

Abstract
Axons of the adult central nervous system exhibit an extremely limited ability to regenerate after spinal cord injury. Experimentally generated patterns of axon growth are typically disorganized and randomly oriented. Support of linear axonal growth into spinal cord lesion sites has been demonstrated using arrays of uniaxial channels, templated with agarose hydrogel, and containing genetically engineered cells that secrete brain-derived neurotrophic factor (BDNF). However, immobilizing neurotrophic factors secreting cells within a scaffold is relatively cumbersome, and alternative strategies are needed to provide sustained release of BDNF from templated agarose scaffolds. Existing methods of loading the drug or protein into hydrogels cannot provide sustained release from templated agarose hydrogels. Alternatively, here it is shown that pH-responsive H-bonded poly(ethylene glycol)(PEG)/poly(acrylic acid)(PAA)/protein hybrid layer-by-layer (LbL) thin films, when prepared over agarose, provided sustained release of protein under physiological conditions for more than four weeks. Lysozyme, a protein similar in size and isoelectric point to BDNF, is released from the multilayers on the agarose and is biologically active during the earlier time points, with decreasing activity at later time points. This is the first demonstration of month-long sustained protein release from an agarose hydrogel, whereby the drug/protein is loaded separately from the agarose hydrogel fabrication process.
1. Introduction
Experimentally induced axonal regeneration in the central nervous system (CNS) after spinal cord injury[1,2] is often disorganized and random, lacking the organization of long linear tracts that normally project through the intact nervous system.[1] To support linear axonal growth into spinal cord lesion sites, arrays of uniaxial channels of uniform diameter, wall thickness, and physical texture similar to normal spinal cord have been patterned with agarose hydrogel.[1] These templated nerve guidance agarose scaffolds were shown to exhibit excellent integration with host tissue. Growth-promoting neurotrophic factors have been shown to promote axonal regeneration into the sites of spinal cord injury.[2–5] Indeed, loading scaffolds with genetically engineered cells that secrete brain-derived neurotrophic factor (BDNF) were shown to significantly promote linear penetrating axons through their channels in vivo for spinal cord injury.[1] However, immobilizing neurotrophic factor secreting cells within a scaffold is cumbersome, and raises the possibility of immune response if non-autologous cells are used. The objective of this study was to provide an alternative strategy to deliver sustained release of growth factors or proteins to enhance axonal regeneration from templated agarose scaffolds.
Agarose hydrogels were chosen as nerve guidance scaffolds because they can be designed to match the mechanical properties of the spinal cord, are biocompatible and bioinert, and more importantly, they are stable for the extended period that is required for regenerating organized axons.[1] Rapidly degrading scaffolds may fail to maintain adequate orientation for regenerating axons.[6,7] Agarose is a weakly ionic hydrogel[8,9] and the pore morphology and porosity are both strongly dependent on agarose concentration.[10,11] Agarose hydrogels can have a broad range of pore diameters, of less than 1 nm up to greater than 500 nm.[8–13] The absence of strong interactions between the proteins and agarose and the relatively large average pore size of the agarose as compared to the size of the protein (e.g., the size of lysozyme is ∼3 × 3 × 4.5 nm[14]) preclude direct impregnation (soaking) of proteins into the agarose hydrogels for sustained release over long periods (see Supporting Information, Fig. S1). This is in contrast to other hydrogels, which can achieve sustained release through entanglements or physical interactions with the drugs.[15–20] Current methods of incorporating drugs into hydrogel structures during the fabrication process[15,21–24] are not compatible with the templated agarose scaffold fabrication process.[1] The latter exposes proteins to harsh organic solvents that are used to selectively etch patterning constituents during the templated scaffold fabrication process, which can denature the proteins. Therefore, a strategy is needed that can load the drug apart from the scaffold fabrication. To our knowledge, sustained drug release over a number of days from an agarose hydrogel, whereby the drug is loaded separately from the fabrication of the agarose hydrogel, has not been achieved.
Layer-by-layer (LbL) assembled multilayers, introduced by Decher,[25] offer promise in the field of controlled drug delivery, due to their tunable film properties, flexibility in choice of assembly components, and ease of processing.[26,27] LbL multilayers can be tuned to incorporate varying amounts of drugs or proteins as well as provide sustained release under specific conditions of pH, salt, or temperature.[26–28] Initial strategies that used the LbL multilayers to control drug release were based on non-degradable polyelectrolyte multilayer capsule formation technology[28–30] However these capsules trigger drug release at non-physiological conditions of pH or ionic concentrations[28] and thereby restrict their in vivo applications. Loading the drug either before or after fabrication of non-degradable multilayers and their subsequent mechanism of release depends on the permeability properties of the film, degree of film swelling, interaction of drug molecules with the polyelectrolytes, that is, the charge and size of the drug molecule, and the type of multilayer assembly. Furthermore, it is limited to small (<5 kDa) molecules that can diffuse under physiological conditions,[28,31–37] thus restricting the type of drug that can be used with non-degradable LbL assemblies for controlled release applications. The LbL methodology has also been shown to provide controlled drug or protein release from synthetic hydrogels; however, these processes again loaded the drug during the hydrogel fabrication process,[38,39] which is not an option with the fabrication process of templated agarose scaffolds that involves harsh organic solvents.
In contrast, degradable multilayer assemblies, based on sequential embedding of drugs during the fabrication, can incorporate any drug independent of the molecular weight of the drug.[40–44] Fabrication of hydrogen bond (H-bond)-based LbL multilayer films was initially reported by Rubner and coworkers[45] and Zhang and co-workers.[46] Subsequently, Sukhishvili and Granick demonstrated the pH controlled assembly and degradation of poly(carboxylic acid)-based H-bonded LbL films,[47,48] which was followed by numerous studies involving these multilayers for different applications, for example, to generate self-standing floating films, solid polymer electrolyte films, or for patterned delivery of nucleic acids to cells.[49–51] Degradable H-bonded films have been used for sustained release of charged compounds, albeit limited to a few hours of controlled release.[52] However, a recent study demonstrated prolonged drug release up to two weeks using a carboxylic acid-based crosslinked H-bonded LbL assembly with a hydrophobic drug contained in amphiphilic block copolymer micelles.[40] Further, formation of degradable[53] or non-degradable[28,38] multilayers over protein impregnated agarose is also not a feasible option since the protein is not easily retained within the agarose gel. The protein leaches from the agarose during the multilayer fabrication, making it difficult to control the amount of protein loaded, and likely results in minimal, if any, protein encapsulation.
Here, we present a simple approach for controlled delivery of proteins from agarose gels, where the proteins are incorporated within the degradable LbL multilayer coatings formed over the agarose. Carboxylic acid (−COOH)-based weak polyelectrolytes form H-bond interactions at low pH (e.g., pH < 3.5 in the case of poly(acrylic acid (PAA)) and deprotonates to carboxylate ions (−COO−) at high pH, which degrades the H-bonded multilayer assembly[48] H-bonded PAA/poly(ethylene oxide) (PEO) multilayer films when built on a planar substrate are known to degrade in about 30 min upon exposure to a pH of 3.5 or higher.[48] However, here we show that the H-bonded films when prepared over agarose as the substrate provided sustained release of the incorporated protein under physiological conditions for a period of more than four weeks. Since nervous system growth factors such as BDNF are rather expensive, a more practical analog, lysozyme, was evaluated because of its similarity in size and isoelectric point to BDNF.[54] Multilayers were formed with either a three component assembly of poly(ethylene glycol) (PEG), PAA and protein, or a two component biocompatible assembly of PEG and protein, under acidic conditions (pH ≤ 3.0). The protein was loaded subsequent to the agarose hydrogel fabrication rather than pre-loaded directly into the hydrogel, avoiding the caustic conditions used in the templated agarose scaffold fabrication. It was determined that there is a close relationship between the weight percent of agarose hydrogel and the amount of released protein. This relationship is believed to be a result of increasing total surface area per unit volume of agarose hydrogel. A variety of drugs or proteins of varying nature, size, and amounts can be explored for controlled release using this LbL approach without the concern or constraints that may be imposed by potential interactions between the drug and hydrogel. Moreover, this approach does not require any specific chemical alterations to the LbL forming polymers, such as copolymerization or covalent bonding.
