

Abstract
Human chromosome 15q11-13 and the syntenic region of mouse chromosome 7 contain multiple imprinted genes necessary for proper neurodevelopment. Due to imprinting, paternal 15q11-13 deficiencies lead to Prader-Willi syndrome (PWS) while maternal 15q11-13 deficiencies cause Angelman syndrome (AS). The mechanisms involved in parental imprinting of this locus are conserved between human and mouse, yet inconsistencies exist in reports of imprinting of the maternally expressed gene Atp10a/ATP10A. Excess maternal 15q11-13 dosage often leads to autism-spectrum disorder therefore further investigation to characterize the true imprinting status of ATP10A in humans was warranted. In this study, we examined allelic expression of ATP10A transcript in 16 control brain samples, and found that 10/16 exhibited biallelic expression while only 6/16 showed monoallelic expression. Contrary to the expectation for a maternally expressed imprinted gene, quantitative RT-PCR revealed significantly reduced ATP10A transcript in Prader-Willi syndrome brains with two maternal chromosomes due to uniparental disomy (PWS UPD). Furthermore, a PWS UPD brain sample with monoallelic ATP10A expression demonstrated that monoallelic expression can be independent of imprinting. Investigation of factors that may influence allelic ATP10A expression status revealed that gender has a major affect, as females were significantly more likely to have monoallelic ATP10A expression than males. Regulatory sequences were also examined, and a promoter polymorphism that disrupts binding of the transcription factor Sp1 also potentially contributes to allelic expression differences in females. Our results show that monoallelic expression of human ATP10A is variable in the population and is influenced by both gender and common genetic variation.
Introduction
Human chromosome 15q11-13 contains a cluster of imprinted genes implicated in multiple neurodevelopmental disorders. Due to the parent-of-origin specific gene expression of this locus, paternal deficiencies of 15q11-13 cause Prader-Willi syndrome (PWS), while maternal deficiencies cause Angelman syndrome (AS) (Knoll et al. 1989). PWS is characterized by hypotonia at birth followed by hyperphagia induced obesity later in life, and mild cognitive impairments with obsessive compulsive behaviors (Bittel and Butler 2005). Contrastingly, AS is characterized by severe neurologic impairments including profound mental retardation, lack of speech, seizures, ataxia, and unprovoked laughter (Cassidy et al. 2000). Duplications of 15q11-13 often lead to the diagnosis of autism-spectrum disorder (ASD), and are also subject to effects of imprinting, as maternally derived 15q11-13 duplications specifically cause the ASD phenotype (Cook et al. 1997). Rett syndrome (RTT), an autism-spectrum disorder caused by mutations in MECP2 (Methyl CpG binding protein 2) (Amir et al. 1999) also has been linked to 15q11-13, as MeCP2 binds throughout the locus (Yasui et al. 2007) and expression of some 15q11-13 genes is altered in RTT brain (Hogart et al. 2007; Samaco et al. 2005).
The cluster of imprinted genes present on human chromosome 15q11-13 and the syntenic region of mouse chromosome 7C have conserved genomic structure and regulation (Nicholls and Knepper 2001). Rare microdeletions mapped in patients with PWS and AS revealed that imprinting throughout the locus is regulated by a bipartite imprinting center consisting of a 4 kb region in the 5′ end of SNRPN (PWS imprinting control region) and an 880 bp region (AS imprinting control region) 35 kb upstream of SNRPN (Nicholls and Knepper 2001). Differential DNA methylation is associated with maintenance of the maternal imprint in paternally expressed genes. While imprinting and differential methylation of the paternally expressed genes appears to be ubiquitous, imprinting of the maternally expressed gene UBE3A is restricted to brain in both human and mouse (Albrecht et al. 1997; Rougeulle et al. 1997; Vu and Hoffman 1997). The mechanism involved in paternal UBE3A silencing is enigmatic, however, some evidence has pointed to a role for a neuron-specific UBE3A antisense transcript that is regulated by promoter sequences at the imprinting center (Landers et al. 2004; Runte et al. 2004; Yamasaki et al. 2003).
