

Abstract
ATP binding cassette (ABC) transporters constitute a ubiquitous superfamily of integral membrane proteins that are responsible for the ATP powered translocation of many substrates across membranes. The highly conserved ABC domains provide the nucleotide dependent engine that drives transport. By contrast, the transmembrane domains that create the translocation pathway are more variable. Recent structural advances with prokaryotic ABC transporters have provided a qualitative molecular framework for deciphering the transport cycle; an important goal is to develop quantitative models that detail the kinetic and molecular mechanisms by which ABC transporters couple the binding and hydrolysis of ATP to substrate translocation.
Introduction
Any object that enters or leaves a cell, whether nutrient, virus or waste product, must penetrate one or more enclosing membranes. The magnitude of this phenomenon may be estimated, for example, by the need of an actively growing Escherichia coli cell to take up ~106 glucose molecules per second1, 2 to support the requisite metabolic demands. Cells must not only be able to import preferred substrates, but they also often have the capability to utilize a wide variety of alternate nutrients when available. With a few exceptions (such as O2 and N2), the movement of small molecules, ions, and even some macromolecules across membranes is mediated by specialized membrane proteins known as transporters. To accommodate the diversity of molecules a cell may need to acquire from the environment, many different transporters are encoded in the genomes of organisms. In E. coli, for example, ~10% of the genome has been classified as participating in transport processes3 and overall, more than 550 different types of transporters have been identified4. The importance of transport activity may be appreciated from the non-trivial metabolic cost of pumping molecules across cell membranes, which can consume a significant fraction (estimated as ~10–60%, depending on conditions) of the ATP requirement of bacteria5, 6 and humans7.
One of the largest classes of transporters is the ABC (ATP binding cassette) transporter superfamily8–10. These transporters use the binding and hydrolysis of ATP to power the translocation of a diverse assortment of substrates, ranging from ions to macromolecules, across membranes. ABC transporters function as either importers, bringing nutrients and other molecules into cells, or as exporters that pump toxins, drugs and lipids across membranes (Box 1). Members of the ABC transporter family are present in organisms from all kingdoms of life; while exporters are found in both eukaryotes and prokaryotes, importers appear to be present exclusively in prokaryotic organisms. ABC transporters constitute the largest protein family in E. coli, including ~80 distinct systems representing 5% of the genome11, while ~50 ABC transporters are present in humans12. Members of the seven families of human ABC transporters13 participate in cholesterol and lipid transport, multidrug resistance, antigen presentation, mitochondrial iron homeostasis, and the ATP-dependent regulation of ion channels (the cystic fibrosis transmembrane conductance regulator and the sulfonyl urea receptors); mutations of these proteins have been associated with a range of disorders including cystic fibrosis, hypercholesterolemia and diabetes.
Box 1. Ins and Outs of ABC transporters.
Cellular survival requires the generation and maintenance of electrical and chemical concentration gradients across the generally impermeable cell membrane. ABC transporters are key participants in this process, and typically use the favorable chemical energy of ATP hydrolysis to translocate molecules across membranes in the thermodynamically unfavorable direction. A given ABC transporter may function as either an importer or an exporter, moving molecules in or out of cells, respectively, but no example is known of an ABC transporter that functions physiologically in both directions. ABC transporters that function as importers are found predominantly in prokaryotes, where they mediate the uptake of essential nutrients, such as amino acids, sugars, and essential metals. Substrates of ABC importers vary greatly in size and chemical nature, ranging from oligopeptides and oligosaccharides to small ions. ABC exporters are found in both prokaryotes and eukaryotes, and their substrates are typically lipophilic. In humans, ABC exporters are crucial participants in lipid, fatty acid and cholesterol export, malfunction of which underlies various diseases. Perhaps the most studied ABC transporter is the human Pgp that maintains cholesterol distribution across the leaflets of the plasma membrane73, 74. Unfortunately, Pgp also extrudes other lipophilic compounds, including chemotherapeutic agents, resulting in multidrug resistance of tumor cells.
Whether functioning as importers or exporters, ABC transporters likely share a high degree of mechanistic similarities. For both exporters and importers, an alternating access model for transport has been suggested17, 31, 48. The key feature of this model is the presence of a substrate binding site that can alternatively access either the extracellular side or the intracellular side of the membrane, corresponding to the “outward” and “inward” facing conformations of the transporter, respectively. ATP binding and hydrolysis drive the conformational changes that result in the alternating exposure of this binding site to the two sides of the membrane; the relative binding affinities for substrate of the two conformations will largely determine the net direction of transport. In particular, the outward facing conformation of an importer is expected to have a higher affinity for substrate than the inward facing conformation, while the opposite relationship will hold for exporters.
ABC transporters have a characteristic architecture, which consists minimally of four domains (Fig. 1a): two transmembrane domains (TMDs) embedded in the membrane bilayer and two ABCs (also designated as the nucleotide binding domains (NBDs)) located in the cytoplasm. At the sequence level, the superfamily of ABC transporters is identified by a characteristic set of highly conserved motifs present in the ABCs; in contrast, the sequences and architectures of the TMDs are quite variable, reflecting the chemical diversity of the translocated substrates. Beyond these four domains, additional elements may be found fused to the TMDs and/or ABCs of ABC transporters that likely serve regulatory functions14. For prokaryotic ABC transporters that function as importers, substrate translocation is also dependent on another protein component, a high affinity binding protein that specifically associates with the ligand in the periplasm for delivery to the appropriate ABC transporter15 (Fig. 1a). As originally recognized by Heppel16, these binding proteins are released upon osmotic shock and hence the associated transport systems, now recognized as ABC transporters, were initially identified as “shock-sensitive”.
Figure 1.