2. Results and Discussion
Agarose is a linear polysaccharide consisting of repeat units of agarobiose (1,3-linked β-D-galactopyranose and 1,4-linked 3,6-anhydro-α-L-galactopyranose).[8,9] Upon cooling the hot solution of agarose in water, it forms a physically crosslinked double-helix 3D gel network of polymer chains interconnected by H-bonding and hydrophobic interactions.[8,9] We capitalized upon the H-bonding and hydrophobic interactions in building the LbL assemblies onto the agarose scaffold. Polymers, such as polyamines and proteins, can provide varying degree of H-bonding and/or hydrophobic interactions with agarose and their varying degree of bonding with agarose can affect the degradation kinetics of the multilayer assembly. Here, branched poly(ethylenimine) (BPEI), linear poly(ethylenimine) (LPEI), and protein were used as the LbL initiating polymers and examined for their affect on the protein release profiles from multilayer-coated agarose hydrogel. LbL initiated with BPEI, LPEI or protein was followed by formation of H-bond multilayer films with the protein as one of the multilayer constituents. Two component- and three component-multilayer assemblies consisting of PAA, PEG and protein were formed, as shown in Scheme 1a–f. The loading of the polymers and the layered structure formation during LbL assembly over agarose was characterized with fluorescence measurements, UV/vis spectroscopy, confocal microscopy, and SEM. The protein release profiles were evaluated for different LbL compositions, varying agarose porosity, LbL initiating polymer, and the number of components in the assembly.
Scheme 1.
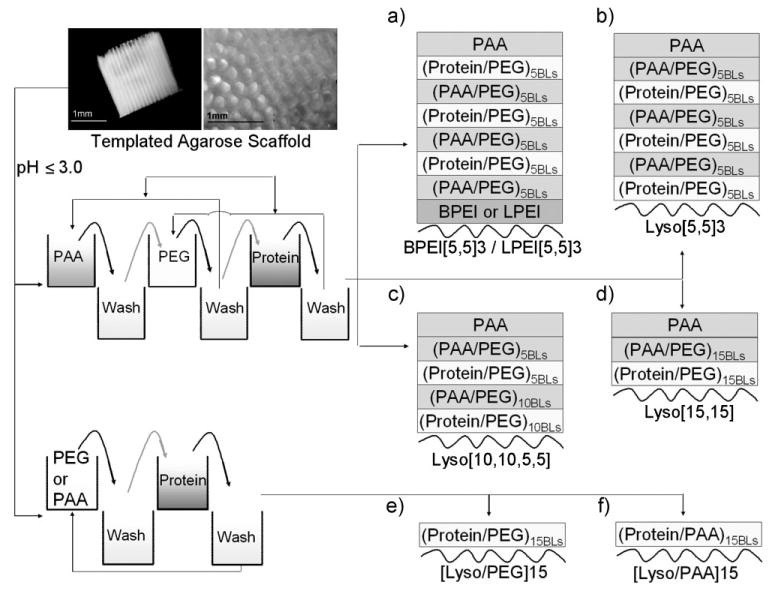
Scheme diagram showing the three- and two-components LbL assembly fabricated onto native agarose as the substrate. Templated agarose scaffolds (as shown in this scheme) were used to characterize film growth, and agarose-filled TCPS plates were used to characterize protein releases. Three component assemblies consisted of PAA, PEG and protein as the multilayer constituents, and two component assemblies consisted of PEG or PAA and protein as the multilayer constituents. BPEI, LPEI, or protein (lysozyme, denoted as Lyso) was used as the LbL initiating polymer in the different cases shown in (a–f). Curved lines in (a–f) represent the agarose, and BLs indicates the bilayers.
2.1. Multilayer Growth on Agarose Substrate
In order to assess the LbL growth of the polymers on agarose, we measured the fluorescence intensity of a LbL polymer component (PEG conjugated with a dye) as the number of bilayers increased. PEG terminated with primary amines at both ends (PEG-Amine) was conjugated to the TRITC dye, which was used to form the LbL assembly with the bovine serum albumin (BSA) protein on agarose. Figure 1a shows the adsorption of PEG during intermediate steps of LbL formation on the agarose. The measured fluorescence intensity of the emission spectrum of TRITC continuously increased as more layers of PEG-Amine-TRITC were adsorbed onto the agarose during the film formation (Fig. 1a). Further, in order to show the adsorption of the second LbL component, BSA, we measured the absorbance of the protein at 280nm as the number of bilayers increased (Fig. 1b). The measured absorbance of the BSA protein continuously increased as more layers of BSA were adsorbed onto the agarose. Therefore, Figure 1a and b shows the increasing deposition of both PEG and protein, respectively, during intermediate steps of the LbL assembly onto agarose. Correspondingly, a continuous decrease in the protein concentration of the loading bulk solution (solution from which the multilayers were formed) was observed (Fig. 1c). There was no decrease in the protein concentration in the bulk solution for the case where the agarose was soaked in the protein solution without LbL assembly (data not shown). However, these measurements do not provide information on the multilayered structure deposited onto the agarose by the LbL process.
Figure 1.
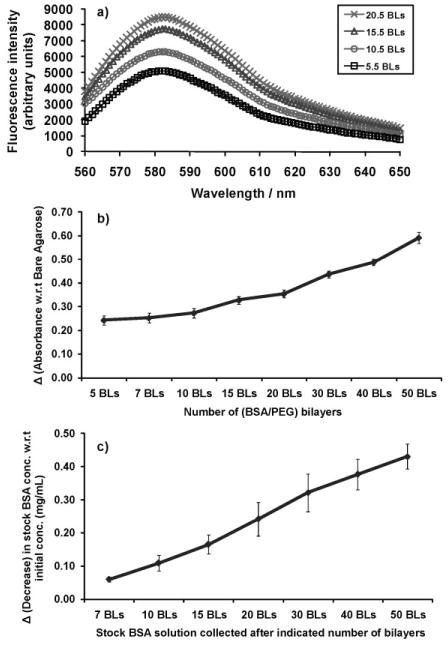
a) Fluorescence intensity measurements of TRITC conjugated to amine terminated PEG (PEG-Amine-TRITC) showed increased adsorption of PEG onto the agarose structure during the LbL deposition of BSA/(PEG-Amine-TRITC) multilayers on agarose hydrogel. b) UV/Vis absorbance measurements at 280 nm showed increased adsorption of bovine serum albumin (BSA) protein onto the agarose structure during the LbL deposition of BSA/(PEG) multilayers. The absorbance values are shown with respect to the absorbance of bare agarose, that is, the difference between the absorbance of LbL coated agarose and the absorbance of bare (non-coated) agarose. Five precursor bilayers of (PAA/PEG) with LPEI as the LbL initiating polymer were built over agarose in each case. BLs denote the number of bilayers. c) Corresponding decrease in lysozyme concentration in the bulk solution from the initial concentration as a function of the number of bilayers.
Next, we characterized the thin film formation of 30 bilayers of PAA/PEG onto 3 wt% templated agarose scaffolds composed of uniaxial channels[1] (as shown in Scheme 1). The nominal film thickness of 30 bilayers of PAA/PEG multilayer film (PAA/PEG)30 is about 600 nm[51] (discussed further in Section 2.3), and the average pore diameter of 3 wt% agarose was determined to be about 20 nm (discussed further in Section 2.2.1). It is expected that during the initial phase of multilayer growth the film formation would occur in the intrinsic micropores of the agarose scaffold. As subsequent layers are deposited, larger and larger pores will fill in to the point where the majority of the intrinsic porosity (all pores small to large) is filled and the films eventually deposit on the superficial surface of the agarose hydrogel scaffolds. To test this hypothesis, 30 bilayers of PAA/PEG and subsequently three additional bilayers of PEG and TRITC conjugated BSA, that is, ((PAA/PEG)30-(TRITC-BSA/PEG)3), were built onto the templated scaffolds. A confocal image section captured below the top surface of the ((PAA/PEG)30-(TRITC-BSA/PEG)3) coated scaffold showed a clear fluorescent ring formed from the (TRITC-BSA/PEG)3 deposited after (PAA/PEG)30, along the inner periphery of the scaffold channels (Fig. 2a, top image). In another case, three bilayers of PEG and TRITC conjugated BSA, that is, (TRITC-BSA/PEG)3, were coated onto the templated scaffolds. A confocal image section captured below the top surface of the (TRITC-BSA/PEG)3 coated scaffold showed the fluorescence in this sample was diffused throughout the agarose structure (Fig. 2a, bottom image). The corresponding intensity profile (Fig. 2a, top spectrum) for the ((PAA/PEG)30-(TRITC-BSA/PEG)3) sample shows a sharp increase in fluorescence along the inner walls of the channels and minimal fluorescence inside the agarose structure. This suggests that the (TRITC-BSA/PEG)3 films formed on the outer surface of the agarose channel wall, contributing to the fluorescent ring formation in the presence of (PAA/PEG)30 layers. On the other hand, the fluorescence intensity profile (Fig. 2a, bottom spectrum) for the (TRITC-BSA/PEG)3 sample is more diffused and does not show the sharp edges along the channel wall. The fluorescence which concentrated along the inner periphery of the ((PAA/PEG)30-(TRITC-BSA/PEG)3) scaffold (with an average intensity of 250 units) channel was more distributed throughout the structure of the (TRITC-BSA/PEG)3 scaffold (reducing the average intensity to 100 units). The three bilayers of (TRITC-BSA/PEG)3 in the absence of the (PAA/PEG)30 layers did not form the ring, suggesting that the initial three bilayers penetrated uniformly throughout the internal pores of the agarose. Comparing these two samples suggests that during the initial phase of multilayer formation (at least up to three bilayers), the film formation was occurring within the intrinsic pores of the agarose. Then, at some stage during the process of multilayer formation (between three and 30 bilayers), the intrinsic pores filled in, causing subsequent layers to be formed predominantly on the outer surface of the agarose.
