

Abstract
Gold nanoparticles were coated with a short peptide to promote intracellular delivery of membrane-impermeable proteins. Through microscopy and enzyme assays we demonstrated the particles were able to transport functional enzymes into variety of cell lines. Significantly, the transported proteins were able to escape from endosomes. Moreover, these particles showed no apparent cytotoxicity.
Introduction
Delivery of functional proteins inside living cells provides a powerful tool for therapeutics and fundamental study of cell biology.1 This technique allows introduction of proteins in a rapid, time-specific and dose-dependent fashion.2 While significant progress in DNA-recombinant technology has lead to availablity of any protein of interest, protein transduction has been limited by the poor membrane permeability of most proteins, and hence requires a transporter.3 In one strategy, cell-penetrating peptides (CPPs) have been attached to proteins genetically or chemically to promote their translocation across plasma membrane.4 Nevertheless, covalent modification can generate challenges such as alteration of activity or possibility of the tag being buried internally, making non-covalent approaches an attractive alternative.5 Recently, nanomaterials including silica nanoparticles, carbon nanotubes, and quantum dots have been shown to provide effective vectors for protein delivery.6 The delivery of enzymes with retention of activity using either covalent and non-covalent strategies, however, remains a major challenge.
Gold colloids are promising candidates in nanomedicine due to their bio-inertness, non-toxicity and cellular imaging ability.7 They have been reported for DNA delivery, however, their use as protein carriers is largely unexplored8 due to challenges in protein recognition and structure retention.9 The large surface area and tunable functionality of gold nanoparticles makes them an excellent scaffold for protein surface recognition.10 Moreover, the conformation of proteins can be preserved by tailoring the monolayer of particles.11 Herein, we report the effective intracellular delivery of β-galactosidase (β-gal), a membrane-impermeable negatively charged protein with high molecular weight (465 kDa) in a variety of cell lines, with endosomal escape of the protein and retention of enzymatic activity inside the cells (Figure 1).
Figure 1.

(a) Schematic representation of intracellular delivery of functional protein using gold nanoparticles. (b) Structure of the nanoparticle, the protein cargo, and the ligand onto particle.
Results and Discussion
We fabricated peptide-coated gold nanoparticles (NP_Pep, core diameter ~2.5 nm) to serve as a protein transporter. Three functional domains were incorporated into particle structure: the interior alkyl chains impart stability to the core, a corona of tetraethylene glycol (TEG) prevents both non-specific interactions with biomolecules and denaturation,11 the exterior peptide-tags serve as a recognition unit. Polyarginine is well-known for translocating molecules across the plasma membrane.12 Therefore, our initial efforts focused on the functionalization of the particles with a short peptide containing three arginine residues (NP-RRR). This strategy, however, yielded particles that were not dispersible in water. In prior studies, we have earlier demonstrated that lysine-coated particles were water-soluble and stable.13 As a result, we chose for our studies a sequence incorporating arginine and lysine,14 with the addition of a histidine residue, as His groups are known to facilitate endosomal escape of cargo.15 Overall, the peptide headgroup consists of strong and weakly basic amino acid residues (Arg, Lys and His) that serve multiple roles: (a) protein surface recognition and plasma membrane association through favorable electrostatic interaction and hydrogen bonding of guanidinium moiety, and (b) “endosomal buffering” due to the presence of the proton-sponge imidazole group of histidine.16 Importantly, these particles (NP_Pep) were well dispersed in aqueous media with a hydrodynamic diameter consistent with their structure (dH 12.9 ± 0.3 nm).17 As expected, these particles were strongly cationic, with a zeta potential of 32 ± 1 mV arising from the ~100 ligands per nanoparticle estimated by particle size.18
Nanoparticle-β-gal complexation was assessed by fluorescent titration using fluorescein isothiocyanate-labeled β-galactosidase (FITC-β-gal). As expected, FITC-fluorescence was quenched by the gold core upon addition of NP_Pep allowing us to quantify the interaction of the cationic particles and anionic β-gal (pI 4.619), with a KD of 1.0 nM observed (Figure 2). Surprisingly, we found that these complexes were stable in 150 mM salt solution as well (KD ~ 11nM), presumably due to additional hydrogen-bonding interactions of the protein with the arginines of nanoparticle (Figure 2).20 Our group has earlier reported the glutathione-mediated release of β-gal adsorbed onto cationic nanoparticles.10 Intracellular glutathione can likewise act as a trigger for release of β-gal by cationic NP_Pep. An alternative possibility is that the acidic environment found during endocytosis might effect the release. To study the release event in acidic conditions, the fluorescence of FITC-β–gal in the presence of NP_Pep was monitored in buffers of pH ranging from 7 to 4. However, sharp decrease in fluorescence of FITC-β–gal itself due to precipitation with lowering pH made it impossible to elucidate any effect of pH change on particle-protein complexation (Figure S3). Analysis of circular dichroism (CD) spectra revealed that there was minimal change in secondary structure in the presence of nanoparticles (Figure 2). Similarly, no shift in λmax of tryptophan-fluorescence of the protein was observed (See SI), further confirming retention of the native structure of the protein.21
Figure 2.

