initially subjected them to 30 min focal ischemia with a preset rectal temperature probe at 37°C. The probe was thermocouple regulated by a heated water pump to maintain constant body temperature during ischemia and up to 1 h of reperfusion. Of 14 control mice, 13 survived up to 48 h, whereas only 7 of 13 hyperlipidemic mice survived for the same time period (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Among the surviving hyperlipidemic mice, infarct size was significantly increased compared with that in normal mice (ND/WT, 45.6 ± 2.9 mm3; HD/KO, 55.5 ± 5.1 mm3; t test, p < 0.05) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Temperature during ischemia and early reperfusion (<1 h) is one of the critical factors affecting the extent of ischemic injury. To investigate whether the high mortality observed in the hyperlipidemic mice may have been associated with an inadvertent high brain temperature during ischemia/recovery, we established a relationship among rectal (YSI probe; YSI, Yellow Springs, OH), abdominal (type J needle probe; Cole-Palmer Instrument, Vernon Hills, IL), and brain temporal muscle (type J needle probe) temperatures. The temperature in the temporal muscle of the head was almost identical to that of abdominal muscle that reflects core temperature of the body. Conversely, rectal temperatures registered on average 1.0° less compared with the abdominal temperatures in our surgical setting (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This indicates that fixing the rectal temperature at 37°C in our initial experiment resulted in a rise in the core temperature of ∼38°C during the procedure. Therefore, a type J probe with a set point of 37°C in the abdomen was used to maintain a mean core temperature at 37.0 ± 0.2°C during ischemia and reperfusion for the study.
Measurement of infarct volume and swelling and assessment of water and lipid contents.
Mice were killed 3 d after MCAO. Brains were excised, frozen, sectioned at a thickness of 30 μm to collect serially (600 μm interval), and stained with cresyl violet. Infarct volume and swelling were determined using Axiovision software (Carl Zeiss, Jena, Germany), and the contribution attributable to swelling was corrected using a method described previously (Lin et al., 1993). To assess water content, wet weight of each hemisphere excluding olfactory and cerebellum was measured. The tissue was then incubated in an oven for 24 h at 100°C to obtain dry weight. Percentage water content for each hemisphere was calculated by [((wet weight − dry weight) × 100)/wet weight]. Increase rate of percentage water in ipsilateral side was obtained by ipsilateral percentage water content/contralateral percentage water content
For lipid content assessment, the dried hemisphere was powdered using liquid nitrogen. Lipid was extracted using a mixture of chloroform/methanol (2:1) and centrifuged at 13,000 rpm for 5 min at room temperature to collect the lower lipid-containing phase. The procedure was repeated three times. Total lipid content was weighed after removal of solvent using a Savant SpeedVac concentrator (Global Medical Instrument, Ramsey, MN).
Tissue preparation for gene and protein measurement.
Infarct normally spans ∼6–7 mm rostrocaudally starting at approximately +2.8 mm and extending to −3.8 mm from bregma. To collect tissue in an unbiased manner that reflect infarct globally, we excised olfactory bulbs and then sliced brain at 1 mm intervals. Each set of alternate brain slices was used for either PCR (slices 1, 3, 5) or Western blot (slices 2, 4, 6) for gene and protein measurement.
Real-time reverse transcription-PCR for CD36, MCP-1, and ABCG1 gene expression.
Relative mRNA levels of CD36, monocyte chemoattractant protein 1 (MCP-1), ATP-binding cassette G1 (ABCG1), CC chemokine receptor 2 (CCR2), interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α), IL-6, and IL-13 were quantified with real-time quantitative reverse transcription-PCR using fluorescent TaqMan technology (Cho et al., 2007). Total RNA was extracted from brain tissues and peritoneal macrophages using TRI Reagent (Molecular Research Center, Cincinnati, OH) or RNeasy RNA purification kit (Qiagen, Valencia, CA), respectively. Total RNA from brain tissues or peritoneal macrophages was reverse transcribed using oligo-dT primers and the SuperScript First-Strand Synthesis System (Invitrogen, Carlsbad, CA), according to the protocol of the manufacturer. PCR primers and probes specific for CD36, MCP-1, ABCG1, and β-actin were obtained as TaqMan predeveloped assay reagents for gene expression (Applied Biosystems, Foster City, CA). β-Actin was used as an internal control for normalization of samples. The PCR reaction was performed using TaqMan Universal PCR Master mix, No AmpErase UNG, and Applied Biosystems 7500 Fast Real-Time PCR system, according to the instructions of the manufacturer. Reactions were performed in 20 μl total volume and incubated at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. The results were analyzed by 7500 Fast Real-Time PCR System software.
Western blot analysis of CD36 protein expression in peritoneal macrophages.
Resident macrophages were collected from mice peritoneal cavities by lavage with 15 ml of PBS. Harvested macrophages were washed with 1× PBS and homogenized in 40 μl volumes of radioimmunoprecipitation assay buffer (50 mm Tris-Cl, pH 8.0, 150 mm NaCl, 5 mm NaF, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm EDTA, and 1% Nonidet P-40) with freshly added protease inhibitor (Sigma, St. Louis, MO). After incubating for 5 min on ice, the homogenate was centrifuged at 8000 rpm for 2 min at 4°C, and the protein concentration of the supernatant fraction was determined using the Lowry method (Bio-Rad, Hercules, CA). CD36 protein expression was determined according to the method described previously (Cho et al., 2005, 2007). Two micrograms of protein were loaded on a gel, electrophoresed, and transferred to polyvinylidene difluoride membranes using an electroblotting apparatus. Membranes were treated for 1 h in blocking buffer (LI-COR, Lincoln, NE) and then incubated with an anti-murine CD36 monoclonal antibody (1:1000; AF2519; R & D Systems, Minneapolis, MN) or anti-β-actin antibody for normalization (1:1000; sc-1615; Santa Cruz Biotechnology, Santa Cruz, CA), followed by mouse Alexa Fluor 680 rabbit anti-goat IgG (Invitrogen). Membranes were washed and exposed using the Odyssey Imaging System (LI-COR).
Measurement of plasma MCP-1.