Figure 2.
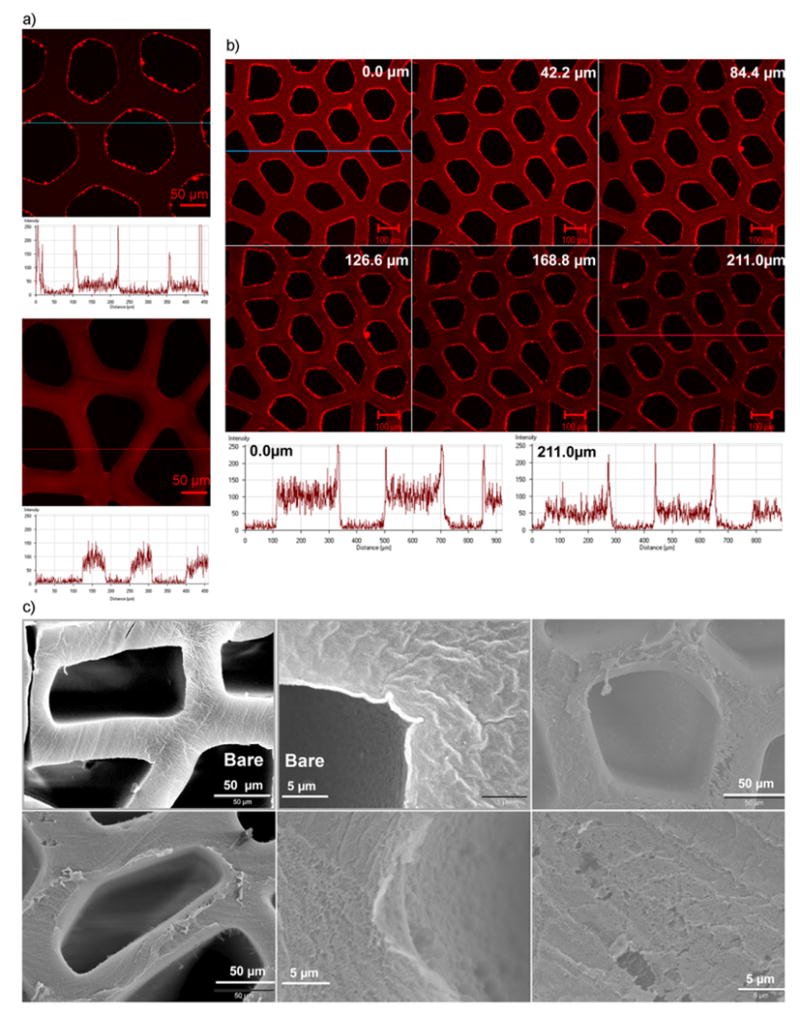
Confocal and scanning electron microscopy images showing the formation of LbL assembled multilayer thin films on agarose hydrogel substrate. a) Confocal microscopy images and associated fluorescence intensity profiles of a section below the top surface of agarose scaffolds coated with 30 bilayers of PAA and PEG followed by three bilayers of PEG and TRITC conjugated BSA (top image), and of agarose scaffolds coated with only 3 bilayers by PEG and TRITC conjugated BSA (bottom image). b) Confocal microscopy acquired z-section images of agarose scaffolds coated with 30 bilayers of PAA and PEG followed by three bilayers of PEG and TRITC conjugated BSA. Fluorescence intensity profiles of the topmost section (top panel: leftmost image) and a middle section (bottom panel: rightmost image) are shown. c) Scanning electron microscopy images of the scaffolds LbL coated with 30 bilayers of PAA and PEG. “Bare” denotes the non-coated scaffolds. BPEI was used as the LbL initiating polyelectrolyte for PAA/PEG assemblies.
Figure 2b illustrates the z-series images captured for the ((PAA/PEG)30-(TRITC-BSA/PEG)3) sample, traversing from the top surface down the structure of the agarose channel. Corresponding intensity profiles of the topmost section (0.0 μm) and a middle section (211 μm) are also shown. A constant ring of fluorescence along the inner wall of the channels and a decrease in the fluorescence intensity through the structure of the scaffold are observed. Therefore, in addition to the discussion of Figure 2a above, this constant ring of fluorescence along the inner wall of the channels further suggests coating on the outer surface of the agarose after formation of the 30 bilayers. The gradual decrease in the intensity profile along the channel of the agarose structure suggests the presence of a diffusion gradient that reduces the polymer deposition within the pores of the agarose down the channel as the building of the bilayers increases on the surface of the agarose. Overall, these results suggest that the LbL assembly process resulted in formation of a thin multilayer structure on the surface of the agarose, if sufficient (∼30) bilayers are built. Figure 2c shows the SEM images of the uncoated scaffolds and scaffolds coated with 30 bilayers of PAA/PEG, the latter shows a film has formed on the agarose surface. It is important to note that although there this is clear evidence that bilayers formed on the outer surface of the scaffolds, the majority of the proteins loaded in the gel are immobilized in the agarose hydrogel intrinsic pores, as explained further in Section 2.2.1.
2.2. Sustained Protein Release Profiles from LbL-Coated Agarose Scaffolds
3 wt% agarose concentration was found to be suitable for stable templated scaffold preparation and better axonal growth in repairing nerve injuries.[1] Figure 3a shows a cumulative profile of the lysozyme released from 3 wt% agarose coated with multilayers in the configuration depicted in Scheme 1a, with BPEI as the LbL initiating polymer. BPEI can provide a sufficient degree of H-bonding due to its multivalent amines, as well as hydrophobic interactions with agarose, and thus was selected as an LbL initiating polyelectrolyte. Multilayer assembly with BPEI as the initiating layer was followed by five bilayers of PAA/PEG, and five bilayers of lysozyme/PEG. These sets of five bilayers of PAA/PEG and lysozyme/PEG were repeated two more times, followed by PAA as the terminating layer (the multilayer assembly is denoted as BPEI[5,5]3). Sustained release was monitored over a period of four weeks, until the concentration of protein released per day was within μg mL−1. Daily releases are expected to continue further in the concentration range of ng mL−1, but are not reported here. This amount of protein released from the agarose hydrogel was within the range required for neurite outgrowth and axonal regeneration[55] (see Supporting Information for more details). The effect of using other polymers, such as LPEI or lysozyme, as the LbL initiating polymers were also evaluated (discussed in Section 2.2.2). In addition, we formed multilayer assemblies with different arrangements and combinations of PEG, PAA and protein, resulting in either three component or two component assemblies (Scheme 1a–f) and analyzed their protein release profiles (discussed below).
Figure 3.
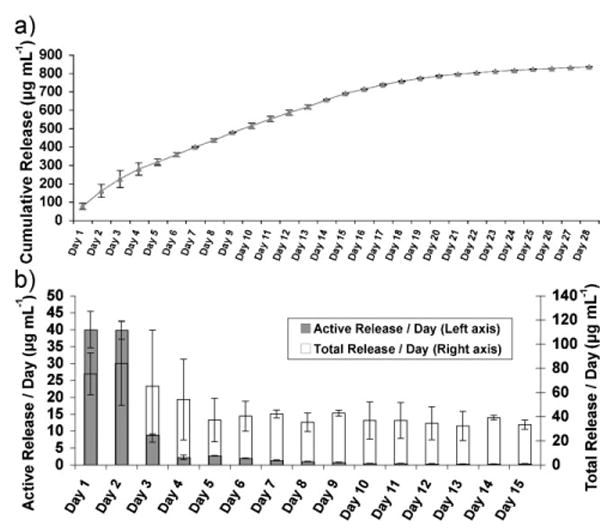
a) Cumulative lysozyme release up to 4 weeks triggered by physiological pH, from LbL multilayer (as shown in Scheme 1a; BPEI initiating) coated 3% agarose hydrogel. b) Comparison between the total and enzymatically active lysozyme released per day, corresponding to the protein released in Figure 3a. Active concentrations were calculated from a standard curve obtained from pure lysozyme used to determine the degree of lysis of Micrococcus lysodeikticus by lysozyme.