In addition to UBE3A, a second maternally expressed imprinted gene, ATP10A, has been identified within 15q11-13. While one report found that ATP10A exhibits imprinted expression similar to UBE3A, with biallelic expression in non-neuronal tissues (Herzing et al. 2001b), another study reported imprinted expression of ATP10A in lymphoblasts as well as brain (Meguro et al. 2001). Imprinting status of the mouse ortholog of ATP10A has been disputed—with one report that Atp10a is paternally imprinted in hippocampus and olfactory bulb (Kashiwagi et al. 2003), and a different study finding that Atp10a is biallelically expressed in all tissues (Kayashima et al. 2003). Given the conflicting evidence for the imprinting status of mouse Atp10a, and the apparent discrepancy in human ATP10A tissue-specific imprinting, we sought to further characterize the imprinting of ATP10A in human brain to better understand the potential role of this gene in multiple neurodevelopmental disorders.
Materials and methods
Genotyping and allelic expression
Post-mortem cerebral cortex samples (Brodmann Area 9) were obtained from the NICHD Brain and Tissue Bank for Neurodevelopmental Disorders. DNA and RNA isolation were performed as previously described (Hogart et al. 2007). Single nucleotide polymorphisms (SNPs) were identified within exons of ATP10A using the UC Santa Cruz Genome Browser (http://genome.ucsc.edu) and the NCBI SNP database (http://www.ncbi.nlm.nih.gov/SNP/). Polymorphisms were screened by NEBcutter (www.neb.com) to find SNPs that disrupted restriction enzyme recognition sequences. Three SNPs within ATP10A exon 18, rs2076741, rs2078743, and rs2076744, are differentially cleaved by AclI, Hpy99I, and AciI, respectively. Approximately 10 ng of brain gDNA was PCR amplified using standard PCR conditions and primers flanking ATP10A exon 18 (Primers listed in Supplementary Table 1). PCR products were digested using an excess of restriction enzyme for 3 hours. RT-PCR amplification of ATP10A cDNA in heterozygous individuals followed previously described methods (Hogart et al. 2007). Restriction enzyme digestion of cDNA products was used to assess allelic expression. Band intensities were quantitatively measured with AlphaEaseFC™ version 4.0 software and only individuals with a single expressed allele were scored as monoallelic. Sequencing of ATP10A cDNA was used to verify allelic expression results. Quantitative RT-PCR of PWS, AS, and control ATP10A brain cDNA was performed according to previously described methods (Hogart et al. 2007). Briefly, crossing point values for ATP10A transcript were normalized to the housekeeping gene GAPDH using the comparative CT method (Applied Biosystems). Significant differences in ATP10A expression in PWS and AS samples compared to control values were evaluated using the Student’s t test.
Analysis of regulatory sequences
Overlapping primers were designed to amplify 2.1kb of the 5′ end of ATP10A with the strongest degree of conservation according to the UCSC Genome Browser. Genomic brain DNA from five individuals with monoallelic ATP10A expression (510, 1027, 1136, 1275, 1321) and five with biallelic ATP10A expression (22, 103, 285, 1029, 4192) was PCR amplified. DNA sequencing was performed by the UC Davis Division of Biological Sciences Sequencing Facility. Sequencing chromatograms were compared to the human genome reference sequence (March 2006 assembly) to identify rare or novel sequence variants. All characterized SNPs in the dbSNP build 128 (http://www.ncbi.nlm.nih.gov/SNP/) within the 2.1 kb region were analyzed. Allele frequencies were calculated within our population by dividing the number of occurrences of each SNP by the total number of alleles for each polymorphism. Transcription factor binding sites were analyzed with the Transcription Element Search System (TESS) TRANSFAC v6.0 database available online (http://www.cbil.upenn.edu/cgi-bin/tess/tess). Additional sequencing was performed to genotype two intronic SNPs (rs4906777 and rs4566129) that potentially disrupt characterized MeCP2 binding sites (Yasui et al. 2007). Fisher’s exact test was used to determine if regulatory and coding SNPs were significantly overrepresented in samples with monoallelic or biallelic ATP10A expression.
Bisulfite Sequencing
Bisulfite sequencing was used to analyze DNA methylation of ATP10A in human brain. First, 1 μg of genomic DNA was converted with the EZ DNA Methylation-Gold kit (Zymo) according to the manufacturer’s protocol. Primers amplifying the 5′ CpG island of ATP10A and the intronic MeCP2 binding site (Figure 3a) were designed using MethPrimer (www.urogene.org/methprimer/index1.htm) (Primers listed in Supplementary Table 1). PCR amplification was performed for 38-40 cycles with standard reaction conditions and approximately 25 ng of bisulfite converted genomic DNA. PCR products were cloned and sequenced according to previously described methods (Hogart et al. 2007).