Plasma MCP-1 levels were determined with a commercially available mouse MCP-1 ELISA kit (R & D Systems). Trunk blood was collected from mice into a heparinized tube and centrifuged. Plasma was kept at −80°C until measurement of MCP-1 levels.
Foam cell assay.
Abundance of foam cells was assessed in the ischemic mouse brain 3 d after MCAO. Brains were removed, frozen, and sectioned at a thickness of 30 μm in a cryostat as described previously (Cho et al., 2007). Brain sections were collected serially on gelatin-coated slides at 600 μm intervals and fixed in methanol for 5 min. Fixed brain sections were washed in 0.1 m phosphate buffer (PB), pH 7.2, followed by methanol, and stained with Oil Red-O for 30 min in a moisturized chamber. Slides were rinsed three times with 0.1 m PB and coverslipped. Initial assessment of foam cell formation was performed in four serial sections spanning rostrocaudal levels (+2.8, +2.2, +1.6, and +1.0 from bregma) by applying the following criteria: 3, >100 foam cells; 2, 20–100 cells; 1, 1–20 cells; and 0, no foam cell. We observed that foam cells were most abundant in the third section (+1.6 from bregma), and the number of foam cells from these sections was counted for quantification. For in vitro foam cell assays, resident peritoneal macrophages from hyperlipidemic ApoE KO and ApoE/CD36 DKO mice were seeded in eight-well chamber slides. After an overnight incubation in Macrophage Serum Free Media (Invitrogen), cells were treated with 25 μg/ml oxLDL (Biomedical Technologies, Stoughton, MA) for 48 h. Cells were prefixed in methanol for 5 min and incubated with Oil Red-O for 30 min in a moisturized chamber at room temperature. To quantify foam cell formation, five separate areas (625 μm2 each) from quadruplicate preparations were blindly selected from each condition. Areas that stained with Oil Red-O were selected and measured using Adobe Photoshop (Adobe Systems, San Jose, CA). Values were expressed as total pixels ± SEM.
Immunohistochemistry.
Mice were perfused transcardially with 5 ml of PBS containing heparin sulfate (1000 U/ml) followed by 4% paraformaldehyde in 0.1 m PB. The brains were postfixed overnight, transferred to a 30% sucrose solution, and sectioned in a cryostat at a 30 μm thickness. The sections were incubated overnight with a polyclonal CD36 antibody (1:1000, sc-9154; Santa Cruz Biotechnology), followed by a tyramide signal amplification kit HRP goat anti-rabbit IgG for 1 h and then Alexa Fluor 488 tyramide (Invitrogen). The specificity of the CD36 immunolabeling was tested by omitting the primary antibody or by processing brain sections of CD36 KO mice.
Statistics.
Data are presented as mean ± SEM. Comparison between two groups was statistically evaluated using Student's t test. Multiple comparisons were made using ANOVA, followed by a post hoc Newman–Keuls test. Differences were considered significant at p < 0.05.
Results
Hyperlipidemia exacerbates ischemic injury and swelling
Using genetic and dietary interventions, we developed three mouse models exhibiting different levels of hyperlipidemia: HD/WT, wild-type mice fed HD; ND/KO, ApoE KO mice fed normal diet; and HD/KO, ApoE KO mice fed HD. Wild-type mice fed ND (ND/WT) were used for the normolipidemic control. In the hyperlipidemic groups (HD/WT, ND/KO, and HD/KO), weight gain and increased serum cholesterol levels occurred faster than in the ND/WT group (Table 1). All mice were subjected to 30 min MCAO at the end of 8 weeks of diet, and infarct size and hemispheric swellings were determined 3 d after ischemia. The severity of ischemia among the groups was similar in that reduction of CBF during ischemia and restoration of CBF at 10 min of reperfusion were comparable (Table 1).
Table 1.
Body weight and total plasma cholesterol levels in ND or HD mice in WT and ApoE KO mice
| ND/WT | HD/WT | ND/KO | HD/KO | |
|---|---|---|---|---|
| Body weight (g) | 25.5 ± 0.79 | 30.4 ± 1.21* | 28.4 ± 0.40** | 36.1 ± 0.93* |
| Plasma cholesterol (mg/dl) | 118.6 ± 6.1 | 253.9 ± 14.0* | 451.4 ± 73.3* | 885.2 ± 28.4* |
| CBF reduction (%) | 85.0 ± 1.2 | 83.7 ± 0.8 | 90.1 ± 1.7** | 87.8 ± 1.7 |
| CBF reperfusion (%) | 136.1 ± 6.7 | 121.8 ± 4.9 | 122.9 ± 4.2 | 119.1 ± 10.7 |
Percentage of CBF reduction during ischemia and percentage of CBF reperfusion at 10 min show comparable ischemic severity among groups. Data are expressed as means ± SEM; n = 8–10 per group. *p < 0.01, **p < 0.05, versus ND/WT.
The degree of hyperlipidemia was positively correlated with increased infarct size and swelling. A significant difference, however, was only observed between the ND/WT and the most hyperlipidemic HD/KO groups attributable to a relatively large variation in infract size and swelling (Fig. 1A–C). Therefore, the remainder of the study uses the HD/KO group and refers to the group as the hyperlipidemic group. Correlation analysis showed that swelling was positively correlated with infarct size within the ND/WT and HD/KO groups (Fig. 1D) (ND/WT, r2 = 0.4715, p = 0.0136; HD/KO, r2 = 0.528, p = 0.0176). Interestingly, the correlation between swelling and infarct size in hyperlipidemic HD/KO mice showed a disproportionately larger contribution of the swelling component compared with that in normal ND/WT mice (Fig. 1D) (slope: ND/WT, 0.202 ± 0.067; HD/KO, 0.946 ± 0.316). To investigate whether the disproportionately larger contribution of the swelling in HD/KO is attributable to the excess lipid, we directly measured water and lipid contents (Fig. 1E). As expected, we observed increased water content in ipsilateral side compared with contralateral side. Interestingly, the magnitude of the increased rate in ND/WT ipsilateral side was not different from that of HD/KO. In ND/WT mice, tissue damage resulted in reduced lipid contents in the ipsilateral side. However, the rate of ipsilateral/contralateral lipid content was significantly higher in HD/KO than that of ND/WT mice. Collectively, the findings suggest that excess lipid is a major component of swelling in the HD/KO hyperlipidemic brain.