The enzymatic activity of the lysozyme released from the multilayers on the agarose was assessed by a bacterial assay (see Experimental Section for further details), and found to be active during the earlier time points, with decreasing activity at later time points. Figure 3b compares the concentration of total lysozyme versus the concentration of active lysozyme released per day, for up to 15 days. The total as well as active lysozyme released were both in the μg mL−1 range for up to a week. Active lysozyme continued to be released after a week in the sub-μg mL−1 and subsequently in the ng mL−1 range. Sukhishvilli and co-workers showed lysozyme forms stable multilayers with carboxylic acid-based weak polyelectrolytes under low pH conditions due to H-bond interactions, whereas the multilayers are disrupted if formed under high pH conditions.[56] They reported that it is likely after dissociation of the H-bonds under physiological conditions and release of lysozyme from the multilayers, the lysozyme (isoelectric point, pI ≈ 11) can interact electrostatically with the ionized PAA. Moreover, these interactions depend on the concentrations of the salts, PAA and lysozyme present in the solution.[56,57] Further, upon release from the multilayers, lysozyme could remain bonded with PEG through H-bonding. Although, the interaction between the released lysozyme with PAA or PEG is possible, it did not appear to have caused an adverse effect on the controlled release of the lysozyme from the agarose (Fig. 3a). However, interactions between the released lysozyme and PAA or PEG cannot be precluded. This interaction may sequester the released lysozyme by forming inter-polyelectrolyte complexes in the solution, and thereby lowering the activity level of the released lysozyme over time (Fig. 3b).
2.2.1. Effect of the Agarose Concentration on the Protein Release Profiles
Multilayers with BPEI as the LbL initiating polymer (Scheme 1a) were formed on different concentrations of agarose. Figure 4a compares the protein release profiles of 1%, 2%, and 3% agarose over five days, and Figure 4b compares the release profiles of 3% and 4% agarose over an extended period of 15 days. The profiles of the protein released were similar across the different agarose concentrations but the amount of proteins released increased with time and agarose concentrations. The amount of protein released is governed by the wt% of the agarose as shown in Figure 4a and b.
Figure 4.
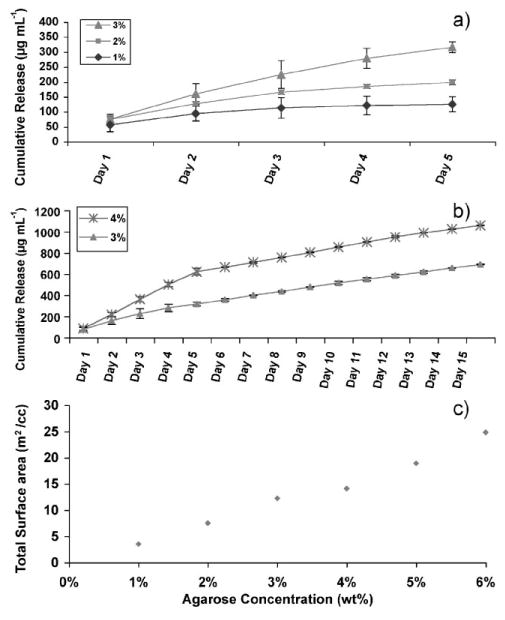
a,b) Cumulative lysozyme release over time triggered by physiological pH from agarose hydrogel of varying concentrations, coated with LbL multilayer assembly (as shown in Scheme 1a; BPEI initiating). a) Comparison between 1%, 2%, and 3% agarose. b) Comparison between 3% and 4% agarose. c) Total surface area per unit volume of pure agarose hydrogel as a function of agarose concentration determined by BET.
To understand the observed increase in protein release from the multilayer-coated agarose with increasing agarose concentration, we measured the total surface area per unit volume available from the intrinsic pores of the agarose. As mentioned above, it is important to note that the agarose hydrogels likely consist of a broad range of pore diameters ranging from <1 nm to several hundred nanometers in diameter.[8–13] However, most of the surface area normalized by the superficial volume of the gels is derived from the high concentration of pores in the tens of nm range or less. Thus, it is believed that the greater the concentration of micropores, the higher the internal surface area for the films to deposit on per unit volume of agarose hydrogel. More surface area available would thus suggest more protein can be loaded into the pores of the hydrogel during LbL assembly. Here, the total surface area of the agarose (prior to multilayer formation) per unit volume of gel was measured using the Brunauer–Emmer–Teller (BET) model on supercritically dried[58] agarose hydrogels. Anincrease in the weight percent of the hydrogel resulted in an increase in the BETsurface area per unit volume of gel (Fig. 4c). This is likely due to the creation of more gel network branches rather than increased branch thickness in agarose. Therefore, at higher concentrations of agarose, more protein would be expected to be loaded into the intrinsic pores of the agarose, resulting in higher protein release.
2.2.2. Effect of the LbL Initiating Polyelectrolyte on the Protein Release Profiles
Three component BPEI[5,5]3 or LPEI[5,5]3 multilayer assemblies (Scheme 1a) were built on agarose with either BPEI or LPEI as the LbL initiating polymer, respectively. To avoid using non-biocompatible BPEI or LPEI, similar multilayer assemblies starting with lysozyme were also prepared (Scheme 1b). Figure 5 shows the protein release profiles with BPEI, LPEI or protein as the initiating LbL, fabricated on 3% agarose scaffolds. BPEI or LPEI as the initiating LbL was also fabricated onto 1% agarose. The profiles of the protein release were similar in all cases, however, there were significant differences in the actual amount of protein released at a given time for the different cases. The amount of protein released with BPEI as the initiating LbL was lower than with LPEI as the initiating LbL, both for the 1% and 3% agarose scaffold. In contrast, the amount of protein released from the multilayers was similar with lysozyme or LPEI as the LbL initiating polymer.
Figure 5.
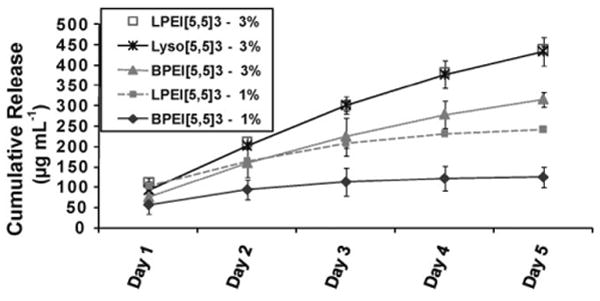
The effect of LbL initiating polymer on lysozyme release triggered by physiological pH, from LbL coated 3% agarose hydrogel. BPEI, LPEI or lysozyme was used as the LbL initiating polymer as shown in Scheme 1a and b.
The difference in the amounts of protein released as a function of the initiating LbL polymer (i.e., BPEI, LPEI or protein) may be due to their degree of binding with the agarose. LPEI, BPEI and lysozyme (protein) are weak polyelectrolytes and their degree of ionization is dependent on the pH. During the LbL assembly, BPEI and LPEI were kept at a high pH, whereas lysozyme was kept at a low pH (see Experimental Section). BPEI contains primary, secondary, and tertiary amines and is a hydrophobic polymer, whereas LPEI which contains only secondary amines is a hydrophilic polymer. Owing to its hydrophobicity BPEI has been shown to preferentially adsorb onto hydrophobic regions on surfaces.[59] Furthermore, due to the presence of the multivalent amines, BPEI can also form more H-bonds with the hydroxyl groups and oxygen on the agarose than LPEI. Depending on the isoelectric point and pH, proteins also can form hydrophobic and H-bonds with agarose. The increased amount of protein released with the lysozyme initiating LbLs in comparison to BPEI as the initiating LbL suggests that at pH of 3.0, lysozyme (pI ≈ 11) binds less strongly than BPEI to the agarose surface, perhaps due to more protonation of the amine groups on lysozyme at low pH, making lysozyme less hydrophobic than BPEI. Therefore, the BPEI initiating LbL would likely bind more strongly to agarose than LPEI or lysozyme initiating LbLs. This would affect the overall degradation kinetics of the multilayer, and consequently the protein released profiles.