Figure 3.

DNA methylation analysis of ATP10A regulatory sequences. (a) Schematic of ATP10A gene organization with vertical lines representing coding exons. Gray boxes indicate the relative locations of the 5′ CpG island and the intronic MeCP2 binding site. (b) Bisulfite sequencing of the 5′ CpG island in PWS 1290 and PWS 1447. Each horizontal line represents an individual clone, with filled circles representing methyled CpG sites and unfilled circles representing unmethylated CpG sites. (c) Bisulfite sequencing results from the MeCP2 binding site with polymorphic CpG sites indicated with stars (site 3 and site 7). A control sample heterozygous for polymorphisms in site 3 and site 7 shows that both sites are normally methylated. PWS 1290 lacks both polymorphic CpG sites while PWS 1447 lacks only site 3.
Results
As ATP10A is proposed to exhibit neuron-specific imprinted expression similar to UBE3A, we performed allelic expression analysis of ATP10A in a panel of control cerebral cortex samples. Individuals analyzed in our study included a wide range of ages (1 day to 46 years old), various ethnic backgrounds, and a relatively equal proportion of males and females (Table 1). Genomic DNA from control brain samples was genotyped for polymorphisms in ATP10A exons. cDNA was synthesized from heterozygous individuals, and quality was verified by RT-PCR amplification of the housekeeping gene GAPDH. Restriction enzyme digestion and sequencing performed on ATP10A cDNA revealed that 37.5% (6/16) of control brain samples exhibited monoallelic expression of ATP10A while 62.5% (10/16) of control samples showed biallelic expression (Table 1 and Figure 1a and 1b). Allele frequencies of SNPs used to detect allelic expression were similar between individuals with monoallelic ATP10A expression and individuals with biallelic ATP10A expression (Table 2). Additionally, exon haplotypes that were transcriptionally silent in some individuals were expressed in other individuals, suggesting that choice of allelic expression is independent of tightly linked genetic factors (Table 2).
Table 1.
Summary of brain samples information and allelic expression status of ATP10A
| Sample | Case # | Age | Ethnicitya | Genderb |
ATP10A Expression |
|---|---|---|---|---|---|
| Control | 1321 | 62 d | AA | F | monoallelic |
| Control | 510 | 2 y | CA | F | monoallelic |
| Control | 1275 | 2 y | AA | F | monoallelic |
| Control | 1711 | 27 y | CA | F | monoallelic |
| Control | 1027 | 22 y | CA | M | monoallelic |
| Control | 1136 | 33 y | CA | F | monoallelic |
| Control | 35 | 1 d | AA | M | biallelic |
| Control | 1055 | 96 d | CA | M | biallelic |
| Control | 22 | 113 d | CA | M | biallelic |
| Control | 390 | 125 d | CA | F | biallelic |
| Control | 103 | 2 y | AA | F | biallelic |
| Control | 629 | 7 y | AA | M | biallelic |
| Control | 1065 | 15 y | CA | M | biallelic |
| Control | 1029 | 29 y | HP | M | biallelic |
| Control | 285 | 30 y | AA | M | biallelic |
| Control | 4192 | 46 y | CA | M | biallelic |
| PWS | 1290 | 44y | CA | F | monoallelic |
| PWS | 1447 | 45y | CA | M | biallelic |
ethnicity identified by donor information, AA, African American, CA, Caucasian, HP, Hispanic
gender, M, male, F, female
Figure 1.

Analysis of ATP10A allelic expression in cerebral cortex. (a) Genomic DNA (upper panel) and cDNA (lower panel) from heterozygous control brain samples was digested with AclI to examine allelic expression. Upper band corresponds to uncut C allele and lower band corresponds to digested G allele. cDNA band quantification shown below the image reflects the percent that each allele contributes to the total densitometry. (b) Sequencing chromatograms of ATP10A cDNA confirm the allelic expression shown in (a). Arrows indicate the SNP differentially cleaved by AclI. (c) qRT-PCR analysis of ATP10A transcript in controls (n = 8), Angelman syndrome deletions (n = 2), Prader-Willi syndrome deletions (n = 2), Prader-Willi syndrome maternal uniparental disomy (n = 2), and Rett syndrome (n = 5). Error bars represent the S.E.M. for each category with significance values, * p < 0.02 and ** p < 0.001.
Table 2.