Figure 1.

Exacerbation of ischemic outcomes in hyperlipidemia. A, Representative cresyl violet-stained sections 3 d after transient focal ischemia in normal (ND/WT) and hyperlipidemic (HD/KO) mice. B, C, Quantification of infarct volume and percentage hemispheric swelling. D, Correlation analysis between infarct size and percentage swelling in the HD/KO group reveals a distinct correlation pattern with a larger swelling component compared with that of ND/WT. n = 8–12 per group. E, Assessment of water and lipid contents 3 d after ischemia. It is expressed by ratios of ipsilateral to contralateral side. KO, ApoE KO mice; Ipsil, ipsilateral; Contral, contralateral. n = 5 per group, *p < 0.05 and **p < 0.01 versus ND/WT
Hyperlipidemia upregulates CD36 and MCP-1 in the postischemic brain
To investigate whether enhanced inflammation is associated with the hyperlipidemia-induced exacerbation of ischemic injury, we first determined CD36, MCP-1, and ABCG1 gene expression in normolipidemic mice after ischemia. Compared with the contralateral side, inflammatory mediators CD36 and MCP-1 mRNA levels were increased. Conversely, gene expression of ABCG1, which mediates the transport of cholesterol from cells to high-density lipoproteins, remained relatively unchanged (Fig. 2). On the basis of temporal gene expression in a normolipidemic brain, we compared gene expressions between the normal and hyperlipidemic conditions before ischemia and at a 6 h after ischemia because we expected additional increase in the expression of CD36 and MCP-1 in the hyperlipidemic condition at this time point. Hyperlipidemic mice exhibited a significant fold reduction in CD36 mRNA level before ischemia (ND/WT, 1.00 ± 0.17; HD/KO, 0.67 ± 0.11; p < 0.05), whereas no significant difference was noted in MCP-1 and ABCG1 mRNA levels (MCP-1: ND/WT, 1.00 ± 0.08; HD/KO, 1.37 ± 0.37, NS; ABCG1: ND/WT, 1.00 ± 0.17; HD/KO, 0.95 ± 0.18, NS). Despite the reduced baseline, hyperlipidemic, but not normolipidemic, mice showed significantly increased ischemia-induced CD36 gene expression (Fig. 3A). Ischemia also significantly induced MCP-1 gene expression in both normal and hyperlipidemic mice, but the effect was much larger in hyperlipidemic mice (Fig. 3B). No observable changes were noted in ABCG1 mRNA levels between the groups (Fig. 3C). Together, the data show that hyperlipidemia further upregulates ischemia-induced proinflammatory mediators in the brain.
Figure 2.
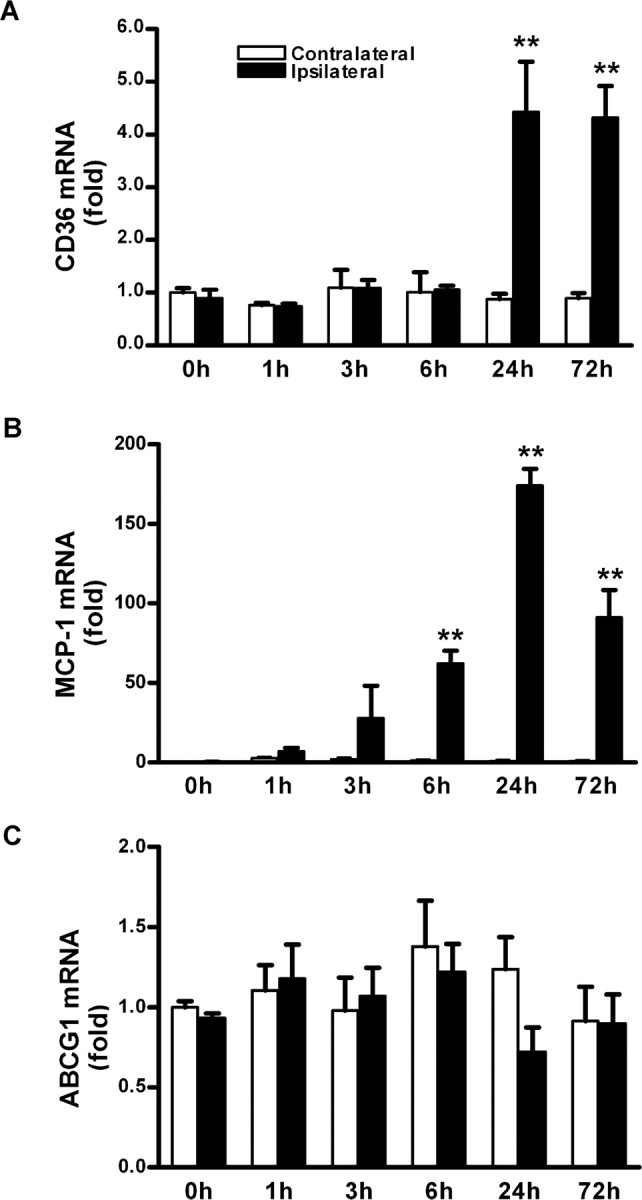
Temporal changes of CD36, MCP-1, and ABCG1 gene expression in the postischemic brain. A–C, mRNA levels of CD36 (A), MCP-1 (B), and ABCG1 (C) were determined before ischemia (0 h) and at different postischemic time points in wild-type mice that were fed normal chow after 30 min transient MCAO. β-Actin was used for an internal control. n = 4 per group. **p < 0.01 versus contralateral.
Figure 3.