2.2.3. Effect of the Stacking Layer Configuration and Assembly Components on the Protein Release Profiles
The previous sections were based on forming [5,5]3 sandwiched multilayers with five bilayers of a three component assembly (Scheme 1a or b). This type of assembly arrangement was chosen initially because the degradation of PAA/PEG multilayers reported by Ono and Decher showed that fewer than seven bilayers of PAA/PEG did not release the upper films as self-standing, floating films, whereas more than seven bilayers allowed the upper films to be released within 30 min.[51] Therefore, we hypothesize that stacking of two components with greater than five bilayers at a time (e.g., Lyso[10,10,5,5] or Lyso[15,15] in Scheme 1c or d) could degrade faster and release more protein at a given time than a five bilayer arrangement of three component assembly (Lyso[5,5]3 in Scheme 1a or b). We formed multilayers using PAA, PEG, and protein, with two different arrangements starting with lysozyme as the LbL initiating polymer (Scheme 1c and d). The total number of layers for each assembly was kept the same as in Scheme 1b. Figure 6a shows the profiles of protein release over five days for the three different arrangements, Lyso[5,5]3 (Scheme 1b), Lyso[10,10,5,5] (Scheme 1c) and Lyso[15,15] (Scheme 1d). No significant difference was observed for the three arrangements at earlier time points; however, the release profiles diverged at later time points. The amount of cumulative protein released was less for the Lyso[5,5]3 than the Lyso[10,10,5,5] assembly and was highest for the Lyso[15,15] assembly. This suggests that a higher number of bilayers stacked together degraded faster and released more protein than the stacking assembly with fewer bilayers. This is in accordance with the previous finding by Ono and Decher[51] that suggested fewer (less than seven) bilayers degraded slower than more bilayers of PAA/PEG. Thus, using fewer bilayers in the stacking assembly may provide sustained release and more control of the amount of protein released over a longer period.
Figure 6.
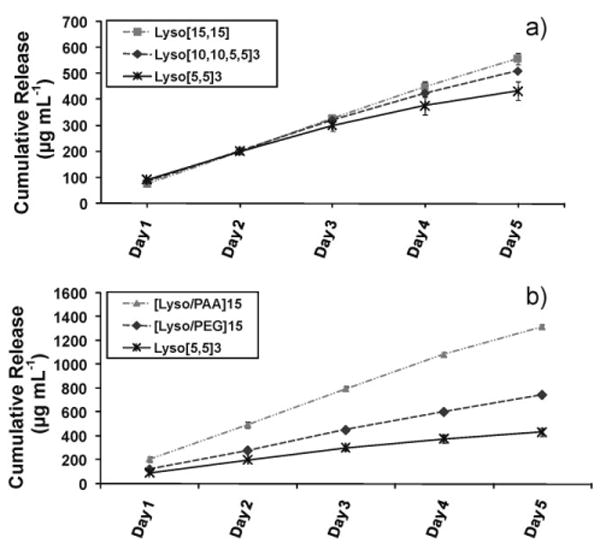
Cumulative lysozyme release from LbL-coated 3% agarose hydrogel, triggered by physiological pH, a) with varying stacking order of the polymers within a multilayer but the same number of cumulative bilayers (as shown in Scheme 1b–d) and b) with two-component assembly of lysozyme and PAA, two-component assembly of lysozyme and PEG, and three component assembly of PAA, PEG and lysozyme (as shown in Scheme 1f, e, and b respectively).
Some studies has reported PAA as a biocompatible and bioinert polymer; however, cytotoxicity has not been explicitly evaluated.[40,51] In addition, PAA is not a FDA approved polymer and thus may not preclude the possibility of being toxic under certain conditions. Nevertheless, PAA toxicity would depend also on the amount of PAA supplied in vitro or in vivo. Therefore, we also evaluated a completely biocompatible multilayer arrangement on agarose consisting of a two component H-bonded assembly of lysozyme and PEG ([Lyso/PEG]15 in Scheme 1e) built at low pH. For comparison, a multilayer assembly of lysozyme and PAA ([Lyso/PAA]15 in Scheme 1f) was also prepared over agarose. In both cases, the total number of layers of protein was kept the same as in the arrangements shown in Schemes 1a–d. Figure 6b shows the protein release followed similar trends as with the three component Lyso[5,5]3 (Scheme 1a) multilayers over agarose (Fig. 6a), but released higher amounts of protein. The amount of protein released from [Lyso/PAA]15 was the highest, followed by [Lyso/PEG]15, and then the three component assembly, Lyso[5,5]3. This difference observed could be due to a stronger H-bonding of the oxygen on PEG with the carboxylic acids and amino groups on the protein than the interactions between the carboxylic acids on PAA and the amino groups on the protein, thereby slowing the degradation of [Lyso/PEG]15 over the [Lyso/PAA]15 assembly.
2.3. Low Molecular Weight H-bonded Multilayer Disintegration on a Planar Substrate
The degradation rate of H-bonded multilayers is one of the many factors that control the protein release from the LbL coated agarose gels. Therefore, as a first step in assessing the degradation of PAA/PEG multilayers, we evaluated the degradation behavior of these H-bonded films fabricated on a planar substrate. The degradation kinetics of high molecular weight PEO and PAA multilayer systems on a planar substrate have been previously investigated in detail, and were reported to degrade in about 30 min upon exposure to a pH of 3.5 or higher.[48]
However, the stability or rate of film degradation will also depend on the choice of constituent polymers and their properties,[48] such as their molecular weight. The effect of molecular weight on the growth profile of PAA/PEO multilayers is known,[49] however to our knowledge, the effect of molecular weight on the degradation kinetics of these H-bonded multilayers has not been studied. Here, the multilayer films on agarose gel were formed using low molecular weight polymers, 15 kDa PAA (sodium salt) and 10 kDa PEG, which differed from previous studies. Therefore, we analyzed these films for their degradation behavior on a planar substrate using SEM. The thickness of 25 bilayers of 15 kDa-PAA/10 kDa-PEG films, as determined by SEM (Fig. 7, t = 0), was approximately 600 nm which is close to previous report with the 25 bilayers of 250 kDa PAA and 15 kDa PEG films.[51] However, we observed that the 15 kDa PAA and 10 kDa PEG films degraded slowly over a period of days (5–10 days; Fig. 7) rather than minutes. It appeared that the low molecular weight PAA/PEG films did not completely degrade even after 10 days. These results suggest that the disintegration rate of the H-bonded PAA/PEG depends also on the molecular weights of the constituent polymers.
Figure 7.

SEM images of H-bonded films composed of 25 bilayers of 10 kDa PEG and 15 kDa PAA formed on a planar substrate and exposed to deionized water (DI) (pH 5.6–6.3) for the time durations indicated. Top panel: SEM images of films after fabrication. Middle panel: SEM images of films after immersion in DI water for five days. Bottom panel: SEM images of films after immersion in DI water for ten days. Same spot on the films before and after degradation were imaged for comparative analysis. Columns 1, 2, and 3 show three different spots on the film.
2.4. Cytophobicity of H-bonded PAA/PEG Multilayers
Previous in vivo investigations revealed that templated agarose scaffolds implanted at the site of a spinal cord injury formed a reactive cell layer of adherent leptomeningeal fibroblasts at the interface of the distal end of the scaffold and the host matrix.[60] This reactive cell layer formed several days after scaffold implantation, and appeared to limit the ability of host axons that have already regenerated into a scaffold to exit the distal end of a scaffold and reinnervate the spinal cord beyond the lesion.[60] Therefore, a strategy that provides sustained release of growth factors for enhanced axonal growth, while simultaneously preventing in vivo fibroblast adhesion onto the outer surface of agarose, is highly desirable.
The low molecular weight H-bonded PAA/PEG films coated onto TCPS substrates were cytophobic towards fibroblast attachment in vitro for at least two weeks. This was possibly due to the slow degradation kinetics of these films, causing the surface to erode gradually and thus changing the surface topography over a period of days. It has been demonstrated previously that varying the surface topography (the physical features on the surface) can regulate adhesion and proliferation of the fibroblasts.[61] As evident from the SEM micrographs (Fig. 7), the initial surface morphology of PAA/PEG films is fairly rough (Fig. 7, t = 0), and becomes rougher as the films degrade over time. Figure 2c (lower panel middle image) of PAA/PEG films coated onto the channel wall of a templated agarose scaffold also clearly shows a rough morphology. The changing surface topography of the gradually eroding PAA/PEG films could lead to a rougher surface over time that may not be favorable to fibroblast adhesion. Further, an eroding/degrading substrate cannot easily support cell adhesion. Moreover, PEG is known to resist protein and cell attachment[62,63] Figure 8a and Figure S2 (Supporting Information) shows the cytophobic behavior of PAA/PEG multilayers coated onto a TCPS substrate with 30.5 and 5.5 bilayers, respectively, for almost two weeks.