Allele frequencies of ATP10A exonic SNPs
| rs2076741 (G/C) |
rs2078743 (T/C) |
rs2076744 (G/A) |
Haplotypes Expressed |
Haplotypes Not Expressed |
|
|---|---|---|---|---|---|
| Monoallelic | G 0.92 (6) | T 0.67 (6) | G 0.8 (5) | GCG, CTG, GTA, GTG |
GTA, GTG, GCG |
| C 0.08 | C 0.33 | A 0.2 | |||
| Biallelic | G 0.77 (11) | T 0.73 (11) | G 0.78 (9) | ||
| C 0.23 | C 0.27 | A 0.22 | |||
| P value | 0.389 | 0.714 | 1 | ||
Numbers in parentheses indicate the number of individuals genotyped for each polymorphism
P values represent comparison of each SNP between monoallelic and biallelic ATP10A expression categories using Fisher’s exact test.
Quantitative RT-PCR analysis of AS and PWS cerebral cortex was used to investigate ATP10A expression from maternal versus paternal chromosomes independently. AS samples with a maternal 15q11-13 deletion (AS del) had significantly reduced ATP10A transcript compared to controls, as expected. Prader-Willi deletion (PWS del) brain samples had reduced transcript but were not significantly different than controls (Figure 1b), similar to observed expression patterns for UBE3A (Hogart et al. 2007). Surprisingly, Prader-Willi syndrome maternal UPD (PWS UPD) samples had significantly reduced ATP10A transcript (Figure 1b) unlike the significantly elevated UBE3A levels observed in the previous report (Hogart et al. 2007). Rett syndrome (RTT) brain samples showed a trend towards reduced ATP10A transcript that was not significantly different from controls, again differing from previously published results for UBE3A (Hogart et al. 2007; Samaco et al. 2005).
To investigate potential explanations for the unexpected ATP10A expression deficiencies in PWS UPD samples, we performed allelic expression analysis of ATP10A cDNA in both PWS samples. Consistent with reports that the majority of PWS maternal UPD cases are heterodisomic due to meiosis I nondisjunction (Fridman and Koiffmann 2000), both PWS UPD samples were heterozygous for coding SNPs within ATP10A (Figure 2). Sequencing chromatograms of ATP10A cDNA revealed that both maternal alleles were equally expressed in PWS 1447. In striking contrast, PWS 1290 expressed only one maternal ATP10A allele (Figure 2). Monoallelic expression of ATP10A in an individual with maternal disomy together with biallelic ATP10A expression in the majority of controls implicate factors other than parental origin in determining monoallelic expression of ATP10A.
Figure 2.
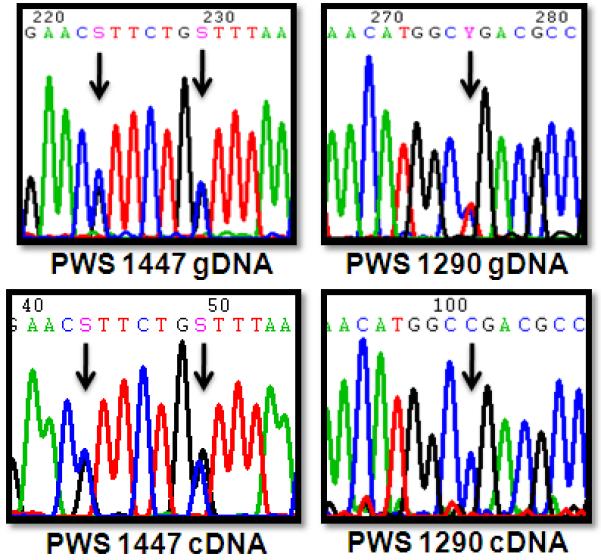
Allelic expression analysis of ATP10A in PWS UPD brain. Sequencing chromatograms of ATP10A brain gDNA (upper panels) show that both PWS UPD individuals are heterozygous for polymorphisms within ATP10A. Sequencing of ATP10A cDNA (lower panels) reveals that PWS 1447 showed biallelic expression while PWS 1290 exhibited monoallelic expression of ATP10A.