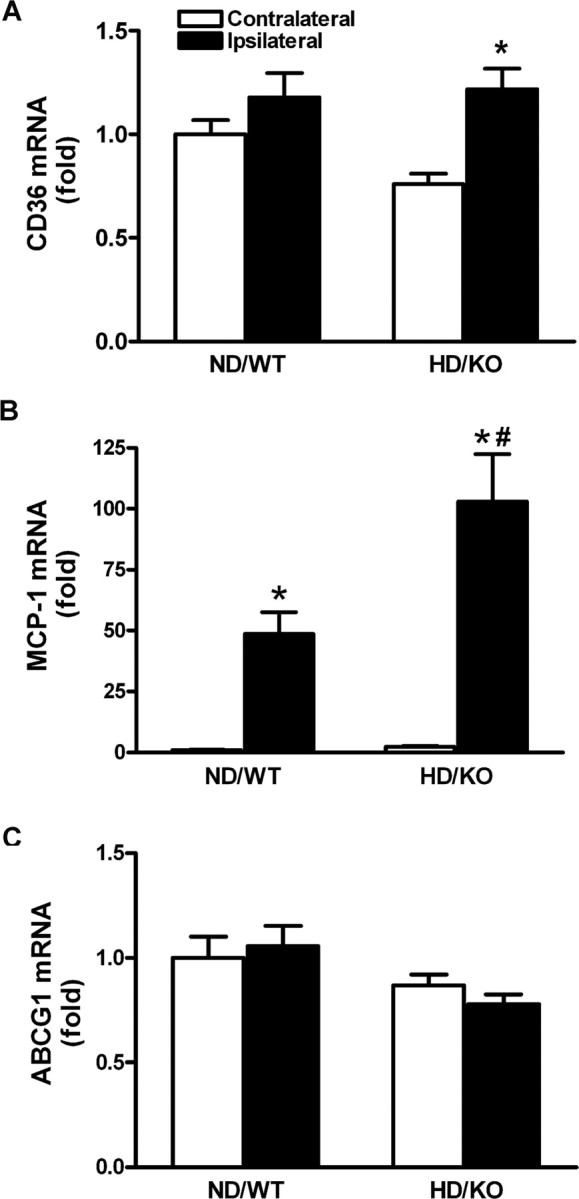
Ischemia induced upregulation of CD36 and MCP-1 gene expression in the hyperlipidemic brain. A–C, Brains from normal (ND/WT) and hyperlipidemic (HD/KO) mice at 6 h after ischemia were collected for determination of CD36 (A), MCP-1 (B), and ABCG1 (C) gene expression. n = 5 per group. *p < 0.05 versus contralateral; #p < 0.05 versus ND/WT ipsilateral.
Hyperlipidemia upregulates CD36 and MCP-1 in periphery
Our previous study also showed that CD36 expression occurs primarily in microglia/macrophages in the infarct territory (Cho et al., 2005). Because infiltrated inflammatory immune cells from the periphery into ischemic territory was implicated in the pathology of ischemic injury (Kochanek and Hallenbeck, 1992; Chen et al., 2003), we further assessed the inflammatory state of peripheral macrophages in the hyperlipidemic condition. There was a slight increase in CD36 mRNA in hyperlipidemic macrophages before ischemia, but no differences were found 3 d after ischemia (Fig. 4A). Conversely, CD36 protein expression was significantly upregulated in hyperlipidemic macrophages before ischemia, as well as 72 h after ischemia (Fig. 4B). Similarly, MCP-1 gene expression was significantly increased before ischemia (Fig. 4C). To further corroborate these genetic responses, plasma MCP-1 levels were likewise upregulated in the hyperlipidemic mice before and after ischemia (Fig. 4D). The data showed that the hyperlipidemic condition upregulates CD36 and MCP-1 in peritoneal macrophages and plasma.
Figure 4.

Enhanced CD36 and MCP-1 expression in the hyperlipidemic periphery. A, CD36 mRNA in normal and hyperlipidemic macrophages. B, Western blot analysis for CD36 protein in normal and hyperlipidemic macrophages. C, MCP-1 gene expression in normal and hyperlipidemic macrophages. D, Plasma MCP-1 level (picograms per milliliter). H, Heart (CD36-positive control); −/−, macrophages from CD36 KO mice (CD36 negative control); 0 h, nonischemic control; 6 or 72 h, postischemic time point; ND/WT, normolipidemic mice; HD/KO, hyperlipidemic mice. n = 4–5 per group. *p < 0.05 and **p < 0.01 versus ND/WT.
Hyperlipidemia promotes foam cell formation in the postischemic brain
The uptake of oxLDL by CD36 in macrophages for the development of atherosclerotic lesions and creation of a proinflammatory environment raise a possibility that hyperlipidemia promotes foam cell formation in the postischemic brain. Although ND/WT mice were completely devoid of foam cells (data not shown), hyperlipidemic mice exhibited numerous foam cells in the postischemic brain 3 d after ischemia in the ipsilateral side (Fig. 5A–C). Interestingly, these foam cells were primarily localized in the penumbra. Because the procedures for tissue preparation and staining for lipid were not compatible with those of CD36 fluorescence immunohistochemistry, we were unable to colocalize foam cells with CD36 immunohistochemistry. Instead, we preformed CD36 immunohistochemistry in adjacent sections. As we reported previously in the sham control brain (Cho et al., 2005), we found basal CD36 staining in the corresponding contralateral area (Fig. 5F). In addition, we found intensely stained CD36-positive (CD36+) cells in the corresponding ipsilateral, but not in contralateral, area (Fig. 5E, arrows). Because hyperlipidemia profoundly exacerbates swelling (Fig. 1C), the unique localization of foam cells in the penumbra and the presence of CD36+ cells in the corresponding area indicate the possibility that CD36+ foam cells may contribute to postischemic swelling.
Figure 5.