Figure 8.
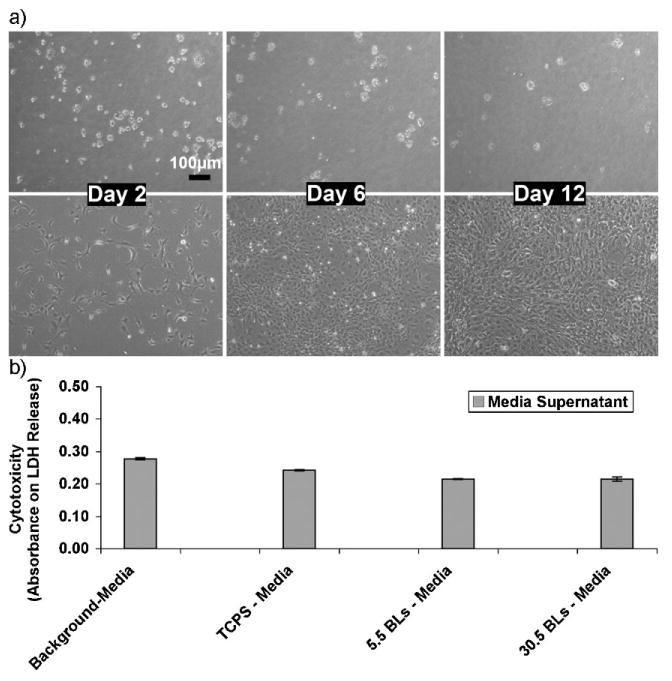
a) Phase contrast microscopy images demonstrating the cytophobicity of the 30.5 bilayers of PAA/PEG multilayers over time (days). Top panel: NIH-3T3 fibroblasts cells on TCPS plates coated with 30.5 bilayers of PAA/PEG. Bottom panel: Fibroblasts on bare TCPS plates (control). b) Cytotoxicity levels of fibroblast cells after 4 days of culturing on (PAA/PEG)5PAA multilayers built onto TCPS (denoted here as 5.5BLs), (PAA/PEG)30PAA multilayers built onto TCPS (denoted here as 30.5 BLs), and bare TCPS plates. Serum in the cell culture medium contains a small amount of LDH, which is shown as “background media”. The absorbance values corresponds to the amount of lactate dehydrogenase (LDH) released into the culture supernatant.
The cytophobic behavior does not indicate cytotoxicity of these films. A quantitative lactate dehydrogenase (LDH) cytotoxicity assay was performed on the fibroblasts seeded onto these films. The LDH released into the supernatant after four days of culture was collected and measured. As shown in Figure 8b, no excess LDH was released into the culture medium from the cells seeded onto these films as compared to the control TCPS substrate after four days. This indicates that the PAA/PEG films, which degrade over time to release PAA and PEG polymers into the culture medium, were not toxic to the cells and the unattached, floating cells on these PAA/PEG films were not due to cytotoxicity. Therefore these films, in addition to providing sustained release of protein from agarose, may help also in preventing the reactive cell layer from forming on the implants in vivo.
3. Conclusions
We fabricated LbL-assembled multilayer films that incorporated proteins into the multilayer structure over agarose, rather than preloading the protein directly into the agarose gel. This allowed the protein to be loaded subsequent to the fabrication, and avoided exposing the protein to harsh organic solvents during fabrication of the templated agarose scaffolds. The LbL multilayers consisting of either a three component assembly of PAA, PEG and protein, or a two component assembly of PEG and protein were fabricated at low pH (pH ≤ 3.0) conditions. These multilayers formed thin films over the surface of the agarose and provided prolonged and sustained protein release at physiological pH conditions for up to a month in certain cases. The lysozyme released was found to be active during the earlier time points, with decreasing activity at later time points. The amount of protein released depended on the concentration of the agarose, the H-bonding and hydrophobic characteristics of the LbL initiating polymers, and the LbL constituents. This suggests that the protein release is a complex phenomenon, governed by both the multilayer degradation kinetics and the agarose porosity. The relative rate of protein released with the three component assemblies was slower than the two component assemblies. The arrangements discussed can be used in clinical trials for axonal regeneration, with either a two component H-bonded biocompatible assembly consisting of PEG and protein, or a three component assembly replacing PAA with another biocompatible polymer such as poly(glutamic acid) or poly(aspartic acid) for slower protein release. The choice of the configuration would depend on the desired rate of protein release. Currently, these combinations are being tested for sustained release of BDNF from templated agarose scaffolds, both in vitro and in vivo, to enhance axonal growth.
4. Experimental
Materials and Methods
Ultrapure agarose (Sigma–Aldrich, USA) was dissolved at a required concentration in 18.2 MΩ cm resistivity deionized (DI) water (MilliQ, Millipore) at near 100°C. For preparing gels for Brunauer–Emmer–Teller (BET) analysis, the solutions were heated in a closed vial. Templated agarose scaffolds were prepared by a phase inversion process using polymer fiber templates supplied by Paradigm Optics Inc. (Vancouver, WA) as described previously [1]. For all the protein release experiments, hot agarose solution (3 mL) was poured into 12-well tissue culture polystyrene (TCPS) plates (Costar, Corning, NY). For fluorescence and ultraviolet/visible (UV/vis) absorbance experiments, the hot agarose solution was filled to the top in a 96-well polystyrene plate (Evergreen Scientific, USA). Agarose was allowed to gel at room temperature. Poly(acrylic acid, sodium salt) solution (PAA, Mw 15 000; catalog no. 416037), poly(ethylene glycol) (PEG, Mw 10000; catalog no. P6667), poly(ethylene glycol) bis(3-aminopropyl) terminated (PEG-Amine) (Mw 1 500; catalog no. 452572), lysozyme (Lyso) from chicken egg white (catalog no. L6876), tetramethylrhodamine isothiocynate (TRITC) (catalog no. 87918) were purchased from Sigma–Aldrich. Cytotoxicity detection kit for LDH release measurements was purchased from Roche Applied Science, Indianapolis, IN.
Lysozyme protein was used for all the protein release experiments, whereas bovine serum albumin (BSA) protein (US Biological, MA) was used for all the LbL growth characterizations (fluorescence, UV/vis absorbance and confocal microscopy). PAA and PEG polymer solutions used to fabricate multilayer assemblies were prepared in DI water to final concentrations of 1 mg mL−1, and their pH was adjusted to 2.0. Protein (lysozyme or BSA) was dissolved in 1 × phosphate buffer saline (PBS) (Invitrogen, USA) at a concentration of 1 mg mL−1 and solution pH was adjusted to 3.0. Different numbers of bilayers of PAA, PEG and protein were prepared as shown in Scheme 1a–f and as explained in Section 2. DI water adjusted to pH 2.0 was used as the wash solution during assembly fabrication. Agarose-filled polystyrene plates were immersed in LbL starting polymer (1 mg mL−1 25 kDa branched polyethylenimine (BPEI) (Sigma–Alrich) at pH 10.5; or 1 mg mL−1 25 kDa linear polyethylenimine (LPEI) (Polysciences Inc.) at pH 7.2–7.4; or protein in PBS at pH 3.0) for 30 min and then rinsed in wash solution for 10 min with agitation. The substrates were then dipped into PAA or PEG solution (depending on multilayer assembly, as shown in Scheme 1a–f) for 30 min followed by 10 min in wash solution with agitation to create one bilayer. The dipping cycle was repeated to form multilayer films. A Carl Zeiss slide stainer was used to form multilayers. Multilayers were fabricated in different bilayer arrangements as shown in Scheme 1a–f. PAA was kept as the terminating layer in each case. In some cases, an additional set of five bilayers of (PAA/PEG) was added before terminating the assembly with PAA, however these additional layers was found to have no significant effect on the protein release profiles. To characterize the LbL adsorption of PEG, PEG-Amine was conjugated to the TRITC dye for fluorescence measurements in 96-well plate. PEG-Amine (300 mg) was dissolved in 1× PBS buffer (300 mL, pH adjusted to ∼9.0), and TRITC dye (∼15 mg, dissolved in DMSO) was added. The mixture was stirred continuously at 4 °C for 24 h and the mixture pH was subsequently reduced to 2.0. Since the molar ratio of dye to PEG-Amine was much less than one, no dialysis or column purification was performed. The mixture was directly used to form multilayers with the BSA protein (pH 3.0) using the slide stainer with extensive intermediate washes (pH 2.0) during LbL. To study the effect of the molecular weight of the polymer on the multilayer degradation behavior, 25 bilayers of PAA/PEG multilayers were prepared onto clean gold substrates, which were coated with mercaptoundecanoic acid (Sigma–Aldrich) followed by BPEI and PAA/PEG deposition. Films, when abbreviated as n.5, denote the PAA/PEG multilayer where n is the number of PAA and PEG bilayers and the “.5” indicates an additional, single terminating layer of PAA. For the fibroblast cytophobicity tests, PAA/PEG multilayers were prepared on O2 plasma-treated (Harrick Plasma Cleaner) and LPEI-coated TCPS plates. NIH-3T3 fibroblasts were purchased from American Type Culture Collection (Rockville, MD), and all cell cultures were maintained in DMEM with 10% FBS and penicillin (100 U mL−1) plus streptomycin (100 μg mL−1) (P/S) at 37°C and 5% CO2.