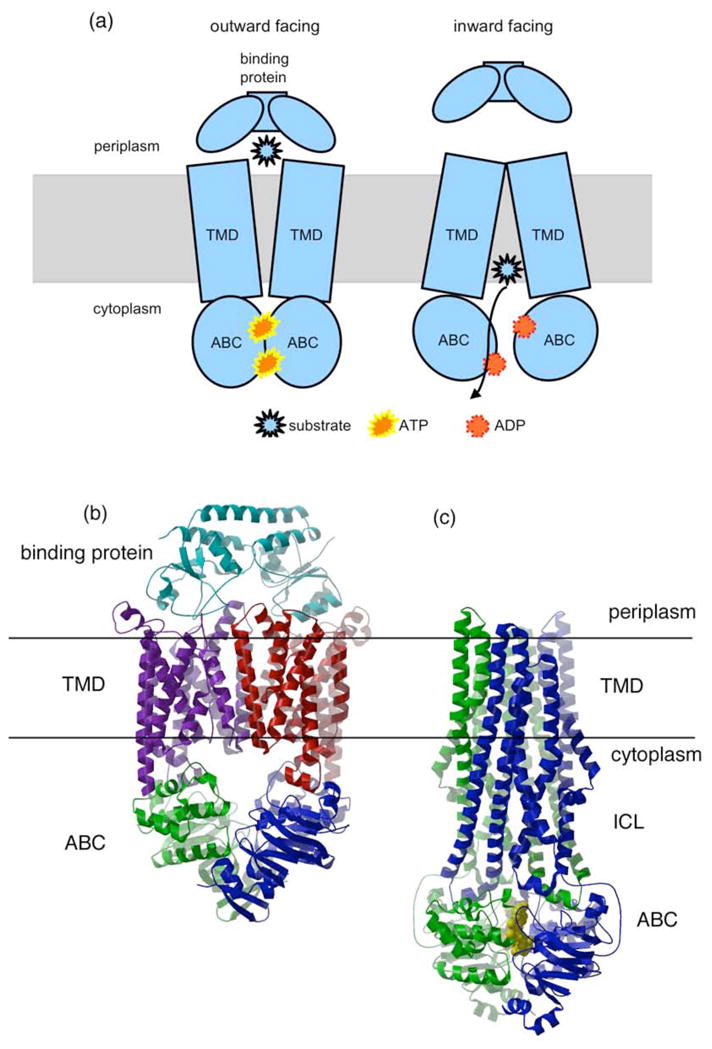
Molecular architecture of ABC transporters. (a) A cartoon representation of the modular organization of ABC transporters, composed of two transmembrane domains (TMD) and two ABC domains. The binding protein component required by importers is also illustrated. Two conformational states of the ABC transporter, outward facing and inward facing, with the substrate binding site oriented towards the periplasmic (extracellular) and cytoplasmic (intracellular) regions, respectively, are depicted to schematically illustrate the alternating access mechanism of transport (Box 1). (b) The E. coli BtuCDF importer (22; PDB 2QI9). The core transporter consists of four subunits, the two membrane spanning subunits, BtuC (purple and red) and the two ABC subunits, BtuD (green and blue). This complex also contains one copy of BtuF, the periplasmic binding protein (cyan). (c) The S. aureus Sav1866 multidrug exporter (18; PDB 2ONJ). Sav1866 consists of two subunits (green and blue), which contain a fused TMD and NBD. The bound nucleotides in this structure are represented by yellow space filling models. The periplasmic and cytoplasmic surfaces oriented towards the top and bottom of the figure, respectively. Molecular figures in this article were prepared with MOLSCRIPT and RASTER3D75, 76 using coordinates from the Protein Data Bank (PDB77).
Following the crystal structure determination of the E. coli vitamin B12 importer BtuCD in 200217, the structural biology of intact ABC transporters has exploded over the past 2 years18–26 (Table 1). Recent reviews highlighting these structural advances may be found in27–33. A brief discussion of developments in the crystallography of ABC transporters may be found in the Supplementary Information (Box S1). In this article, we build on this structural foundation and focus on comparative and mechanistic aspects of ABC transporters, particularly emphasizing developments involving prokaryotic members of this superfamily.
Table 1.
Structurally characterized ABC transporter systems, classified by TMD fold. The nomenclature of the fold classification is from33. Note that while the outward facing conformation generally corresponds to the ATP (or suitable analogue) bound state, exceptions are evident in the structures of ADP bound to Sav1866 and nucleotide free BtuCD.
| transporter | organism | nucleotide state | inward/outward | PDB ID | resolution | reference |
|---|---|---|---|---|---|---|
| Type I ABC importer fold | ||||||
| ModABC | A. fulgidus | apo | inward | 2ONK | 3.1 Å | 22 |
| MalFGK | E. coli | ATP | outward | 2R6G | 2.8 Å | 23 |
| ModAB | M. acetivorans | apo | inward | 3D31 | 3.0 Å | 25 |
| MetNI | E. coli | apo | inward | 3DHW | 3.7 Å | 26 |
| Type II ABC importer fold | ||||||
| BtuCD | E. coli | apo or (VO2)4 | outward | 1L7V | 3.2 Å | 17 |
| HI1470/1 | H. influenzae | apo | inward | 2NQ2 | 2.4 Å | 20 |
| BtuCDF | E. coli | apo | occluded | 2QI9 | 2.6 Å | 21 |
| ABC exporter fold | ||||||
| Sav1866 | S. aureus | ADP | outward | 2HYD | 3.0 Å | 18 |
| AMPPNP | outward | 2ONJ | 3.4 Å | 19 | ||
| MsbA | S. typhimurium | AMPPNP | outward | 3B60 | 3.7 Å | 24 |
| ADP- vanadate | outward | 3B5Z | 4.2 Å | |||
| E. coli | apo | inward | 3B5W | 5.3 Å | ||
| V. cholerae | apo | inward | 3B5X | 5.5 Å | ||
Structural organization
The overall architecture of ABC transporters can be illustrated by two structures that were solved in the group of Kaspar Locher: the BtuCD importer in complex with the associated binding protein BtuF (Fig 1b;21) and the multidrug exporter Sav1866 from Staphylococcus aureus (Fig 1c;18). The four domains of the BtuCD transporter consist of four individual polypeptide chains: two copies of the BtuC (membrane-spanning) and two copies of the BtuD (ABC) subunits. By contrast, each subunit of Sav1866 contains a TMD and an ABC fused together, so that the complete transporter is a dimer. In both cases, the transporter exhibits a molecular two-fold axis that relates the pairs of TMDs and the pairs of ABCs, respectively. The straddling of the TMDs around the two-fold axis creates the translocation pathway across the membrane, whereas the two ABCs are oriented such that the conserved sequence motifs (Fig 2a) are positioned at the dimer interface (Figs 2b, 2c). As discussed below, distinct quaternary structure arrangements have been observed that correspond to outward or inward facing conformational states (Box 1).
Figure 2.

Structure and dimer interactions of an ABC subunit. (a) A linear representation of the protein sequence of an ABC domain, illustrating the relative positions along the polypeptide chain of the conserved amino acid motifs. (b) Stereoview of the ABC subunit BtuD (17; PDB 1L7V). The P-loop, Walker B, Q-loop, H-motif and ABC signature motif, labeled as ‘P’, ‘B’, ‘Q’, ‘H’ and ‘ABC’, respectively, are positioned along one surface of the subunit. A cyclotetravandate bound to the P-loop is shown as a ball-and-stick model. (c) The nucleotide mediated ABC dimer from Sav1866 (18; PDB 2ONJ). The two NBDs are represented as ribbons (green and blue), while the sandwiched AMPPNP nucleotides are in yellow space filling models. The P-loops and the signature motifs are depicted in red and cyan space filling models, respectively. The coupling helices of the TMDs in contact with the ABCs are shown as purple ribbons. The view is down the molecular two-fold axis.