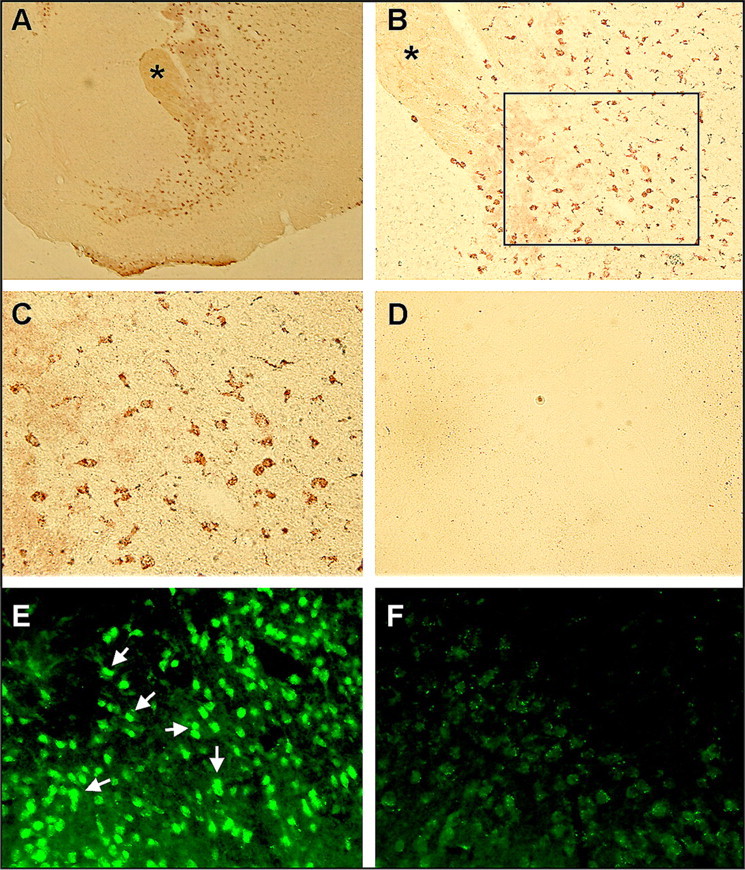
Foam cell formation in the hyperlipidemic brain. A–D, Oil Red-O staining in the ipsilateral (A–C) and contralateral (D) side of a hyperlipidemic brain 3 d after ischemia. No foam cells were observed in the normolipidemic brain, and Oil Red-O staining in the ipsilateral side of the normolipidemic brain was similar to D. The asterisks in A (40×) and B (100×) indicate the same region. The outline in B indicates the area of photomicrograph C (200×). E, Photomicrograph of CD36 immunohistochemistry performed in the adjacent section of C. F, Photomicrograph of CD36 immunohistochemistry (200×) performed in the adjacent section of D.
CD36 mediates hyperlipidemia-induced exacerbation in ischemic outcomes
To evaluate a critical involvement of CD36 and CD36+ foam cells in hyperlipidemia-induced exacerbation, we compared ischemic outcomes in hyperlipidemic ApoE KO and ApoE/CD36 DKO mice that were fed HD for 8 weeks. Despite no differences in ischemic severity as indicated by percentage CBF reduction during ischemia (ApoE KO, 87.8 ± 1.7%; DKO, 86.3 ± 1.7%) and percentage CBF restoration at 10 min reperfusion (ApoE KO, 116.9 ± 8.4%; DKO, 115.2 ± 9.2%), DKO mice showed smaller infarct volume and marked attenuation in percentage swelling (Fig. 6A,B). Correlation analysis showed that percentage swelling was positively correlated with infarct size within ApoE KO and DKO groups (ApoE KO, r2 = 0.5278, p = 0.0173; DKO, r2 = 0.5274, p = 0.0413). However, the disproportionately larger contribution of the swelling component in ApoE KO mice was reduced in DKO mice (Fig. 6C) (slope: ApoE KO, 0.9461 ± 0.3164; DKO, 0.2047 ± 0.07912). The reversal of phenotype in DKO mice indicates that CD36 is involved in the postischemic swelling. The improved ischemic outcome in DKO mice was not attributable to plasma cholesterol levels or MCP-1 expression in the periphery (Fig. 6D–F).
Figure 6.

Reduced ischemic injury in the absence of CD36. Ischemic outcome in ApoE KO and ApoE/CD36 DKO mice fed HD for 8 weeks. A, B, Infarct volume (A) and percentage swelling (B) were determined 3 d after ischemia. C, Correlation analysis between infarct volume and percentage swelling shows that a disproportionately larger contribution of the swelling component in ApoE KO mice was reduced in DKO mice. D–F, Between ApoE KO and DKO mice, cholesterol levels before ischemia (D), plasma MCP-1 levels (in picograms per milliliter) 3 d after ischemia (E), and MCP-1 gene expression from macrophages 3 d after ischemia (F) were comparable. **p < 0.01, and ***p < 0.001 versus ApoE KO.
We next investigate whether CD36 is involved in promoting inflammation in a hyperlipidemic condition at 6 h after ischemia. Compared with normolipidemic WT mice, ischemia-induced MCP-1, CCR2 (cognate receptor for MCP-1), interleukin IL-1β, TNF-α, and IL-6 mRNA levels were significantly increased in hyperlipidemic ApoE KO mice. The enhanced inflammation was reduced in DKO mice (Fig. 7A–E), whereas anti-inflammatory cytokine IL-13 mRNA levels were not different among groups (Fig. 7F). Together, the data suggest that CD36 is involved in the hyperlipidemia-induced inflammation and exacerbation of ischemic injury in the brain.
Figure 7.

Reduced ischemia-induced proinflammatory cytokines and chemokine gene expression in the CD36-deficient brain. A–F, mRNA levels for proinflammatory MCP-1 (A), CCR2 (B), TNF-α (C), IL-1β (D), IL-6 (E), and anti-inflammatory IL-13 (F) were measured at 6 h after ischemia. Proinflammatory chemokine and cytokine gene expression was reduced in the absence of cd36. WT, Normal-diet-fed wild type; ApoE KO, high-fat-fed ApoE KO; DKO, high-fat-fed ApoE/CD36 double KO. n = 4–5 per group. *p < 0.05 versus contralateral; #p < 0.05 versus ApoE KO ipsilateral.