(a) Fluorescence titration of 100 nM FITC-β-gal with NP_Pep in 5 mM phosphate buffer (without and with NaCl of 150 mM) at 25 °C. (b) The CD spectra of the protein (100 nM) before and after addition of nanoparticles in 5 mM phosphate buffer at 25 °C. In the inset, the percent of secondary structures calculated using DICHROWEB indicating minimal conformational change upon nanoparticle addition.
Cellular delivery of β-gal protein was first monitored using FITC-β–gal as a fluorescent probe. Human cervical carcinoma cells (HeLa) were treated with 2:1 molar ratio NP_Pep/FITC-β-gal complexes, as precipitation occurred at higher particle/protein ratios in media. Following 3 h of incubation, cells were washed and cultured for another 3 h. Green fluorescence was observed when cells were treated with particle-protein complexes, while cells incubated with FITC-β-gal alone displayed no fluorescence (Figure 3a–b). Cells treated with the complexes also showed presence of gold from inductively coupled plasma mass spectrometry (ICP-MS), consistent with the expected nanoparticle-promoted co-translocation of this membrane-impermeable protein (Figure 3c). The delivery process was likewise effective in serum (see Figure S5 for micrographs).
Figure 3.

Fluorescence micrographs of Hela cells transfected with FITC-β-gal (50 nM) (a) in absence or (b) in presence of NP_Pep (100 nM). (c) ICP-MS measurements after treating cells with NP_Pep/β-gal (100 nM/50 nM) complex at different temperatures, and cells treated with β-gal alone as a control. (d–f) CLSM images of HeLa cells after protein transfection (NP_Pep/FITC-β-gal: 100 nM/50 nM): (d) bright field, (e) fluorescence and (f) the merged image.
We next performed confocal laser scanning microscope (CLSM) experiments to confirm that the observed fluorescence was coming from inside the cells as opposed to adsorbed on cell surface. As shown in micrographs, punctate fluorescence was present in the perinuclear region of the cells, indicating protein internalization, presumably via endocytosis (Figure 3d–f and Figure S6).22 This mechanism was supported by substantial decrease in fluorescence (Figure S7) coupled with a decrease in gold (via ICP, Figure 3c) with uptake experiments performed at 4 °C.23
Current research has been focused on cytoplasmic delivery of proteins. As the complex presumably entered into cells via endocytic pathway, we investigated whether the cargo was able to escape from endosome. For this purpose, cells were treated with FM 4–64, a red endosome-specific marker.24 FITC-fluorescence that arises from delivered FITC-β–gal was monitored along with red-fluorescence from FM 4–64 using CLSM (Figure 4a–b). After merging the green and the red channels, we observed the presence of significant amount of green sites within the cells, indicating proteins that were outside the endosomes (Figure 4c). Relatively few yellow areas indicative of proteins trapped in endosomes (overlapped of green and red dyes) were observed.
Figure 4.
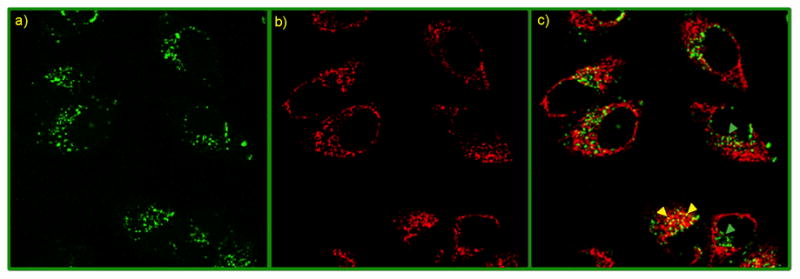
A colocalization study using CLSM after protein transfection (NP_Pep/FITC-β-gal: 100 nM/50 nM) of HeLa cells in presence of FM 4–64: (a) green fluorescence from FITC-β-gal, (b) red fluorescence from FM 4–64, an endosome-specific marker, and (c) overlap of the green and the red channels. In the merged image, green spots (shown with green arrowheads) are indicating proteins outside endosomes, while entrapped proteins inside endosomes appear as yellow dots (shown with yellow arrowheads).