ABC subunits
ABC subunits are dominated by the NBD, which may be further divided into two constituent domains: a catalytic core domain that contains the conserved P-loop or Walker A motif (GXXGXGK(S/T)), Walker B motif (φφφφD, where φ is a hydrophobic residue), Q-loop and H-motif (switch region), together with a more structurally diverse α-helical domain34 that contains the ABC signature motif LSGGQ (Figs 2a, 2b). The relative orientation of the helical and catalytic domains is sensitive to the nucleotide state35. In an intact transporter, the ABC subunits pack together in a “head-to-tail” fashion such that the P loop of one subunit is oriented towards the signature motif of the other (Fig 2c). An important consequence, explored in more detail below, is that the binding site for nucleotide is positioned at the subunit-subunit interface, in a manner originally proposed from modeling studies36 and crystallographically characterized for the non-transporting ABC protein Rad5037. This arrangement was first observed structurally in an isolated ABC subunit by Hunt and coworkers38, and subsequently in the intact ABC transporters Sav186618 and MalFGK23. It is generally observed that the ATP bound state is associated with the most extensive interface between ABC domains, while the structures of nucleotide-free transporters exhibit conformations with greater separations between the ABCs (although the correlation between an extensive interface and the presence of bound ATP (or analogues) does have exceptions, such as the ADP bound structure of Sav186618).
Transmembrane domains
Although it is not infrequently commented that the TMDs of “typical” ABC transporters contain a total of 12 TM helices (6 per domain), in reality, the membrane spanning subunits are structurally heterogeneous, with three distinct sets of folds currently recognized. These folds have been designated33 as Type I ABC importer (Fig 3a), Type II ABC importer (Fig 3b), and ABC exporter (Fig 3c); the Type I and Type II importer nomenclature reflects the historical sequence in which members of the relevant families were characterized in sufficient detail to recognize they had distinct TMD folds.
Figure 3.

The polypeptide folds of a single transmembrane domain from representative ABC transporters. (a) The Type I ABC importer fold of MetI (PDB 3DHW), (b) the Type II ABC importer fold of BtuC (PDB 1L7V), (c) the ABC exporter fold of Sav1866 (PDB 2ONJ). At the top are the traces of the polypeptide fold for each structure, illustrated with a color gradient ranging from red at the N-terminus through yellow and green to blue at the C-terminus. The molecular two-fold axis (corresponding to the normal to the membrane plane) is vertical. At the bottom are ribbons representations of the TM helices in these structures, viewed from the periplasmic surface rotated 90° from the top view. The position of the molecular two-fold axis is depicted in red in the lower figures. In (a), all the TM helices (TM1-5) are represented for MetI; in (b) the BtuC helices are divided into two groups, TM1-5 and TM6-10; and in (c) the Sav1866 helices are divided into groups TM1-3 and TM4-6. Within each set, the helices are colored red to blue proceeding along the polypeptide chain from the N- to C-terminus to illustrate the internal symmetry present in BtuC and Sav1866. These internal repeats are related by a rotation axis oriented approximately vertically in the plane of the figure.
The Type I ABC importer fold was originally observed in the ModB TM subunit of the molybdate transporter22 and subsequently found in the maltose transporter MalF and MalG TM subunits23 and the methionine transporter MetI26. This fold is organized around a minimal set of 5 TM helices as observed in MetI (Fig 3a), with additional helices present in ModB, MalF and MalG, for a total of 6, 8 and 6 TM helices, respectively, in each subunit. Consequently, for the complete transporters, a total of 10, 12 and 14 TM helices are found in the methionine, molybdate and maltose transporters, respectively. Using the helix numbering of MetI, helices TM2-TM5 adopt a “up-down” topology lining the permeation pathway, while TM1 wraps around the outer (membrane facing) surface and contacts the other four helices. The additional helices in ModB, MalF and MalG primarily interact with the other subunit.
The Type II ABC importer fold, found in BtuC17 and a homologous transporter from Haemophilus influenzae, HI147120, contains 10 TM helices per subunit, or a total of 20 for the complete transporter (Fig. 3b). These helices pack together in a rather intricate topology that positions helix TM2 through the center of the subunit in proximity to most of the other helices. The N- and C-terminal halves of the BtuC subunit exhibit similar helix packing, particularly for the sets of helices TM2-TM5 and TM6-TM10, but with opposite polarities through the membrane. Helices TM5 and TM10 dominate the interface between TMDs.
The ABC exporter fold, originally established for the Sav1866 exporter18 and subsequently in the homologous MsbA24, contains 6 TM helices per subunit, or 12 for the complete transporter (Fig. 3c). In the outward facing conformation adopted by Sav1866, the transmembrane spanning region is organized into two “wings” composed of helices TM1-2 from one subunit, and TM3-6 of the other in an unanticipated domain-swapped arrangement. A notable aspect of this arrangement is that TM1-3 are related to TM4-6 by an approximate two-fold rotation about an axis in the membrane plane. A further point of distinction from the importers is the presence of long intracellular loops (ICLs) that extend the transmembrane helices ~25 Å beyond the membrane surface into the cytoplasm.
Although the detailed folds of the TMDs may vary, they share the common feature of interacting with the helical domains of the ABC domain through “coupling helices” located in the loops between TM helices17. For importers, the coupling helices contain the consensus “EAA” motif39, 40 that was recognized prior to the structural studies as contributing to the interface between the TMD and the NBD. Although the arrangements are distinct, structurally equivalent coupling helices are present in the loops between TM3-4 and TM6-7 of the Type I and Type II ABC importer folds, respectively. The equivalent region in exporters involves the ICL2 between helices TM3-4 that contacts the NBD of the other subunit. In all structures, the region of the NBD that interacts with the TMD primarily involves the Q-loop in the α-helical domain. The Q-loop is conformationally variable, with the conserved glutamine participating in nucleotide binding; changes in this region have been proposed to be involved in the coupling of nucleotide hydrolysis to the conformational states of the TMD41.
Are there additional classes of TMD folds in ABC transporters yet to be identified? The answer is almost certainly “yes”. One indication of this comes from a phylogenetic classification based on the protein sequences of ABC domains that delineated three basic classes of ABC transporters29, 42. Two of these classes contain membrane spanning domains with either fused ABC and TMDs (sequence class 1) and or with the TMDs and ABCs on independent polypeptide chains (sequence class 3). A comparison between this sequence derived classification and the structurally characterized families of ABC transporters reveals some interesting features relevant to the existence of structurally uncharacterized TMD folds.
Proteins present in the sequence class 1 of ABC transporters typically function as exporters, and as the sequence similarities are relatively high, it is likely that the membrane topology of all these proteins corresponds to the structure of the ABC exporter fold found in Sav186618. In contrast, the sequence divergence between the binding protein dependent importers found in the sequence class 3 is significantly greater. On the basis of the sequence comparisons of the ABC domains, 12 families of importers have been identified29, 42. Structurally characterized transporters with the Type I ABC importer fold (maltose, molybdate and methionine), correspond to the sequence derived OSP, MOI and DLM families of transporters (nomenclature of ref.29), respectively, and are present in one branch of the sequence derived phylogenetic tree. In contrast, transporters with the structurally characterized Type II ABC importer fold correspond to the sequence derived ISVH family, which occupies a separate branch of the sequence derived phylogenetic tree from proteins with the Type I ABC importer fold.