CD36 mediates hyperlipidemia-induced foam cell formation
To investigate whether hyperlipidemia-promoted foam cell formation is mediated by CD36, we assessed the degree of foam cell formation in the ApoE KO and DKO ischemic brain. In contrast to the presence of many foam cells in the peri-infarct area of ApoE KO mice, DKO mice exhibited significantly less number of foam cells in the corresponding area (Fig. 8A) (ApoE KO, 227.1 ± 69.7; DKO 29.7 ± 16.9; p < 0.001). Similar to the previous report in 12 weeks of HD (Febbraio et al., 2000), 8 weeks of HD intervention in the current study also revealed reduced oxLDL-induced foam cell formation in peritoneal macrophages from DKO mice (Fig. 8B) (ApoE KO, 10199 ± 865; DKO, 5099 ± 432; p < 0.01). The data indicate that CD36 mediates hyperlipidemia-induced foam cell formation in the postischemic brain.
Figure 8.

Reduced foam cell formation in the brain and macrophages in the absence of cd36. A, Oil Red-O-stained foam cells in the brain 3 d after ischemia. Sections are at the level of striatum (+1.6 mm from bregma) from hyperlipidemic ApoE KO and DKO mice. The DKO photomicrographs in the top were taken from the DKO brain that exhibited the most foam cells. Numerous foam cells were present in the penumbra of ApoE KO mice, whereas the majority of DKO mice were devoid of foam cells. Arrows indicate cells in insets from which high-power photomicrographs were taken. B, oxLDL-induced foam cell formation in mouse peritoneal macrophages obtained from KO and DKO mice fed 8 weeks of HD. Note less foam cells in DKO macrophages compared with the KO macrophages. KO, ApoE KO mice; DKO, ApoE/CD36 DKO mice; V, vehicle.
Discussion
The current study suggests that hyperlipidemia exacerbates ischemic brain injury by promoting CD36-mediated inflammation. We generated mice with different degrees of hyperlipidemia by genetic and/or dietary interventions. HD in the same strains of mice was shown previously to elevate total plasma cholesterol, free fatty acids, and triglycerides levels (Febbraio et al., 2000). The total plasma cholesterol levels apparently dictate the extent of infarct size and swelling, resulting in a significant difference between ND/WT and the most hyperlipidemic HD/KO mice. The HD/KO mice showed no difference in relative CBF reduction during ischemia and reperfusion after ischemia compared with other groups as shown in Table 1, suggesting that genetic background and/or 8 weeks diet intervention produced no major hemodynamic changes. Thus, hyperlipidemia-induced exacerbation of ischemic injury likely involves a novel mechanism other than vascular or hemodynamic effects in our ischemic model.
Hyperlipidemia-promoted inflammation was investigated by first determining expression of CD36 in the brain. We found that ischemia-induced CD36 and MCP-1, but not ABCG1, gene expression was higher in hyperlipidemic brain (Fig. 3). There was a moderate but significant reduction (33%) in CD36 mRNA before ischemia. The reason for the lower basal level of CD36 mRNA in the hyperlipidemic mice is likely caused by the absence of apoe gene during development and/or a chronic high-fat diet intervention. Similarly, the absence of significant difference in CD36 mRNA between normal versus hyperlipidemic ipsilateral side is likely attributable to the reduced basal CD36 mRNA levels in the hyperlipidemic mice. The reduced baseline, however, does not negate the fact that ischemia significantly upregulates CD36 mRNA levels only in hyperlipidemic, but not normolipidemic, ipsilateral side.
Despite a previous study showing ischemia-induced changes in CD36 protein by Western blot, immunohistochemistry data from the same study showed that the expression of CD36 protein occurs primarily in microglia/macrophages in the postischemic brain (Cho et al., 2005). Because the amount of CD36 in this subset of infiltrating immune cells in the brain was likely below the sensitivity to detect a difference between normal and hyperlipidemic ipsilateral side by Western blot (especially compared with real-time PCR), we determined CD36 protein expression in the peripheral macrophages as a reflection of the population of cells that infiltrate into the injury site after ischemia. We reported a measurable CD36 protein difference between normal and hyperlipidemic macrophages without a parallel increase in peripheral macrophages mRNA (Fig. 4). Whereas the changes of CD36 gene expression in the brain were the result of ischemic insult, macrophage changes may reflect the effect of chronic peripheral hyperlipidemia. It has been shown that CD36 expression is regulated at the level of translation (Griffin et al., 2001). The increased CD36 protein without a parallel increase in peripheral macrophage mRNA, as found in the current study, was reported previously in diabetic ob/ob macrophages (Liang et al., 2004).
Studies indicate that MCP-1 and its receptor CCR2 are involved in ischemic injury. Overexpression of MCP-1 exacerbates ischemic injury and enhances recruitment of inflammatory cells to the injury sites (Chen et al., 2003), whereas the absence of ccr2 or mcp-1 gene has been shown to reduce ischemia-reperfusion injury (Hughes et al., 2002; Dimitrijevic et al., 2007). MCP-1 is also shown to induce the expression of CD36 and promote monocyte differentiation into foam cells (Tabata et al., 2003). Furthermore, monocytes isolated from hypercholesterolemic patients show increased CCR2 expression and are hyper-responsive to chemotactic stimuli (Han et al., 1999). We also found increased CD36, MCP-1, and CCR2 proinflammatory cytokine gene expression in the hyperlipidemic ischemic brain. Profound shifts to the reduced proinflammatory status in the DKO mice suggest that CD36 is involved in promoting hyperlipidemia-induced inflammation and injury in the postischemic brain.
The identities of cell type(s) that account for the exacerbation of stroke outcome are not clear. CD36 expression in the postischemic brain occurs mainly in CD11b+ microglia or monocytes/macrophages (Cho et al., 2005). However, ischemia-induced breakdown of the blood–brain barrier makes it difficult to distinguish resident microglia from infiltrated monocytes/macrophages. The role of microglia versus macrophages in ischemic injury is controversial. For instance, selective ablation of proliferating microglia exacerbates ischemic injury in the brain, suggesting that proliferating resident microglia are beneficial (Lalancette-Hebert et al., 2007). In contrast, microglia and monocyte/macrophages are the main source of excess cytokines and chemokines that elicit inflammatory responses. A recent report suggests a specific contribution from a monocyte subset in myocardial ischemic injury (Nahrendorf et al., 2007), which implicates additional complexity in identifying role(s) for the subset of monocytes in cerebral ischemia. Because we showed that hyperlipidemia enhances the expression of CD36 and MCP-1 in peritoneal macrophages and plasma even before ischemia, the finding indicates that the primed subpopulation of the infiltrated immune cells may serve as a mediator for peripheral hyperlipidemic effects. Consistent with our result, Podrez et al. (2007) demonstrated recently that a novel family of oxidized choline glycerophospholipids, which exhibit high affinity for CD36, is significantly increased in hyperlipidemic plasma and indicated a prothrombotic role for platelet CD36 in hyperlipidemia. The relative contribution of monocytes/macrophages compared with that of other CD36-expressing cells such as microglia, microvascular endothelial cells, and platelets remains to be answered. However, upregulated CD36 and MCP-1 in macrophages from hyperlipidemic mice strongly suggests a contributing role of macrophage CD36 in ischemic injury in hyperlipidemia.