To address the question of whether ATP10A monoallelic expression is due to polymorphisms in regulatory regions, we sequenced 2.1 kb of the promoter in control individuals to look for promoter variants that may correlate with monoallelic expression of ATP10A. While no novel variants were identified through sequencing, three characterized SNPs were observed. Allele frequencies for promoter SNPs rs2076748, rs4906796, and rs2076749 are shown in Table 3. Allele frequencies for rs4906796 and rs2076749 were similar between individuals with monoallelic and bialleic ATP10A expression, however the G allele of rs2076748 was absent from samples with monoallelic expression (Table 3). Interestingly, the A allele disrupts an Sp1 binding site while the ancestral G allele preserves the consensus sequence for this ubiquitous transcription factor. This single promoter polymorphism does not completely explain the variability in monoallelic expression, as some individuals with biallelic ATP10A expression were also homozygous for the A allele. Interestingly, all four females homozygous for the A allele exhibited monoallelic expression.
Table 3.
Allele frequencies of ATP10A regulatory polymorphisms
| Promoter SNPs | Intron SNPs | |||||
|---|---|---|---|---|---|---|
| rs2076748 (A/G) |
rs4906796 (C/G) |
rs2076749 (T/A) |
rs4906777 (A/G) |
rs4566129 (T/C) |
Gender (M/F) |
|
| Monoallelic | A 1 (5) | C 0.8 (5) | T 0.5 (3) | A 0.83 (6) | T 0.67 (6) | M 0.14 (7) |
| G 0 | G 0.2 | A 0.5 | G 0.17 | C 0.33 | F 0.86 | |
| Biallelic | A 0.6 (5) | C 0.7 (5) | T 0.67 (4) | A 0.59 (11) | T 0.41(11) | M 0.82 (11) |
| G 0.4 | G 0.3 | A 0.33 | G 0.41 | C 0.59 | F 0.18 | |
| P value | 0.087 | 1 | 0.58 | 0.252 | 0.282 | 0.013 |
Numbers in parentheses indicate the number of individuals genotyped for each polymorphism
P values represent comparison of each SNP or gender between monoallelic and biallelic ATP10A expression categories using Fisher’s exact test.
In addition to the proximal promoter, we investigated polymorphisms in an intronic ATP10A sequence that was shown by chromatin immunoprecipitation in human cells to be a strong binding site for the neuronal epigenetic factor MeCP2 (Yasui et al. 2007). MeCP2 binds to methylated CpG dinucleotides (Meehan et al. 1992) and functions as a modulator of gene expression (Chahrour et al. 2008; Yasui et al. 2007), therefore we hypothesized that SNPs that disrupt CpG sites may affect allelic expression. Genotyping of polymorphisms that abolish CpG dinucleotides (rs4906777 and rs4566129) revealed that individuals with monoallelic expression showed a trend towards higher frequencies of SNPs that disrupt CpG sites (A allele for rs4906777 and T allele for rs4566129). In addition, all four males homozygous for the A allele of rs4906777 were biallelic for ATP10A transcript, while all three females homozygous for the A allele were monoallelic. While the allele distributions for SNPs rs4906777 and rs4566129 were not significantly different between individuals with monoallelic and bialellic ATP10A expression, the female PWS UPD sample with monoallelic expression lacked both polymorphic CpG sites (Figure 3c). Interestingly, ethnicity did not correlate with monoallelic expression (Table 1), however we found a significant correlation based on gender (p = 0.0128), with female samples being significantly overrepresented in the monoallelic expression category (Table 3). While we included the PWS UPD samples in our gender analysis to increase sample size, analysis of gender exclusively in control samples also yielded a significant association (p = 0.035), with females significantly overrepresented in the monoallelic ATP10A category.
The correlation between gender and monoallelic expressed led us to hypothesize that gender-specific epigenetic differences may contribute to allelic expression of ATP10A. Bisulfite sequencing was used to analyze DNA methylation of the ATP10A promoter CpG island in each brain sample (Figure 3). While overall ATP10A promoter methylation was low, the female PWS UPD sample (1290) with monoallelic expression exhibited one clone with elevated promoter methylation compared to the male PWS UPD sample (1447) (Figure 3b).While this evidence does not strongly support an epigenetic basis for the allelic expression differences, the presence of a highly methylated clone in the female PWS UPD sample does suggest that DNA methylation may contribute to the silencing of one maternal allele. Bisulfite sequencing confirmed that the MeCP2-bound sequence of the ATP10A intron including the polymorphic CpG sites was normally heavily methylated, and normal methylation levels were present in both PWS UPD brains (Figure 3c). Bisulfite sequencing was also used to analyze promoter methylation in control brain samples (Figure 4). All samples showed variable low levels of promoter methylation, ranging from 1-12% but individuals with monoallelic ATP10A expression (light grey bars) did not have increased promoter methylation compared to individuals with biallelic ATP10A expression (dark grey bars). Furthermore, no significant differences were observed between males and females for ATP10A promoter methylation. These results suggest that promoter methylation is not a general mechanism that accounts for variable monoallelic expression of ATP10A.