Intriguingly, a third branch of the sequence derived phylogenetic tree exists, distinct from these other two groups, which contains the HAA (hydrophobic amino acids, such as the leucine, isoleucine, valine system (Liv)) and MOS (monosaccharides, such as ribose (Rbs)) families of ABC transporters. As the HAA and MOS families are well separated from the branches containing transporters with either Type I or Type II importer folds, they represent potential candidates for a distinct TMD fold. One member of the HAA family, the RbsC protein, likely contains 10 TM helices and no obvious mapping of these helices on the 10 TM helices of BtuC was reported43 (it should be noted, however, that an “EAA” sequence is present in the loop between putative TM helices 6 and 7 of RbsC that corresponds to the location of this motif in BtuC17). In addition to the sequence comparisons of the ABC domains, there are also hints from the structures of the binding proteins that the HAA and MOS branch could exhibit a distinct TMD fold. The structures of the binding proteins for ABC transporters vary in the topological threading between domains44–46 and may be classified into three distinct categories (designated I, II and III, which unfortunately overlaps with the nomenclature used for the TMD folds). There is a tantalizing correlation that transport systems with a Type I ABC importer fold for the TMD have a binding protein Type I fold (ModA, MalE, MetQ), while transport systems with a Type II ABC importer fold have a binding protein Type III fold (BtuF). Suggestively, transport systems exhibiting the binding protein Type II fold (RbsB, LivJ and LivK) correspond to the HAA and MOS sequence families of ABC domains, indicative, perhaps, that they also exhibit a distinct structural architecture for the TMDs. The validity of this speculation will of course ultimately be established by the crystal structure determination of an intact transporter from one of these families.
Kinetic mechanism of ABC transporters
The similarities in ABC structure support a common mechanism by which ABC transporters, both importers and exporters, orchestrate a series of nucleotide- and substrate-dependent conformational changes that result in substrate translocation across the membrane27. The “alternating access” model47, 48, in which the substrate-binding site alternates between outward and inward facing conformations, provides a productive framework for this mechanistic analysis (Box 1). The key to successful operation of transporters moving molecules across membranes against a concentration gradient is the elimination of short-circuiting by preventing the uncoupled processes from occurring in their energetically favorable directions, that is leakage of accumulated substrate across the membrane or the futile cycling of ATP hydrolysis.
These general considerations may be illustrated through an idealized kinetic model for transport described in the Supplementary Information (Box S2). In this model, it is assumed that ATP and ADP are bound by the outward and inward facing conformations of the transporter, respectively, and that ATP hydrolysis drives the conversion from outward to inward facing states. The net rate of substrate translocation is given by the difference between the rates of import and export. To function as a pump that imports substrates across the membrane, the rate of the forward reaction (influx into the cell) needs to be maximized while the back reaction (efflux) is minimized. For importers, optimization of the substrate uptake rate, while minimizing ATP hydrolysis, can be kinetically achieved through a combination of effects involving (i) a higher affinity for substrate in the outward facing conformation than in the inward facing conformation, (ii) stimulation of ATPase activity when substrate is bound to the outward facing conformation of the transporter and (iii) a higher rate of nucleotide exchange of ATP for ADP in the unliganded state of the transporter, thereby resetting the transporter. For exporters, the opposite set of relationships would hold. The key to minimizing the futile cycling of nucleotide is to keep the rate of ATP hydrolysis minimal until the proper liganded state of the transporter is achieved, at which point ATP hydrolysis will drive substrate translocation.
ATP hydrolysis
A key mechanistic question for ABC transporters concerns the stoichiometry of ATP hydrolyzed per translocated substrate, and whether or not there is complete coupling of these processes under physiological conditions. While the mechanistically relevant stoichiometry for ABC transporters is likely 2 ATP per substrate transported49, this ratio has only been reliably observed in an in vitro system for the tightly coupled OpuA transporter50. More typically, the ATPase and transport rates differ by one to three orders of magnitude49, 51–53, and completely uncoupled ATPase activity can be observed in the absence of substrate49. While two ABC domains are always required for activity (discussed in more detail below), only one functional ATPase site can support transport activity in at least some systems, including the histidine permease54, although this is not a universal property of ABC transporters29.
The enzymatic hydrolysis of ATP requires the presence of two sets of properly positioned groups for (i) binding the phosphates and (ii) catalysing the attack of water on the γ-phosphate. For a large family of nucleotide-binding proteins, including ABC transporters, the conserved Walker A and Walker B motifs55 participate in the first capacity by binding the nucleotide phosphates and the Mg+2 coordinated to the nucleotide, respectively. These interactions are illustrated for Sav1866 in Fig 4a. The Mg+2 interaction involves the Asp in the Walker B motif, which is Asp502 in Sav1866. In addition to these binding interactions, two sets of residues are commonly found in the active site to catalyze the rate of ATP hydrolysis: one group serves as a general base that promotes the attacking water, while the other electrostatically stabilizes the β-phosphate56, 57. Candidates for the general base include Glu503 (adjacent to the Walker B motif), Gln422 from the Q-loop, and His534 in the H-motif region, which all cluster in the vicinity of the cleaved phosphate (Fig 4a). The residue serving as the (primary) catalytic residue remains ambiguous. While there is strong evidence in some systems that the Glu adjacent to the Walker B motif is the crucial catalytic residue38, 58, 59, this may not be universally true, as an important role in other systems for the H-motif His has been observed60.
Figure 4.

Nucleotide-protein interactions in the active sites of various ATPases. (a) AMPPNP bound to the ABC transporter Sav1866 (19; PDB 2ONJ), (b) the ADP-AlF4 site of the F1-ATPase (61, PDB 1H8E), (c) the ADP-AlF4 state of the nitrogenase Fe-protein (78, PDB 1M34). The nucleotide is depicted with yellow bonds, while the Mg+2 (or equivalent) is cyan and the AlF4 (when present) has purple atoms. The P-loop is represented by the green Cα trace at the back of each structure, while the Walker B Asp from the same subunit is shown with red bonds on the left. In Sav1866 (a), potential catalytic residues are Glu503 (adjacent to the Walker B), Gln422 in the Q-loop and His534 of the H-motif. The backbone atoms of the LSGGQ signature motif provided from the dimer-related ABC subunit are depicted with blue bonds. For the F1-ATPase (b), the catalytic residue Gluβ188 occupies the same spatial location as the Q-loop, while Argα373 from the adjacent subunit coincides with the signature motif. For nitrogenase (c), Asp39 in the Switch I region, and Gly128 correspond to the Q-loop and H-motif region, respectively; the catalytic residue Asp129 from the adjacent subunit has no obvious counterpart in the ABC transporters. Lys10 from the adjacent subunit is positioned similarly to the signature motif in the ABC transporters.