Enzymatic activity provides a particularly stringent test for the retention of bioactivity after transduction, a key concern in protein delivery. The NP_Pep/protein complex was incubated with HeLa cells for 3 h, then washed and cultured for additional 3 h followed by treatment with X-gal, a colorless substrate for β-gal that turns blue upon enzymatic hydrolysis. Significantly, blue precipitates appeared inside cells transfected with the particle-protein complex, demonstrating preservation of enzymatic activity after delivery (Figure 5b). No color was observed, as expected, when cells were treated with protein alone (Figure 5a). Consistent with the CLSM images, X-gal staining of the cells after trypsin digestion also indicated that the proteins were internalized by the cells as opposed to adsorbed onto cell surface (Figure S9). In a control experiment, cells were treated with NP_Pep alone (without any β-gal), and no blue spot was observed thereby ensuring that the observed staining was not due to lysosomal galactosidases (Figure S11). β-Gal delivery experiments were also performed with NP_TEG (a neutral nanoparticle with a hydroxyl group replacing the peptide) and NP_TTMA (a non-peptidic, cationic nanoparticle19 with a trimethylammonium group replacing the peptide). The superior efficacy of β-gal delivery with NP_Pep was evident from X-gal assay (enzyme activity: NP_Pep>NP_TTMA>NP_TEG) coupled with ICP-MS experiment (nanoparticle uptake: NP_Pep>NP_TTMA>NP_TEG) (See figure S12). The peptide tag enhanced the native uptake capability of these gold nanoparticles as measured by ICP-MS, mirroring the more efficient protein delivery using NP_Pep (See figure S12d). With longer incubation times (24 h) after transfection, we observed a decrease in enzymatic activity (Figure S10),25 potentially due to protein degradation by intracellular proteases.
Figure 5.

(a–c) X-gal staining after transfection. (a) HeLa with protein only. Transfected (b) HeLa and (c) C2C12 cells with NP_Pep/β-gal (100 nM/50 nM). (d) The percent of transfection with NP_Pep/β-gal (100 nM/50 nM) in defferent cell lines. (e) Dose-dependent protein delivery into HeLa cells at 2:1 NP_Pep/β-gal.
We next studied the dose-dependent response of the delivery system, a useful tool for controlled protein delivery. Cells were treated with varied concentration of the enzyme, and protein delivery was assessed by X-gal histochemical staining. The percentage of transfected cells, judged by positively stained cells, increases with dose, with quantitative transfection (~98%) achieved at 100 nM of protein concentration (Figure 5e). Significantly, efficient transfection (>80%) was observed at 50 nM of protein in a diverse array of cell lines, including COS-1 (monkey kidney cells), MCF7 (human breast cancer cells), and even hard-to-transfect muscle cells (C2C12) (Figure 5c–d).26
Concurrent with our delivery studies we investigated the cytotoxicity of the NP_Pep particles. From trypan blue exclusion test, we observed no cell death after 3 h of transfection as well as 24 h later (Figure 6a and S14a). This lack of toxicity was also validated from calcein AM assay (Figure S13). Importantly, full retention of cell vitality was observed from alamar blue assay (Figure 6b and S14b).
Figure 6.
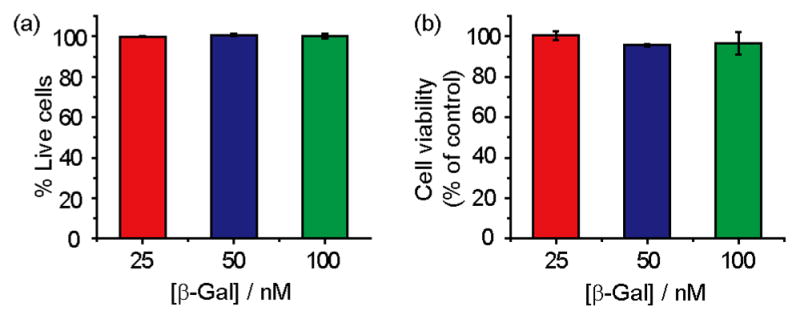
Nanoparticles showed no toxicity after 24 h of protein transfection as measured by (a) trypan blue exclusion test and (b) alamar blue assay.