Figure 4.

ATP10A CpG island methyation in control brain. Bisulfite sequencing was used to assess methylation of the ATP10A 5′ CpG island in control brain samples with monoallelic (light grey) and biallelic (dark grey) ATP10A expression. Percent methylation represents the number of methylated CpG sites divided by the total number of possible CpG sites for all clones analyzed (10-12 clones per individual).
Discussion
Discrepancies in the imprinting status of the mouse Atp10a gene led us to question if human ATP10A imprinting was similarly variable. In our survey of ATP10A allelic expression in human control brain only a minority of individuals exhibited monoallelic ATP10A expression and allelic expression status appeared to be influenced by gender. Due to the anonymous nature of tissue donation we were unable to determine if the preferentially expressed allele in controls was always of maternal origin. Interestingly, monoallelic ATP10A expression in PWS UPD brain demonstrated that maternal alleles can be silenced and raises the possibility that ATP10A monoallelic expression is independent of parental chromosome origin.
Quantitative RT-PCR analysis of ATP10A transcript in PWS and AS brain samples also did not support a strict maternal basis for ATP10A expression. AS deletion samples with a single paternal 15q11-13 allele had significantly reduced ATP10A expression compared to controls, while PWS deletion samples were not significantly different from controls. This maternal chromosome bias in expression is in agreement with the original reports that concluded that ATP10A is a maternally expressed imprinted gene (Herzing et al. 2001a; Meguro et al. 2001). A parental chromosome bias in gene expression in PWS and AS brain was also observed for the neighboring gene GABRB3, although this gene is not imprinted (Hogart et al. 2007) demonstrating that this evidence alone is insufficient proof of imprinting. Contrasting to the elevated ATP10A in PWS UPD lymphoblasts reported by Bittel et al (Bittel et al. 2003), we observed significantly reduced ATP10A in PWS UPD brain compared to control brain. Tissue specific differences in ATP10A regulation, biological differences between different PWS UPD individuals, and technical differences between microarray expression profiling and qRT-PCR analysis may all contribute to the different observations between our reports. Consistent with Bittel et al (Bittel et al. 2003) we previously reported elevated UBE3A expression in the same PWS UPD brain samples (Hogart et al. 2007). Opposite directional changes in the two maternally expressed genes ATP10A and UBE3A in PWS UPD brain samples (Figure 1b and (Hogart et al. 2007) suggests that regulation of ATP10A is distinct from UBE3A imprinting in brain.
Unexpectedly, we found a significant association between gender and allelic expression status, with females being more likely than males to exhibit monoallelic ATP10A expression. Additionally, the A allele of the ATP10A promoter polymorphism rs2076748 disrupts a consensus binding site for the ubiquitous transcription factor Sp1, and may contribute to allelic expression differences between individuals. Interestingly the single male control sample with monoallelic ATP10A expression (1027) was homozygous for the A allele of the promoter variant rs2076748, and a female with biallelic expression was homozygous for the G allele of this variant, suggesting that gender combined with differences in genetic background influences monoallelic expression in the population. As ATP10A is physically located between the strictly biallelically expressed gene GABRB3 and an actively imprinted gene UBE3A, we hypothesize that this gene is influenced by neighboring imprints, but unlike GABRB3 (Hogart et al. 2007), ATP10A does not appear to have a strong overriding mechanism to maintain biallelic expression over monoallelic expression.
Recently a genome-wide study of allelic expression revealed that 5% of autosomal genes are subject to random monoallelic expression (Gimelbrant et al. 2007). Interestingly, although ATP10A was not mentioned in the study, a closely related gene, ATP11A, was found to be subject to random monoallelic expression. Consistent with our variable allelic expression results in control brain, analysis of ATP10A allelic expression in human embryonic stem cell lines found that 8 of 12 informative cell lines had biallelic ATP10A expression while only 4 of 12 had monoallelic expression (Kim et al. 2007). Strict monoallelic expression of other 15q11-13 imprinted genes, SNRPN, NDN, and MAGEL2 in the stem cell lines and our observation of frequent biallelic expression of ATP10A in brain, favors the interpretation that the variability in ATP10A allelic expression in stem cells is due to normal differences between individuals.