An interesting comparison may be made with other ATPases, including the F1-ATPase (Fig 4b;61) and the nitrogenase Fe-protein (Fig 4c;62) that are central participants in the processes of ATP synthesis and biological nitrogen fixation, respectively. The F1-ATPase shares structural similarity to the fold of the catalytic domain of the ABC transporters, while the Fe-protein is in a distinct, although related, branch of nucleotide-binding domains that includes G-proteins and ras p2163, 64. The interaction of Mg+2 ATP with the Walker A and B motifs is similar among ABC transporters, F1-ATPase and nitrogenase Fe-protein (although the conformation of the ATP varies between these structures). The catalytic residue, Gluβ188, of the F1-ATPase is spatially analogous to the Q-loop Gln, while there are no counterparts to Glu503 or His534 in this protein. Gly128 of nitrogenase, which has been implicated in stabilizing the γ-phosphate, superimposes with the H-motif His534, while Asp39 in the Switch I region superimposes with Glu503. No counterpart to the residue likely promoting the attack of water in nitrogenase, Asp129 of the other subunit in the Fe-protein dimer, is present in Sav1866. Thus, in these other characterized ATPases, no consistent pattern of catalytic relevance may be assigned to potential residues in the ABC transporter, suggesting that there is not a unique solution to the organization of the active site residues in Walker A- and Walker B-containing enzymes.
The conformation of ABC transporters that is catalytically competent for nucleotide hydrolysis involves ATP bound at the interface between two ABCs, with the terminal phosphates of the nucleotide bound between the P-loop on one ABC and the LSGGQ signature motif of the other (illustrated for Sav1866 with AMPPNP in Fig 2b and Fig 4a). Consequently, ATP binding and hydrolysis involves the participation of both ABCs. This arrangement explains why two ABCs are required, since individual subunits are unable to productively bind and hydrolyze ATP. Again, comparisons of the active site arrangement for ATP hydrolysis in other ATPases are quite informative. The residues that correspond to the LSGGQ signature motif in the F1-ATPase and nitrogenase ATPase active sites (the positively charged residues Argα373 and Lys 10′, respectively) are provided by neighboring subunits (Figs 4a, 4b, 4c). This bipartite organization of the active site, in which essential catalytic groups are positioned on different subunits, is found in such diverse systems as G-proteins65, AAA+ ATPases64 and helicases66 and has the important implication that the rate of nucleotide hydrolysis can be controlled by the process of assembling the active site67, 68.
Nucleotide-dependent changes in transporter NBDs
The key to the functioning of ABC transporters is that the positional relationships between the two ABC domains depends on the nucleotide state through the types of interactions described in the previous section. As the transporter cycles through the stages of ATP binding, hydrolysis, and product dissociation, the interface between the two ABCs switches from a closed interface, with the bound ATP sandwiched between the conserved sequence motifs of different domains, to a more open interface characteristic of the non-ATP states; these nucleotide dependent conformational transitions ultimately drive the conformational changes in the TMDs resulting in substrate translocation. The magnitude of the variability in the relative positions of ABC domains may be illustrated through a comparison of the ABCs from different transporters (Fig 5). Some considerations relevant to the superposition and comparison of structures are discussed in Supplementary information Box S3. It should also be emphasized that the assumption is made in this type of comparison that the observed distribution of structures reflects conformations mechanistically relevant to the transport cycle, rather than the idiosyncrasies of the different transporters; while common themes are observed, one must keep this caveat in mind when considering specific details. Upon superposition of one ABC domain from each of a set of different transporters, substantial variation is evident in the positioning of the second ABC domain. A measure of the separation between ABC domains may be provided by the distance between the P-loop and LSGGQ signature motifs on different subunits (defined here as the distance between the Cα positions corresponding to BtuD residues Gly38 and Gly129, respectively). The intersubunit separations between the two conserved motifs range from ~11 Å in the ATP-bound form of Sav1866, to 14 Å, 16 Å and ~28 Å in the nucleotide free forms of BtuD, HI1470 and MetN, respectively 26. A more detailed analysis indicates relationships between the non-superimposed subunits in the different structures may be approximately described in terms of two basic operations: (i) a “hinge” or tweezer type rotation that juxtaposes the ABC domains by closing up the interface69 and (ii) a “twist” type rotation corresponds to a sliding or translational shift of ABC domains along the interface20.
Figure 5.

Relationships between dimeric ABC structures. The polypeptide fold of BtuD (PDB 1L7V) is shown with the two subunits traced in different shades of blue. The positions of the P-loop and ABC signature motifs (defined by the Cα positions of Gly38 and Gly129, respectively) in BtuD are denoted by the smaller and larger blue spheres, respectively. Following the superposition onto the right-most BtuD (subunit 1) of one ABC domain of Sav1866 (red, 2ONJ), MalK (yellow, 1Q12), HI1470 (cyan; 2NQ2); and MetN (green, 3DHW), the positions of the P-loop and ABC signature motifs in these structures are designated by the appropriately colored spheres. The bound AMPPNPs at the ABC dimer interface of Sav1866 (PDB 2ONJ) are shown in purple. The closed interface characteristic of the ATP bound state, as observed in Sav1866 and MalK, is associated with an intersubunit separation between the P-loop and ABC signature motifs of ~11 Å; in the nucleotide-free structures, the separation increases from 14 Å in BtuD to 16 Å in HI1470 to ~28 Å in MetN. The large separation between ABC subunits in MetN likely underlies the phenomenon of transinhibition, in which methionine binding to a regulatory domain of MetI stabilizes an ATPase inactive form by sterically preventing the formation of the catalytically essential closed interface between these domains.
Interconversion of inward and outward facing conformations
The interconversion between the closed ABC interface in the ATP state and the more open interfaces in non-ATP states drives the switching of the translocation pathway between the outward (ATP) and inward facing (ADP or nucleotide free) conformations, respectively52 (although it should be notied that exceptions to this generalization are evident in the available crystal structures (Table 1), particularly the outward facing conformations of Sav1866 and BtuCD in the ADP and nuclelotide-free states, respectively). Specifically, these nucleotide dependent transitions may be coupled to the TMDs through the interaction between the helical domains and the coupling helices of the ABC domains and TMDs, respectively (Fig 2c), thus connecting the conformation of the permeation pathway with the nucleotide state of the transporter. For the three sets of TMD folds in transporters of currently defined structure (Table 1), an ensemble of structural states are available that can be used to identify the nature of these conformational transitions in the TMDs, at least in broad outline.