Conclusion
In summary, we have reported a highly efficient strategy for intracellular protein delivery of anionic proteins. Supramolecular complexes with engineered nanoparticles were shown to translocate exogeneous proteins into a variety of cells without exhibiting any cytotoxicity. Crucially, the transported enzyme was able to escape from endosomes and retained its biological activity, providing the potential for fundamental and therapeutic applications of this strategy. We are currently investigating the effect of peptide structure on protein delivery.
Supplementary Material
Acknowledgments
This research was supported by the NIH (GM077173) SA acknowledges a Graduate School Fellowship and RA was supported through a Chemistry-Biology Interface training grant (T32 GM008515).
Footnotes
Supporting Information Available: Synthesis of nanoparticles, experimental procedures, micrographs, and figures of NP_Pep-β–gal binding and toxicity. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Mitragotri S, Blankschtein D, Langer R. Science. 1995;269:850. doi: 10.1126/science.7638603. [DOI] [PubMed] [Google Scholar]; (b) Kobsa S, Saltzman WM. Pediatr Res. 2008;63:513. doi: 10.1203/PDR.0b013e318165f14d. [DOI] [PubMed] [Google Scholar]; (c) Gombotz WR, Pettit DK. Bioconjug Chem. 1995;6:332. doi: 10.1021/bc00034a002. [DOI] [PubMed] [Google Scholar]
- 2.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science. 1999;285:1569. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 3.Murthy N, Xu MC, Schuck S, Kunisawa J, Shastri N, Frechet JMJ. Proc Natl Acad Sci USA. 2003;100:4995. doi: 10.1073/pnas.0930644100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Phelan A, Elliott G, O’Hare P. Nat Biotechnol. 1998;16:440. doi: 10.1038/nbt0598-440. [DOI] [PubMed] [Google Scholar]
- 5.(a) Morris MC, Depollier J, Mery J, Heitz F, Divita G. Nat Biotechnol. 2001;19:1173. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]; (b) Myrberg H, Lindgren M, Langel U. Bioconjug Chem. 2007;18:170. doi: 10.1021/bc060266g. [DOI] [PubMed] [Google Scholar]
- 6.(a) Slowing II, Trewyn BG, Lin VSY. J Am Chem Soc. 2007;129:8845. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]; (b) Kam NWS, Jessop TC, Wender PA, Dai HJ. J Am Chem Soc. 2004;126:6850. doi: 10.1021/ja0486059. [DOI] [PubMed] [Google Scholar]; (c) Medintz IL, Pons T, Delehanty JB, Susumu K, Brunel FM, Dawson PE, Mattoussi H. Bioconjug Chem. 2008;19:1785. doi: 10.1021/bc800089r. [DOI] [PubMed] [Google Scholar]
- 7.(a) Bhattacharya R, Mukherjee P. Adv Drug Deliv Rev. 2008;60:1289. doi: 10.1016/j.addr.2008.03.013. [DOI] [PubMed] [Google Scholar]; (b) Ghosh P, Han G, De M, Kim CK, Rotello VM. Adv Drug Deliv Rev. 2008;60:1307. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]; (c) De M, Ghosh PS, Rotello VM. Adv Mater. 2008;20:4225. [Google Scholar]; (d) Nativo P, Prior IA, Brust M. Acs Nano. 2008;2:1639. doi: 10.1021/nn800330a. [DOI] [PubMed] [Google Scholar]; (e) Verma A, Uzun O, Hu YH, Hu Y, Han HS, Watson N, Chen SL, Irvine DJ, Stellacci F. Nat Mater. 2008;7:588. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Pujals S, Bastus NG, Pereiro E, Lopez-Iglesias C, Punte VF, Kogan MJ, Giralt E. Chembiochem. 2009;10:1025. doi: 10.1002/cbic.200800843. [DOI] [PubMed] [Google Scholar]
- 8.(a) Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA. Science. 2006;312:1027. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]; (b) Ghosh PS, Kim CK, Han G, Forbes NS, Rotello VM. Acs Nano. 2008;2:2213. doi: 10.1021/nn800507t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Paciotti GF, Kingston DGI, Tamarkin L. Drug Develop Res. 2006;67:47. [Google Scholar]