ATP10A is a member of the P-type ATPase family of proteins that are involved in active transport of molecules across the phospholipid membrane (Flamant et al. 2003). Mice heterozygous for a deletion spanning Atp10a exhibit increased body fat and obesity specifically when the deletion is maternally inherited (Dhar et al. 2000). Wildtype, maternal deletion, and paternal deletion mice showed similar levels of Atp10a transcript, leading to the conclusion that the maternal-specific inheritance of the obesity phenotype was most likely due to maternal environment effects instead of imprinting (Dhar et al. 2000). Further characterization of the Atp10a maternal-deletion mice revealed gender differences in severity of the obesity-related phenotypes with females being more significantly affected than males (Dhar et al. 2004). In light of our observation of gender influencing monoallelic expression of ATP10A in humans, it is interesting to speculate that similar effects may be influencing Atp10a allelic expression in mice.
Multiple human disorders are caused by abnormal dosage of 15q11-13, therefore understanding how genes within this locus are expressed is critical to understanding the molecular pathogenesis related to abnormal 15q11-13 karyotypes. Genotype-phenotype analyses of PWS and AS patients have revealed that many genes throughout 15q11-13 contribute to the clinical manifestations of patients (Lossie et al. 2001; Torrado et al. 2007). Additionally, as maternally derived 15q11-13 duplications often lead to autism-spectrum disorder, the two maternally expressed imprinted genes ATP10A and UBE3A are prime candidate genes for the 15q11-13 associated autism phenotype. Further studies are required to determine if any behavioral consequences result from abnormal ATP10A expression. Our analysis of ATP10A imprinting reveals that this gene is subject to complex regulation that is influenced by gender as well as parental-origin and genetic factors. Given this result, dysregulation of ATP10A may occur in previously unexpected ways, and potentially contributes to some of the obesity-related complications in PWS and some individuals with 15q11-13 duplications (Dennis et al. 2006; Ungaro et al. 2001).
Supplementary Material
Acknowledgements
We are grateful for access to human brain samples through the NICHD Brain and Tissue Bank for Developmental Disorders and the Harvard Brain Tissue Resource Center (supported in part by R24MH068855). We would like to thank Heather Rowe for valuable discussion of this manuscript and Amy George and Michelle Martin for technical assistance with bisulfite sequencing. This work was supported by NIH F31MH078377 (A.H.) and NIH 1R01HD048799 (J.M.L.).
References
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–8. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7:1–20. doi: 10.1017/S1462399405009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittel DC, Kibiryeva N, Talebizadeh Z, Butler MG. Microarray analysis of gene/transcript expression in Prader-Willi syndrome: deletion versus UPD. J Med Genet. 2003;40:568–74. doi: 10.1136/jmg.40.8.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy SB, Dykens E, Williams CA. Prader-Willi and Angelman syndromes: sister imprinted disorders. Am J Med Genet. 2000;97:136–46. doi: 10.1002/1096-8628(200022)97:2<136::aid-ajmg5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–9. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Jr., Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928–34. [PMC free article] [PubMed] [Google Scholar]
- Dennis NR, Veltman MW, Thompson R, Craig E, Bolton PF, Thomas NS. Clinical findings in 33 subjects with large supernumerary marker(15) chromosomes and 3 subjects with triplication of 15q11-q13. Am J Med Genet A. 2006;140:434–41. doi: 10.1002/ajmg.a.31091. [DOI] [PubMed] [Google Scholar]
- Dhar M, Webb LS, Smith L, Hauser L, Johnson D, West DB. A novel ATPase on mouse chromosome 7 is a candidate gene for increased body fat. Physiol Genomics. 2000;4:93–100. doi: 10.1152/physiolgenomics.2000.4.1.93. [DOI] [PubMed] [Google Scholar]
- Dhar MS, Sommardahl CS, Kirkland T, Nelson S, Donnell R, Johnson DK, Castellani LW. Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. J Nutr. 2004;134:799–805. doi: 10.1093/jn/134.4.799. [DOI] [PubMed] [Google Scholar]