For Type I ABC importers, structures are available that represent a progression of conformations of the translocation pathway ranging from the occluded/outward (maltose) to inward (molybdate) to wide-open, inward facing (methionine). Superimposing the structurally conserved TM2-TM4 helices (MetI labeling) onto the MalF subunit of the maltose transporter, the transformations required to superimpose the appropriate helices of MalG with the corresponding regions of ModB and MetI correspond to rigid body rotations of ~20° and 32°, respectively26. This is equivalent to a hinge-type motion, where the rotation axis is approximately perpendicular to the symmetry axis of the transporter and passes near a conserved proline (Pro 67 in MetI) at a kink in TM2 near the periplasmic surface.
Structures that represent outward (BtuCD), occluded (BtuCDF), and inward (HI1470/1) facing states of homologous Type II ABC importers are also available that exhibit differences in their tertiary and quaternary arrangements of subunits20, 22. At the tertiary structure level, the main difference between the outward and occluded structures are the local rearrangements in the three helices, TM3-5, of one subunit that are not part of the rigid core. Of these, the most significant difference is a 20° shift in the helix axis of TM5, because this helix is part of the dimer interface between BtuC subunits. The conversion to the inward facing conformation involves a twist of ~9° about an axis normal to the molecular two-fold axis. As a result of the repositioning of helices TM3-5 and the overall twist, the translocation pathway shifts from outward to inward facing.
For ABC exporters, Sav1866 and MsbA structures are available that adopt outward-facing conformations corresponding to the ATP-bound state. Two lower resolution structures of MsbA homologues have also been solved that adopt inward facing conformations described as closed and open, the latter with the NBDs widely separated. A comparison of the MsbA structures indicates that the transition from the open to closed inward conformation corresponds to a ~30° hinge axis rotation; formation of the tight ABC interface involve a distinct twist of TM4-5 that displaces the ABC domains along the subunit-subunit interface until the closed ATP state is achieved. During these rearrangements, the packing of the TM helices changes such that while TM1-2-3-6 and TM4-5 from different subunits are together in the inward state, TM1-2 and TM3-6 from different subunits are juxtaposed in the outward state24.
Regulation of ABC transporter activity
The activity of ABC transporters may be regulated at the level of protein function through the action of domains fused to the NBDs and/or TMDs14; it should also be considered that non-covalently associating subunits could also be involved. Trans-inhibition is one example of such a regulatory process, whereby the uptake of an external ligand is inhibited by intracellular concentrations of the same ligand, as observed for the uptake of methionine by Kadner70. Recent crystallographic analyses of the ModBC25 and MetNI26 systems have established the structural basis for this effect as involving ligand binding to the C-terminal domain of the ABC subunits. Although the domains in the two systems are distinct, they have each been found to participate in the binding of allosteric ligands to other proteins, and hence appear to have been recruited by ABC transporters for these properties. In each case, the consequences of ligand binding to the regulatory domain are quite similar in that the liganded C-terminal domains sterically separate the ABCs and prevent their association. Because the ABC subunits must associate to form the ATP bound state, keeping them separated will reduce the rate of ATP binding, thereby inhibiting the overall transport cycle.
Intriguingly, a large family of soluble ABC proteins containing two NBDs has been found to participate in antibiotic resistance by certain pathogens, and of particular note, the C terminus of the S. aureus Vga(A) protein has been implicated in regulating the response to antibiotics71. This observation suggests a mechanism similar to that underlying the phenomenon of trans-inhibition by ABC transporters may be relevant to this system.
Future Directions
Our current understanding of ABC transporters may be likened to that of a play where we know the names of essentially all the actors, what some of them do, what some of them look like, and we also have a synopsis of the plot. Missing, however, is the detailed script (or scripts, if there is more than one basic plot) that describes what all the actors are doing, and how and why they are doing it. Reconstruction of this script will require progress in many directions.
Given the prevalence of ABC transporters throughout life, surprisingly few systems have been characterized, whether biochemically, structurally or mechanistically. As a consequence, broad generalizations about the functioning of these systems are extrapolated from relatively few observations, and so a pressing need exists to extend these characterizations to other transporters. Furthermore, although the basic features of the nucleotide binding sites are understood, less is known concerning the binding modes of the translocated substrate and how this is coupled to the nucleotide state. For importers, the binding protein plays a crucial role; one or more binding sites may also be present in the translocation pathway as observed in the MalFGK structure23. For exporters, particularly multidrug resistance proteins, the details of the substrate binding site have yet to be crystallographically identified18, 24.
The high resolution structural characterization of eukaryotic ABC transporters, particularly human ones, remains a fertile area for research, not only for the intrinsic mechanistic interest, but also for the therapeutic potential evolving from characterization of substrate binding, nucleotide and regulatory sites in these systems. Furthermore, ABC subunits are not intrinsically specialized to function exclusively in transport systems, but are also integrated into a range of other processes including DNA repair and chromosome maintenance72. It has been proposed in these systems that ATP binding and hydrolysis drives the reversible association and dissociation of these subunits to act as rings or snaps to tether together substrates such as DNA or chromosomes. These mechanistic similarities with transporters are important areas to explore.
Mechanistic studies of the translocation reaction of ATP transporters are complicated by the challenges of working with membranes and membrane proteins, particularly when addressing such fundamental questions as the stoichiometry of ATP hydrolyzed to substrate translocated, the timing between ATP hydrolysis and translocation, and how substrate binding is coupled to ATP hydrolysis. This is an area where single molecule methods have great potential to follow the behaviour of individual transporters without the effects of ensemble averaging.
The goal of these efforts should be to understand in quantitative detail the molecular and kinetic mechanism of ABC transporter function and ABC proteins more generally. Not only will such insights be informative for the development of therapeutically useful agents, but they will also help us appreciate how one basic engine can generate the power to change the conformations of a variety of distinct protein systems to achieve the movement of molecules in, out and around cells.
Supplementary Material
Acknowledgments
We would like to thank A.T. Lee and J. B. Howard for discussions and their long term contributions to our research in this area. The support of the Fulbright Foundation and Jane Coffin Childs Memorial Funds for Medical Research (O.L.) and NIH grant GM45162 (D.C.R) is gratefully acknowledged.