- 9.Jones S, Thornton JM. Proc Natl Acad Sc USA. 1996;93:13. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) You CC, Verma A, Rotello VM. Soft Matter. 2006;2:190. doi: 10.1039/b517354j. [DOI] [PubMed] [Google Scholar]; (b) Verma A, Simard JM, Worrall JWE, Rotello VM. J Am Chem Soc. 2004;126:13987. doi: 10.1021/ja046572r. [DOI] [PubMed] [Google Scholar]
- 11.Hong R, Fischer NO, Verma A, Goodman CM, Emrick T, Rotello VM. J Am Chem Soc. 2004;126:739. doi: 10.1021/ja037470o. [DOI] [PubMed] [Google Scholar]
- 12.(a) Holowka EP, Sun VZ, Kamei DT, Deming TJ. Nature Mat. 2007;6:52. doi: 10.1038/nmat1794. [DOI] [PubMed] [Google Scholar]; (b) Rothbard JB, Jessop TC, Wender PA. Adv Drug Deliv Rev. 2005;57:495. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]; (c) Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. J Pep Res. 2000;56:318. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]; (d) Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, Wender PA, Khavari PA. Nature Med. 2000;6:1253. doi: 10.1038/81359. [DOI] [PubMed] [Google Scholar]; (e) Koch AM, Reynolds F, Merkle HR, Weissleder R, Josephson L. Chembiochem. 2005;6:337. doi: 10.1002/cbic.200400174. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh PS, Han G, Erdogan B, Rosado O, Krovi SA, Rotello VM. Chem Biol Drug Des. 2007;70:13. doi: 10.1111/j.1747-0285.2007.00534.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang KW, Yan XH, Cui Y, He Q, Li JB. Bioconjug Chem. 2007;18:1735. doi: 10.1021/bc060315p. [DOI] [PubMed] [Google Scholar]
- 15.(a) Benns JM, Choi JS, Mahato RI, Park JS, Kim SW. Bioconjug Chem. 2000;11:637. doi: 10.1021/bc0000177. [DOI] [PubMed] [Google Scholar]; (b) Leng QX, Mixson AJ. Nuc Acids Res. 2005;33:e40. doi: 10.1093/nar/gni040. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pichon C, Goncalves C, Midoux P. Adv Drug Deliv Rev. 2001;53:75. doi: 10.1016/s0169-409x(01)00221-6. [DOI] [PubMed] [Google Scholar]
- 16.Futaki S. Adv Drug Deliv Rev. 2005;57:547. doi: 10.1016/j.addr.2004.10.009. [DOI] [PubMed] [Google Scholar]; (b) Midoux P, Monsigny M. Bioconjug Chem. 1999;10:406. doi: 10.1021/bc9801070. [DOI] [PubMed] [Google Scholar]; (c) Wang KW, Yan XH, Cui Y, He Q, Li JB. Bioconjug Chem. 2007;18:1735. doi: 10.1021/bc060315p. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh PS, Verma A, Rotello VM. Chem Commun. 2007:2796. doi: 10.1039/b705554d. [DOI] [PubMed] [Google Scholar]
- 18.Hostetler MJ, Templeton AC, Murray RW. Langmuir. 1999;15:3782. [Google Scholar]
- 19.Miranda OR, You CC, Phillips R, Kim IB, Ghosh PS, Bunz UHF, Rotello VM. J Am Chem Soc. 2007;129:9856. doi: 10.1021/ja0737927. [DOI] [PubMed] [Google Scholar]
- 20.(a) Blondeau P, Segura M, Perez-Fernandez R, de Mendoza J. Chem Soc Rev. 2007;36:198. doi: 10.1039/b603089k. [DOI] [PubMed] [Google Scholar]; (b) Salvatella X, Martinell M, Gairi M, Mateu MG, Feliz M, Hamilton AD, de Mendoza J, Giralt E. Angew Chem Int Ed. 2004;43:196. doi: 10.1002/anie.200352115. [DOI] [PubMed] [Google Scholar]
- 21.You CC, De M, Han G, Rotello VM. J Am Chem Soc. 2005;127:12873. doi: 10.1021/ja0512881. [DOI] [PubMed] [Google Scholar]
- 22.Kosuge M, Takeuchi T, Nakase I, Jones AT, Futaki S. Bioconjug Chem. 2008;19:656. doi: 10.1021/bc700289w. [DOI] [PubMed] [Google Scholar]
- 23.Khalil IA, Kogure K, Akita H, Harashima H. Pharma Rev. 2006;58:32. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 24.Vida TA, Emr SD. J Cell Biol. 1995;128:779. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciechanover A. Cell Death & Diff. 2005;12:1178. doi: 10.1038/sj.cdd.4401692. [DOI] [PubMed] [Google Scholar]
- 26.Yamanouchi D, Wu J, Lazar AN, Kent KC, Chu CC, Liu B. Biomaterials. 2008;29:3269. doi: 10.1016/j.biomaterials.2008.04.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.