Characterization
Emission spectra of TRITC in 96-well plate were acquired using a SPECTRA-MAX GEMINI-EM fluorescence plate reader (Molecular Devices, CA) at an excitation wavelength of 529 nm with bottom-read option. UV/vis peaks in 96-well plate were measured at 25 °C at 280 nm using SPECTRAmax Plus 384 (Molecular Devices). Confocal laser scanning microscopy (CLSM) images of the scaffolds were obtained using Zeiss Pascal laser scanning confocal microscope. Excitation wavelength used for TRITC dye for confocal microscopy was 543 nm, and emission spectrum was obtained at wavelengths greater than 560 nm using a long pass filter. Gain and offset settings were kept the same during sample and control imaging. Phase contrast optical microscopy images were collected using Leica DM IL inverted microscope (Bannockburn, IL) equipped with SPOT RT color camera (Diagnostics Instruments, MI).
For scanning electron microscopy (SEM) imaging of the scaffolds, LbL-coated and non-coated scaffolds were dehydrated through a series of 30 min immersions in ethanol solutions of 25%, 50%, 75%, 95%, and 100% concentration; supercritically dried using CO2 (Balzers CPD, Lichtenstein) followed by a 7–10 nm gold sputter coating (EMSCOPE SC500 Sputter coater, Ashford, Great Britain). SEM images were obtained with field emission JEOL 6400 electron microscope. The series of dehydration before SEM imaging likely caused the defects in the coatings, as observed in Figure 2c. In order to visualize the coating formation on the agarose surface, particular areas of defects were selectively identified for imaging. For BET experiments, hydrogels were dehydrated using pure ethanol (at 10 times the volume of the gel) and the ethanol was replaced twice, each time immersing for 24 h. This was followed by CO2 supercritical drying [58] and stored in a desiccator. BET surface area characterization was performed with a nitrogen-adsorption testing apparatus (Micromeritics ASAP 2020), using helium to measure the free space and nitrogen to measure the surface area and pore-size distribution. Each sample was fractured into small pieces using a scalpel in order to fit into the test chamber and reduce the amount of time spent waiting for diffusion of the analysis gas. Before analysis, samples were degassed under vacuum at 90 °C for eight hours followed by a nitrogen backfill, run through a short analysis to obtain the free space of the test chamber, then degassed again under the same conditions for 24 h with no backfill prior to the actual analysis. This second degassing step allows the microporosity to be measured accurately by removing any previously adsorbed gasses.
Unless otherwise specified, for all protein release experiments, LbL-coated agarose in 12-well plates were incubated in 1X-PBS(pH 7.4)/well (1 mL) at 37 °C, the buffer was replaced with fresh PBS (1 mL) after each sampling interval (to obtain the cumulative release profiles), and the collected buffer was stored either at −20 °C (or 4°C for short term) until further analysis. Additionally, lysozyme releases were also characterized by incubating the LbL-coated agarose in fibroblast cell culture medium (1 mL) at 37°C (see Supporting Information, Fig. S3). Concentrations of the lysozyme released from the LbL-coated agarose were measured by the BCA or micro-BCA protein assay (Pierce, Rockford, USA) according to the manufacturer's instruction. The releases obtained at each sampling interval were added to obtain the cumulative release profiles. Lysozymal enzymatic activity was determined spectrophotometrically by measuring the degree of lysis of Micrococcus lysodeikticus bacteria (Sigma–Aldrich) [64]. Active lysozyme damages the cell wall of the bacteria and thus its activity is characterized by the decrease in the amount of live, intact bacteria in the solution. An M. lysodeikticus suspension (3 mg mL−1) was prepared in an assay buffer containing phosphate buffer (50 mM, pH adjusted to 7.4), sodium azide (0.1%) (Sigma–Aldrich) and BSA (1 mg mL−1). An aliquot (200 μL) of collected sample was added to assay buffer (1.8 mL) with M. lysodeikticus at a final concentration of 300 μg mL−1 (1:10 dilution), and incubated at 37 °C for 2 h. Change in turbidity was monitored at a wavelength of 450 nm with the assay buffer as a blank. A standard curve for the lysozyme activity was obtained using pure lysozyme dissolved in 1× PBS to calculate the active lysozyme concentration released from the agarose. All data shown as mean ± SD are from at least three independent sets of experiments.
Supplementary Material
Acknowledgments
The work was supported in part by the National Institute of Health (R01GM079688, R21CA126136, R21RR024439 and R01NS049881), the Veterans Administration RR&D service, the National Science Foundation (CMMI-0832730), the University Research Corridor and the MSU Foundation. Supporting Information is available online from Wiley InterScience or from the author.
Contributor Information
Sumit Mehrotra, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA).
Daniel Lynam, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA).
Ryan Maloney, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA).
Kendell M. Pawelec, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA)
Mark H. Tuszynski, Director-Center for Neural Repair, Department of Neurosciences, University of California, San Diego 92093, CA (USA)
Prof. Ilsoon Lee, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA)
Prof. Christina Chan, Email: krischan@egr.msu.edu, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA) Department of Biochemistry and Molecular Biology Michigan State University East Lansing, Michigan 48824, MI (USA).