- Flamant S, Pescher P, Lemercier B, Clement-Ziza M, Kepes F, Fellous M, Milon G, Marchal G, Besmond C. Characterization of a putative type IV aminophospholipid transporter P-type ATPase. Mamm Genome. 2003;14:21–30. doi: 10.1007/s00335-002-3032-3. [DOI] [PubMed] [Google Scholar]
- Fridman C, Koiffmann CP. Origin of uniparental disomy 15 in patients with Prader-Willi or Angelman syndrome. Am J Med Genet. 2000;94:249–53. doi: 10.1002/1096-8628(20000918)94:3<249::aid-ajmg12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318:1136–40. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- Herzing LB, Kim SJ, Cook EH, Jr., Ledbetter DH. The human aminophospholipid-transporting ATPase gene ATP10C maps adjacent to UBE3A and exhibits similar imprinted expression. American Journal of Human Genetics. 2001a;68:1501–5. doi: 10.1086/320616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzing LBK, Kim S-J, Cook EH, Ledbetter DH. The human aminophospholipid-transporting ATPase gene ATP10C maps adjacent to UBE3A and exhibits similar imprinted expression. American Journal of Human Genetics. 2001b;68:1501–1505. doi: 10.1086/320616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogart A, Nagarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16:691–703. doi: 10.1093/hmg/ddm014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi A, Meguro M, Hoshiya H, Haruta M, Ishino F, Shibahara T, Oshimura M. Predominant maternal expression of the mouse Atp10c in hippocampus and olfactory bulb. J Hum Genet. 2003;48:194–8. doi: 10.1007/s10038-003-0009-3. [DOI] [PubMed] [Google Scholar]
- Kayashima T, Yamasaki K, Joh K, Yamada T, Ohta T, Yoshiura K, Matsumoto N, Nakane Y, Mukai T, Niikawa N, Kishino T. Atp10a, the mouse ortholog of the human imprinted ATP10A gene, escapes genomic imprinting. Genomics. 2003;81:644–7. doi: 10.1016/s0888-7543(03)00077-6. [DOI] [PubMed] [Google Scholar]
- Kim KP, Thurston A, Mummery C, Ward-van Oostwaard D, Priddle H, Allegrucci C, Denning C, Young L. Gene-specific vulnerability to imprinting variability in human embryonic stem cell lines. Genome Res. 2007;17:1731–42. doi: 10.1101/gr.6609207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll JH, Nicholls RD, Magenis RE, Graham JM, Jr., Lalande M, Latt SA. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am J Med Genet. 1989;32:285–90. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- Landers M, Bancescu DL, Le Meur E, Rougeulle C, Glatt-Deeley H, Brannan C, Muscatelli F, Lalande M. Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nucleic Acids Res. 2004;32:3480–92. doi: 10.1093/nar/gkh670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, Theriaque D, Hutson A, Nicholls RD, Zori RT, Williams CA, Driscoll DJ. Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet. 2001;38:834–45. doi: 10.1136/jmg.38.12.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085–92. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meguro M, Kashiwagi A, Mitsuya K, Nakao M, Kondo I, Saitoh S, Oshimura M. A novel maternally expressed gene, ATP10C, encodes a putative aminophospholipid translocase associated with Angelman syndrome. Nat Genet. 2001;28:19–20. doi: 10.1038/ng0501-19. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–75. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain [letter] [In Process Citation] Nat Genet. 1997;17:14–5. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- Runte M, Kroisel PM, Gillessen-Kaesbach G, Varon R, Horn D, Cohen MY, Wagstaff J, Horsthemke B, Buiting K. SNURF-SNRPN and UBE3A transcript levels in patients with Angelman syndrome. Hum Genet. 2004;114:553–61. doi: 10.1007/s00439-004-1104-z. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrado M, Araoz V, Baialardo E, Abraldes K, Mazza C, Krochik G, Ozuna B, Leske V, Caino S, Fano V, Chertkoff L. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am J Med Genet A. 2007;143:460–8. doi: 10.1002/ajmg.a.31520. [DOI] [PubMed] [Google Scholar]
- Ungaro P, Christian SL, Fantes JA, Mutirangura A, Black S, Reynolds J, Malcolm S, Dobyns WB, Ledbetter DH. Molecular characterisation of four cases of intrachromosomal triplication of chromosome 15q11-q14. J Med Genet. 2001;38:26–34. doi: 10.1136/jmg.38.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu T, Hoffman A. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nature Genet. 1997;17:12–13. doi: 10.1038/ng0997-12. [DOI] [PubMed] [Google Scholar]
- Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–47. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–21. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.