Previous studies attempted to limit ischemic injury by attenuating postischemic inflammation (Becker, 1998; Barone and Feuerstein, 1999; del Zoppo et al., 2000). Based on the role of foam cells in eliciting inflammation and in the development of atherosclerotic lesions (Febbraio et al., 2000), we investigated a possible contribution of CD36-bearing foam cells to postischemic inflammation in the brain. Infarct size is typically reported as “indirect infarct volume,” which is calculated after swelling is subtracted from the direct measurement of injured volume. It is generally believed that the postischemic swelling is attributable to water retention. We found that the effect of hyperlipidemia on swelling was much more prominent than on infarct size. The profound increase in swelling was associated with a conspicuous presence of numerous foam cells in the hyperlipidemic but not in the normolipidemic, brain. Interestingly, these foam cells were predominantly localized in the penumbra, a primary site that accounts for postischemic inflammation. Swelling was attributed to a significant increase in lipid, not water, content in the HD/KO mice, indicating that the lipid and/or lipid-laden foam cell is a major contributor to the swelling in the hyperlipidemic brain.
Previous studies collectively suggest that CD36 functions as an inflammatory mediator in a setting of acute ischemia (Cho et al., 2005, 2007). The current study directly explores hyperlipidemia-promoted inflammatory responses by determining the expression of inflammatory markers. Compared with normal mice, ApoE KO brains showed increased mRNA levels of proinflammatory cytokines and MCP-1/CCR2. The mRNA levels were reduced in the absence of cd36 at 6 h after ischemia, a time when the infarct is histologically not visible. Reversing the proinflammatory phenotype in the DKO brain resulted in a smaller infarct, reduced swelling, and less foam cell formation at 3 d after ischemia despite similar peripheral plasma cholesterol levels and macrophage MCP-1 levels. Therefore, CD36-mediated inflammation apparently plays a role in the hyperlipidemia-induced exacerbation of ischemic injury and swelling.
The close association between swelling/ischemic injury and foam cells raises a possible role of CD36 function in injury development. It is possible that uptake of excess oxidized lipids and sequestration of membrane debris to cope with the insult may be initially beneficial. Nevertheless, excessive accumulation of foam cells in the hyperlipidemic brain may cause secondary cytotoxic effects by creating a proinflammatory environment. In the absence of CD36, the swelling was significantly reduced to the level of normolipidemic mice (Figs. 1D, 6C). The inhibition and/or delay in the uptake of excess ligands in the DKO brain perhaps minimize secondary inflammation around the injury site. The significant reduction of foam cells formation in the brain and macrophages of DKO mice further supports the idea that CD36 promotes foam cell formation in hyperlipidemic conditions. Our observation that foam cells were significantly but not completely reduced in an in vivo and in vitro study implicates the involvement of other scavenger receptors (e.g., scavenger receptor A) in the uptake of oxLDL in addition to CD36 (de Winther et al., 2000). Although our study reports a protective effect in the absence of CD36 in an acute setting of ischemia, the lack of CD36 on a long-term stroke recovery requires additional investigation.
In summary, the present study demonstrates that hyperlipidemia produced by genetic and diet intervention exacerbates ischemic brain injury. The exacerbation is associated with increased inflammation and enhanced CD36 and MCP-1 gene and/or protein expression in the postischemic brain as well as in the periphery. In addition, the hyperlipidemic condition promotes foam cell formation into the penumbra, a probable cause of the increased swelling and enhanced postischemic inflammation. Reduced proinflammatory markers in DKO brain at early hours of reperfusion suggest a critical role of CD36-mediated inflammation in hyperlipidemia-induced exacerbation. Collectively, the results of this study suggest that targeting CD36 could have therapeutic value in minimizing acute-stroke injury in hyperlipidemic subjects. Furthermore, the study suggests that inclusion of clinically predisposing conditions, such as hyperlipidemia or other lifestyle factors in experimental animal models of stroke, may improve the translation of information from the laboratory to the clinic.
Footnotes
This work was supported by National Institutes of Health Grant HL82511 (S.C.).