glossary/abbreviations
- AAA+
ATPases associated with diverse cellular activities: a large family of structurally conserved ATPases where assembly of the ATPase active site requires subunit oligomerization
- ABC
ATP binding cassette, a set of conserved sequence motifs defining an eponymous family of ATPases
- BtuCD
the vitamin B-twelve uptake system composed of the integral membrane protein and ABC subunits BtuC and BtuD, respectively
- F1-ATPase
a stable, water-soluble subassembly of the membrane bound ATP synthase with the active site for ATP synthesis and hydrolysis located at the interface between homologous α and β subunits
- G-protein
a family of GTP binding proteins involved in signal transduction, where GTP binding and hydrolysis and the subsequent nucleotide exchange drives a series of conformational changes used in signaling cascades
- MalFGK
the maltose transporter composed of two homologous integral membrane subunits MalF and MalG, and the ABC subunit MalK
- MetNI
the methionine transporter composed of the integral membrane protein and ABC subunits MetI and MetN, respectively
- NBD
nucleotide binding domain, often used interchangeably with ABC in ABC transporters
- nitrogenase
the enzyme system catalyzing the ATP dependent reduction of dinitrogen to ammonia during the process of biological nitrogen fixation; the nitrogenase iron (Fe-) protein contains the binding site for ATP
- Sav1866
a bacterial ABC exporter from Staphylococcus aureus homologous to eukaryotic multidrug resistance transporters
- TM(D)
transmembrane (domain)
Footnotes
Competing interests statement
The authors declare no competing financial interests
References
- 1.Monod J. Recherches sur la croissance des culture bactériennes. Hermann, Editeurs des Sciences et des Arts; Paris: 1942. [Google Scholar]
- 2.Phillips R, Quake SR. The biological frontier of physics. Physics Today. 2006;59:38–43. [Google Scholar]
- 3.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 4.Busch W, Saier MHJ. The transporter classification (TC) system. Crit Rev Biochem Mol Biol. 2002;27:287–337. doi: 10.1080/10409230290771528. [DOI] [PubMed] [Google Scholar]
- 5.Stouthamer AH. The search for correlation between theoretical and experimental growth yields. Int Rev Biochem. 1979;21:1–47. [Google Scholar]
- 6.Neijsel OM, Teixeira de Mattos MJ, Tempest DW. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt FC, editor. ASM Press; Washington, D.C: 1996. pp. 1283–1692. [Google Scholar]
- 7.Skou JC. The identification of the sodium-potassium pump (Nobel Lecture) Angew Chem Intl Ed. 1998;37:2321–2328. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2320::AID-ANIE2320>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 9.Ames GF, Mimura CS, Holbrook SR, Shyamala V. Traffic ATPases: a superfamily of transport proteins operating from Escherichia coli to humans. Adv Enzymol Relat Areas Mol Biol. 1992;65:1–47. doi: 10.1002/9780470123119.ch1. [DOI] [PubMed] [Google Scholar]
- 10.Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. ABC proteins: From Bacteria to Man. Academic Press; London: 2003. [Google Scholar]
- 11.Linton KJ, Higgins CF. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13. doi: 10.1046/j.1365-2958.1998.00764.x. [DOI] [PubMed] [Google Scholar]
- 12.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 13.Dean M, Annilo T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu Rev Genomics Hum Genet. 2005;6:123–142. doi: 10.1146/annurev.genom.6.080604.162122. [DOI] [PubMed] [Google Scholar]
- 14.Biemans-Oldehinkel E, Deoven MK, Poolman B. ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 2006;580:1023–1035. doi: 10.1016/j.febslet.2005.11.079. [DOI] [PubMed] [Google Scholar]
- 15.Ames GFL. Bacterial periplasmic transport systems - structure, mechanism and evolution. Annu Rev Biochem. 1986;55:397–425. doi: 10.1146/annurev.bi.55.070186.002145. [DOI] [PubMed] [Google Scholar]
- 16.Heppel LA. The effect of osmotic shock on release of bacterial proteins and on active transport. J Gen Physiol. 1969;54:95–113. doi: 10.1085/jgp.54.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–8. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 18.Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 19.Dawson RJP, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 2007;581:935–938. doi: 10.1016/j.febslet.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 20.Pinkett HW, Lee AT, Lum P, Locher KP, Rees DC. An inward-facing conformation of a putative metal-chelate type ABC transporter. Science. 2007;315:373–377. doi: 10.1126/science.1133488. [DOI] [PubMed] [Google Scholar]
- 21.Hvorup RN, et al. Asymmetry in the structure of the ABC transporter binding protein complex BtuCD-BtuF. Science. 2007;317:1387–1390. doi: 10.1126/science.1145950. [DOI] [PubMed] [Google Scholar]
- 22.Hollenstein K, Frei DC, Locher KP. Structure of an ABC transporter in complex with its binding protein. Nature. 2007;446:213–216. doi: 10.1038/nature05626. [DOI] [PubMed] [Google Scholar]
- 23.Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–522. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 24.Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci USA. 2007;104:19005–19010. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerber S, Comellas-Bigler M, Goetz BA, Locher KP. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science. 2008;321:246–250. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]
- 26.Kadaba NS, Kaiser JT, Johnson E, Lee A, Rees DC. The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation. Science. 2008;321:250–253. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollenstein K, Dawson RJP, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Linton KJ. Structure and function of ABC transporters. Physiology. 2007;22:122–130. doi: 10.1152/physiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- 29.Davidson AL, Dassa E, Orelle C, Chen J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev. 2008;72:317–364. doi: 10.1128/MMBR.00031-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davidson AL, Maloney PC. ABC transporters: how small molecules do a big job. Trends in Microbiol. 2007;15:448–455. doi: 10.1016/j.tim.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Dawson RJP, Hollenstein K, Locher KP. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol Micro. 2007;65:250–257. doi: 10.1111/j.1365-2958.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 32.Oldham ML, Davidson AL, Chen J. Structural insights into ABC transporter mechanism. Curr Opin Struct Biol. 2008 doi: 10.1016/j.sbi.2008.09.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locher KP. Structure and mechanism of ATP-binding cassette transporters. Phil Trans R Soc B. 2008 doi: 10.1098/rstb.2008.0125. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitt L, Benabdelhak H, Blight MA, Holland IB, Stubbs MT. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J Mol Biol. 2003;330:333–342. doi: 10.1016/s0022-2836(03)00592-8. [DOI] [PubMed] [Google Scholar]
- 35.Karpowich N, et al. Crystal structures of the MJ1267 ATP binding cassette reveal an induced-fit effect at the ATPase active site of an ABC transporter. Structure. 2001;9:571–86. doi: 10.1016/s0969-2126(01)00617-7. [DOI] [PubMed] [Google Scholar]
- 36.Jones PM, George AM. Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol Lett. 1999;179:187–202. doi: 10.1111/j.1574-6968.1999.tb08727.x. [DOI] [PubMed] [Google Scholar]