Prof. Jeffrey Sakamoto, Email: jsakamot@egr.msu.edu, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, Michigan 48824, MI (USA)
References
- 1.Stokols S, Sakamoto J, Breckon C, Holt T, Weiss J, Tuszynski MH. Tissue Eng. 2006;12:2777. doi: 10.1089/ten.2006.12.2777. [DOI] [PubMed] [Google Scholar]
- 2.Tuszynski MH, Gabriel K, Gage FH, Suhr S, Meyer S, Rosetti A. Exp Neurol. 1996;137:157. doi: 10.1006/exnr.1996.0016. [DOI] [PubMed] [Google Scholar]
- 3.Blesch A, Tuszynski MH. J Comp Neurol. 2003;467:403. doi: 10.1002/cne.10934. [DOI] [PubMed] [Google Scholar]
- 4.Bradbury EJ, Khemani S, King VR, Priestley JV, McMahon SB. Eur J Neurosci. 1999;11:3873. doi: 10.1046/j.1460-9568.1999.00809.x. [DOI] [PubMed] [Google Scholar]
- 5.Menei P, Montero-Menei C, Whittemore SR, Bunge RP, Bunge MB. Eur J Neurosci. 1998;10:607. doi: 10.1046/j.1460-9568.1998.00071.x. [DOI] [PubMed] [Google Scholar]
- 6.Doolabh VB, Hertl MC, Mackinnon SE. Rev Neurosci. 1996;7:47. doi: 10.1515/revneuro.1996.7.1.47. [DOI] [PubMed] [Google Scholar]
- 7.Fournier E, Passirani C, Montero-Menei CN, Benoit JP. Biomaterials. 2003;24:3311. doi: 10.1016/s0142-9612(03)00161-3. [DOI] [PubMed] [Google Scholar]
- 8.Fatin-Rouge N, Milon A, Buffle J, Goulet RR, Tessier A. J Phys Chem B. 2003;107:12126. [Google Scholar]
- 9.Liang SM, Xu J, Weng LH, Dai HJ, Zhang XL, Zhang LN. J Controlled Release. 2006;115:189. doi: 10.1016/j.jconrel.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Maaloum M, Pernodet N, Tinland B. Electrophoresis. 1998;19:1606. doi: 10.1002/elps.1150191015. [DOI] [PubMed] [Google Scholar]
- 11.Pernodet N, Maaloum M, Tinland B. Electrophoresis. 1997;18:55. doi: 10.1002/elps.1150180111. [DOI] [PubMed] [Google Scholar]
- 12.Gutenwik J, Nilsson B, Axelsson A. AIChE J. 2004;50:3006. [Google Scholar]
- 13.Xiong JY, Narayanan J, Liu XY, Chong TK, Chen SB, Chung TS. J Phys Chem B. 2005;109:5638. doi: 10.1021/jp044473u. [DOI] [PubMed] [Google Scholar]
- 14.Blake CCF, Koenig DF, Mair GA, North ACT, Phillips DC, Sarma VR. Nature. 1965;206:757. doi: 10.1038/206757a0. [DOI] [PubMed] [Google Scholar]
- 15.Hoare TR, Kohane DS. Polymer. 2008;49:1993. [Google Scholar]
- 16.Lynch I, de Gregorio P, Dawson KA. J Phys Chem B. 2005;109:6257. doi: 10.1021/jp0502149. [DOI] [PubMed] [Google Scholar]
- 17.Nakamae K, Nishino T, Kato K, Miyata T, Hoffman AS. J Biomater Sci Polym Ed. 2004;15:1435. doi: 10.1163/1568562042368095. [DOI] [PubMed] [Google Scholar]
- 18.Park H, Temenoff JS, Holland TA, Tabata Y, Mikos AG. Biomaterials. 2005;26:7095. doi: 10.1016/j.biomaterials.2005.05.083. [DOI] [PubMed] [Google Scholar]
- 19.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Adv Mater. 2006;18:1345. [Google Scholar]
- 20.Peppas NA, Leobandung W. J Biomater Sci Polym Ed. 2004;15:125. doi: 10.1163/156856204322793539. [DOI] [PubMed] [Google Scholar]
- 21.Bouhadir KH, Kruger GM, Lee KY, Mooney DJ. J Pharm Sci. 2000;89:910. doi: 10.1002/1520-6017(200007)89:7<910::AID-JPS8>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 22.Nuttelman CR, Tripodi MC, Anseth KS. J Biomed Mater Res Part A. 2006;76:183. doi: 10.1002/jbm.a.30537. [DOI] [PubMed] [Google Scholar]
- 23.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Nat Biotechnol. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 24.Sutter M, Siepmann J, Hennink WE, Jiskoot W. J Controlled Release. 2007;119:301. doi: 10.1016/j.jconrel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 25.Decher G. Science. 1997;277:1232. [Google Scholar]
- 26.Lynn DM. Adv Mater. 2007;19:4118. [Google Scholar]
- 27.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203. [Google Scholar]
- 28.De Geest BG, Sanders NN, Sukhorukov GB, Demeester J, De Smedt SC. Chem Soc Rev. 2007;36:636. doi: 10.1039/b600460c. [DOI] [PubMed] [Google Scholar]
- 29.Donath E, Sukhorukov GB, Caruso F, Davis SA, Mohwald H. Angew Chem Int Ed. 1998;37:2202. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2201::AID-ANIE2201>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 30.Sukhorukov GB, Donath E, Lichtenfeld H, Knippel E, Knippel M, Budde A, Mohwald H. Colloids Surf A. 1998;137:253. [Google Scholar]
- 31.Antipov AA, Sukhorukov GB, Donath E, Mohwald H. J Phys Chem B. 2001;105:2281. [Google Scholar]
- 32.Berg MC, Zhai L, Cohen RE, Rubner MF. Biomacromolecules. 2006;7:357. doi: 10.1021/bm050174e. [DOI] [PubMed] [Google Scholar]
- 33.Chung AJ, Rubner MF. Langmuir. 2002;18:1176. [Google Scholar]
- 34.Qiu XP, Donath E, Mohwald H. Macromol Mater Eng. 2001;286:591. [Google Scholar]
- 35.Shi XY, Caruso F. Langmuir. 2001;17:2036. [Google Scholar]
- 36.Sukhorukov GB, Antipov AA, Voigt A, Donath E, Mohwald H. Macromol Rapid Commun. 2001;22:44. [Google Scholar]
- 37.Sukhorukov GB, Brumen M, Donath E, Mohwald H. J Phys Chem B. 1999;103:6434. [Google Scholar]
- 38.De Geest BG, Dejugnat C, Sukhorukov GB, Braeckmans K, De Smedt SC, Demeester J. Adv Mater. 2005;17:2357. [Google Scholar]
- 39.Matsusaki M, Sakaguchi H, Serizawa T, Akashi M. J Biomater Sci Polym Ed. 2007;18:775. doi: 10.1163/156856207781034160. [DOI] [PubMed] [Google Scholar]
- 40.Kim BS, Park SW, Hammond PT. ACS Nano. 2008;2:386. doi: 10.1021/nn700408z. [DOI] [PubMed] [Google Scholar]
- 41.Picart C, Schneider A, Etienne O, Mutterer J, Schaaf P, Egles C, Jessel N, Voegel JC. Adv Funct Mater. 2005;15:1771. [Google Scholar]
- 42.Serizawa T, Yamaguchi M, Akashi M. Angew Chem Int Ed. 2003;42:1115. doi: 10.1002/anie.200390293. [DOI] [PubMed] [Google Scholar]
- 43.Wood KC, Boedicker JQ, Lynn DM, Hammond PT. Langmuir. 2005;21:1603. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 44.Wood KC, Chuang HF, Batten RD, Lynn DM, Hammond PT. Proc Natl Acad Sci USA. 2006;103:10207. doi: 10.1073/pnas.0602884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stockton WB, Rubner MF. Macromolecules. 1997;30:2717. [Google Scholar]
- 46.Wang LY, Wang ZQ, Zhang X, Shen JC, Chi LF, Fuchs H. Macromol Rapid Commun. 1997;18:509. [Google Scholar]
- 47.Sukhishvili SA, Granick S. J Am Chem Soc. 2000;122:9550. [Google Scholar]
- 48.Sukhishvili SA, Granick S. Macromolecules. 2002;35:301. [Google Scholar]
- 49.DeLongchamp DM, Hammond PT. Langmuir. 2004;20:5403. doi: 10.1021/la049777m. [DOI] [PubMed] [Google Scholar]
- 50.Mehrotra S, Lee I, Chan C. Acta Biomater. 2009;5:1474. doi: 10.1016/j.actbio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ono SS, Decher G. Nano Lett. 2006;6:592. doi: 10.1021/nl0515504. [DOI] [PubMed] [Google Scholar]
- 52.Kharlampieva E, Sukhishvili SA. Langmuir. 2004;20:9677. doi: 10.1021/la048763d. [DOI] [PubMed] [Google Scholar]
- 53.De Geest BG, Vandenbroucke RE, Guenther AM, Sukhorukov GB, Hennink WE, Sanders NN, Demeester J, De Smedt SC. Adv Mater. 2006;18:1005. [Google Scholar]
- 54.Koennings S, Sapin A, Blunk T, Menei P, Goepferich A. J Controlled Release. 2007;119:163. doi: 10.1016/j.jconrel.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Hollis ER, Jamshidi P, Low K, Blesch A, Tuszynski MH. Proc Natl Acad Sci USA. 2009;106:7215. doi: 10.1073/pnas.0810624106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Izumrudov VA, Kharlampieva E, Sukhishvili SA. Biomacromolecules. 2005;6:1782. doi: 10.1021/bm050096v. [DOI] [PubMed] [Google Scholar]
- 57.Izumrudov VA, Lim SH. Polym Sci Ser A. 2002;44:484. [Google Scholar]
- 58.Brinker C, Scherer G. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing. Ist. Academic Press; San Diego CA USA: 1990. pp. 501–505. [Google Scholar]
- 59.Jiang XP, Clark SL, Hammond PT. Adv Mater. 2001;13:1669. [Google Scholar]
- 60.Gros T, Sakamoto J, Blesch A, Havton L, Tuszynski MH. doi: 10.1016/j.biomaterials.2010.04.035. unpublished. [DOI] [PubMed] [Google Scholar]
- 61.Kidambi S, Udpa N, Schroeder SA, Findlan R, Lee I, Chan C. Tissue Eng. 2007;13:2105. doi: 10.1089/ten.2006.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kidambi S, Chan C, Lee I. Langmuir. 2008;24:224. doi: 10.1021/la702925r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang MQ, Desai T, Ferrari M. Biomaterials. 1998;19:953. doi: 10.1016/s0142-9612(98)00026-x. [DOI] [PubMed] [Google Scholar]
- 64.Selsted ME, Martinez RJ. Anal Biochem. 1980;109:67. doi: 10.1016/0003-2697(80)90011-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.