References
- Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Becker KJ. Inflammation and acute stroke. Curr Opin Neurol. 1998;11:45–49. doi: 10.1097/00019052-199802000-00008. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hallenbeck JM, Ruetzler C, Bol D, Thomas K, Berman NE, Vogel SN. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab. 2003;23:748–755. doi: 10.1097/01.WCB.0000071885.63724.20. [DOI] [PubMed] [Google Scholar]
- Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G, Silverstein RL, Iadecola C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci. 2005;25:2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10:95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Winther MP, van Dijk KW, Havekes LM, Hofker MH. Macrophage scavenger receptor class A: a multifunctional receptor in atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:290–297. doi: 10.1161/01.atv.20.2.290. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkind MS. Inflammation, atherosclerosis, and stroke. Neurologist. 2006;12:140–148. doi: 10.1097/01.nrl.0000215789.70804.b0. [DOI] [PubMed] [Google Scholar]
- Engström G, Lind P, Hedblad B, Stavenow L, Janzon L, Lindgärde F. Effects of cholesterol and inflammation-sensitive plasma proteins on incidence of myocardial infarction and stroke in men. Circulation. 2002;105:2632–2637. doi: 10.1161/01.cir.0000017327.69909.ff. [DOI] [PubMed] [Google Scholar]
- Engström G, Stavenow L, Hedblad B, Lind P, Eriksson KF, Janzon L, Lindgärde F. Inflammation-sensitive plasma proteins, diabetes, and mortality and incidence of myocardial infarction and stroke: a population-based study. Diabetes. 2003;52:442–447. doi: 10.2337/diabetes.52.2.442. [DOI] [PubMed] [Google Scholar]
- Evans M, Roberts A, Davies S, Rees A. Medical lipid-regulating therapy: current evidence, ongoing trials and future developments. Drugs. 2004;64:1181–1196. doi: 10.2165/00003495-200464110-00003. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. 2001;108:785–791. doi: 10.1172/JCI14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Guy E, Silverstein RL. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:2333–2338. doi: 10.1161/01.ATV.0000148007.06370.68. [DOI] [PubMed] [Google Scholar]
- Griffin E, Re A, Hamel N, Fu C, Bush H, McCaffrey T, Asch AS. A link between diabetes and atherosclerosis: glucose regulates expression of CD36 at the level of translation. Nat Med. 2001;7:840–846. doi: 10.1038/89969. [DOI] [PubMed] [Google Scholar]
- Guy E, Kuchibhotla S, Silverstein R, Febbraio M. Continued inhibition of atherosclerotic lesion development in long term Western diet fed CD36o/apoEo mice. Atherosclerosis. 2007;192:123–130. doi: 10.1016/j.atherosclerosis.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Han KH, Han KO, Green SR, Quehenberger O. Expression of the monocyte chemoattractant protein-1 receptor CCR2 is increased in hypercholesterolemia. Differential effects of plasma lipoproteins on monocyte function. J Lipid Res. 1999;40:1053–1063. [PubMed] [Google Scholar]
- Hirano K, Kuwasako T, Nakagawa-Toyama Y, Janabi M, Yamashita S, Matsuzawa Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc Med. 2003;13:136–141. doi: 10.1016/s1050-1738(03)00026-4. [DOI] [PubMed] [Google Scholar]
- Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab. 2002;22:308–317. doi: 10.1097/00004647-200203000-00008. [DOI] [PubMed] [Google Scholar]
- Hutter CM, Austin MA, Humphries SE. Familial hypercholesterolemia, peripheral arterial disease, and stroke: a HuGE minireview. Am J Epidemiol. 2004;160:430–435. doi: 10.1093/aje/kwh238. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol. 2001;14:89–94. doi: 10.1097/00019052-200102000-00014. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Cho S, Feuerstein GZ, Hallenbeck J. Stroke: pathophysiology, diagnosis, and management. Philadelphia: Churchill Livingstone; 2004. Cerebral ischemia and inflammation; pp. 883–894. [Google Scholar]
- Janabi M, Yamashita S, Hirano K, Sakai N, Hiraoka H, Matsumoto K, Zhang Z, Nozaki S, Matsuzawa Y. Oxidized LDL-induced NF-kappa B activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol. 2000;20:1953–1960. doi: 10.1161/01.atv.20.8.1953. [DOI] [PubMed] [Google Scholar]
- Kernan WN, Inzucchi SE. Type 2 diabetes mellitus and insulin resistance: stroke prevention and management. Curr Treat Options Neurol. 2004;6:443–450. doi: 10.1007/s11940-004-0002-y. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CP, Han S, Okamoto H, Carnemolla R, Tabas I, Accili D, Tall AR. Increased CD36 protein as a response to defective insulin signaling in macrophages. J Clin Invest. 2004;113:764–773. doi: 10.1172/JCI19528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Medeiros LA, Khan T, El Khoury JB, Pham CL, Hatters DM, Howlett GJ, Lopez R, O'Brien KD, Moore KJ. Fibrillar amyloid protein present in atheroma activates CD36 signal transduction. J Biol Chem. 2004;279:10643–10648. doi: 10.1074/jbc.M311735200. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- Pinto A, Tuttolomondo A, Di Raimondo D, Fernandez P, Licata G. Cerebrovascular risk factors and clinical classification of strokes. Semin Vasc Med. 2004;4:287–303. doi: 10.1055/s-2004-861497. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Gugiu B, Fox PL, Hoff HF, Salomon RG, Hazen SL. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J Biol Chem. 2002a;277:38503–38516. doi: 10.1074/jbc.M203318200. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Poliakov E, Shen Z, Zhang R, Deng Y, Sun M, Finton PJ, Shan L, Febbraio M, Hajjar DP, Silverstein RL, Hoff HF, Salomon RG, Hazen SL. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J Biol Chem. 2002b;277:38517–38523. doi: 10.1074/jbc.M205924200. [DOI] [PubMed] [Google Scholar]
- Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4:211–221. doi: 10.1016/j.cmet.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J. Recognition and phagocytosis of cells undergoing apoptosis. Br Med Bull. 1997;53:491–508. doi: 10.1093/oxfordjournals.bmb.a011626. [DOI] [PubMed] [Google Scholar]
- Strandgaard S. Hypertension and stroke. J Hypertens Suppl. 1996;14:S23–S27. doi: 10.1097/00004872-199610003-00005. [DOI] [PubMed] [Google Scholar]
- Stuart LM, Bell SA, Stewart CR, Silver JM, Richard J, Goss JL, Tseng AA, Zhang A, El Khoury JB, Moore KJ. CD36 signals to the actin cytoskeleton and regulates microglial migration via a p130Cas complex. J Biol Chem. 2007;282:27392–27401. doi: 10.1074/jbc.M702887200. [DOI] [PubMed] [Google Scholar]
- Tabata T, Mine S, Kawahara C, Okada Y, Tanaka Y. Monocyte chemoattractant protein-1 induces scavenger receptor expression and monocyte differentiation into foam cells. Biochem Biophys Res Commun. 2003;305:380–385. doi: 10.1016/s0006-291x(03)00771-x. [DOI] [PubMed] [Google Scholar]
- Willerson JT, Ridker PM. Inflammation as a cardiovascular risk factor. Circulation. 2004;109:II2–II10. doi: 10.1161/01.CIR.0000129535.04194.38. [DOI] [PubMed] [Google Scholar]