- 37.Hopfner KP, et al. Structural biology of rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- 38.Smith PC, et al. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell. 2002;10:139–149. doi: 10.1016/s1097-2765(02)00576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saurin W, Koster W, Dassa E. Bacterial binding protein-dependent permeases - characterization of distinctive signatures for functionally related integral cytoplasmic membrane proteins. Mol Micro. 1994;12:993–1004. doi: 10.1111/j.1365-2958.1994.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 40.Mourez M, Hofnung N, Dassa E. Subunit interactions in ABC transporters: A conserved sequence in hydrophobic membrane proteins of periplasmic permeases defines an important site of interaction with the ATPase subunits. EMBO J. 1997;16:3066–3077. doi: 10.1093/emboj/16.11.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones PM, George AM. Mechanism of ABC transporters: A molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc Natl Acad Sci USA. 2002;99:12639–12644. doi: 10.1073/pnas.152439599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dassa E, Bouige E. The ABC of ABCs: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol. 2001;152:211–229. doi: 10.1016/s0923-2508(01)01194-9. [DOI] [PubMed] [Google Scholar]
- 43.Steward JB, Hermodson MA. Topology of RbsC, the membrane component of the Escherichia coli ribose transporter. J Bact. 2003;185:5234–5239. doi: 10.1128/JB.185.17.5234-5239.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukami-Kobayashi K, Tateno Y, Nishikawa K. Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J Mol Biol. 1999;286:279–290. doi: 10.1006/jmbi.1998.2454. [DOI] [PubMed] [Google Scholar]
- 45.Quiocho FA, Ledvina P. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variations of common themes. Mol Microbiol. 1996;20:17–25. doi: 10.1111/j.1365-2958.1996.tb02484.x. [DOI] [PubMed] [Google Scholar]
- 46.Wilkinson AJ, Verschueren KHG. In: ABC Proteins: From Bacteria to Man. Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. Academic Press; London: 2003. p. 647. [Google Scholar]
- 47.Widdas WF. Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J Physiology. 1952;118:23–39. doi: 10.1113/jphysiol.1952.sp004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jardetsky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- 49.Poolman B, et al. Functional analysis of detergent solubilized and membrane-reconstituted ABC transporters. Methods Enzymol. 2005;400:429–459. doi: 10.1016/S0076-6879(05)00025-X. [DOI] [PubMed] [Google Scholar]
- 50.Patzlaff JS, van der Heide T, Poolman B. The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J Biol Chem. 2003;278:29546–29551. doi: 10.1074/jbc.M304796200. [DOI] [PubMed] [Google Scholar]
- 51.Davidson AL, Nikaido H. Overproduction, solubilization, and reconstitution of the maltose transport system from Escherichia coli. J Biol Chem. 1990;265:4254–4260. [PubMed] [Google Scholar]
- 52.Chen J, Sharma S, Quiocho FA, Davidson AL. Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc Natl Acad Sci U S A. 2001;98:1525–30. doi: 10.1073/pnas.041542498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borths EL, Poolman B, Hvorup RN, Locher KP, Rees DC. In vitro functional characterization of BtuCD-F, the Escherichia coli ABC transporters for vitamin B12 uptake. Biochemistry. 2005;44:16301–16309. doi: 10.1021/bi0513103. [DOI] [PubMed] [Google Scholar]
- 54.Nikaido K, Ames GFL. One intact ATP-binding subunit is sufficient to support ATP hdryolysis and translocation in an ABC transporter, the histidine permease. J Biol Chem. 1999;274:26727–26735. doi: 10.1074/jbc.274.38.26727. [DOI] [PubMed] [Google Scholar]
- 55.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. Embo J. 1982;1:945–51. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maegley KA, Admiraal SJ, Herschlag D. Ras-catalyzed hydrolysis of GTP: a new perspective from model studies. Proc Natl Acad Sci USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Matte A, Tari LW, Delbaere LTJ. How do kinases transfer phosphoryl groups. Structure. 1998;6:413–419. doi: 10.1016/s0969-2126(98)00043-4. [DOI] [PubMed] [Google Scholar]
- 58.Geourjon C, et al. A common mechanism for ATP hdryolysis in ABC transporter and hlicase superfamilies. Trends in Biochem Sci. 2001;25:539–544. doi: 10.1016/s0968-0004(01)01907-7. [DOI] [PubMed] [Google Scholar]
- 59.Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ. Cooperative, ATP-dependent association of the nucleotide binding casettes during the catalytic cycle of ATP-binding cassette transporters. J Biol Chem. 2002;277:21111–21114. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zaitseva J, et al. H662 is the linchpin of ATP hydrolysis in the nucleotide-bidning domain of the ABC transporter HlyB. EMBO J. 2005;24:1901–1910. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Menz RI, Walker JE, Leslie AGW. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell. 2001;106:331–341. doi: 10.1016/s0092-8674(01)00452-4. [DOI] [PubMed] [Google Scholar]
- 62.Schindelin H, Kisker C, Schlessman JL, Howard JB, Rees DC. Structure of ADP-AlF4− stabilized nitrogenase complex and its implications for signal transduction. Nature. 1997;387:370–376. doi: 10.1038/387370a0. [DOI] [PubMed] [Google Scholar]
- 63.Leipe DD, Koonin EV, Aravind L. Evolution and classification of P-loop kinases and related proteins. J Mol Biol. 2003;333:781–815. doi: 10.1016/j.jmb.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 64.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 65.Scheffzek K, et al. The Ras-RasGAP complex: Structural basis for GTPase activiation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 66.Enemark EJ, Joshua-Tor L. On helicases and other motor proteins. Curr Opin Struct Biol. 2008;18:243–257. doi: 10.1016/j.sbi.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wittinghofer A. Signaling mechanistics: Aluminum fluoride for molecule of the year. Curr Biol. 1997;7:R682–R685. doi: 10.1016/s0960-9822(06)00355-1. [DOI] [PubMed] [Google Scholar]
- 68.Rees DC, Howard JB. Structural bioenergetics and energy transduction mechanisms. J Mol Biol. 1999;293:343–350. doi: 10.1006/jmbi.1999.3005. [DOI] [PubMed] [Google Scholar]
- 69.Chen J, Lu G, Lin J, Davidson AL, Quiocho FA. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol Cell. 2003;12:651–61. doi: 10.1016/j.molcel.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Kadner RJ. Regulation of methionine transport activity in Escherichia coli. J Bact. 1975;122:110–119. doi: 10.1128/jb.122.1.110-119.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacquet E, et al. ATP hydrolysis and pristinamycin IIA inhibition of the Staphylococcus aureus Vga(A), a dual ABC protein involved in streptogramin A resistance. J Biol Chem. 2008;283:25332–25339. doi: 10.1074/jbc.M800418200. [DOI] [PubMed] [Google Scholar]
- 72.Hopfner KP, Tainer JA. Rad50/SMC proteins and ABC transporters: unifying concepts from high-resolution structures. Curr Opin Struct Biol. 2003;13:249–255. doi: 10.1016/s0959-440x(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 73.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 74.Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 75.Kraulis PJ. MOLSCRIPT - a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 76.Merritt EA, Murphy MEP. Raster3D Version 2.0 - a program for photorealistic molecular graphics. Acta Crystallogr. 1994;D50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 77.Berman HM, et al. The Protein Data Bank. Nuc Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmid B, et al. Biochemical and structural characterization of the crosslinked complex of nitrogenase: comparison to the ADP-AlF4− stabilized structure. Biochemistry. 2002;41:15557–15565. doi: 10.1021/bi026642b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.