

Abstract
Origins of regioselectivity and alkene-directing effects in nickel-catalyzed reductive coupling reactions were investigated by density functional calculations. Regioselectivity of simple alkynes is controlled by steric effects, while conjugated enynes and diynes are predicted to have increased reactivity and very high regioselectivities, placing alkenyl or alkynyl groups distal to the forming C–C bond. The reactions of enynes and diynes involve 1,4-attack on the conjugated enyne of the Ni-carbonyl complex on the conjugated enyne or diyne.
Introduction
Nickel-catalyzed reductive coupling reactions between alkynes and aldehydes provide an efficient way to prepare synthetically useful allylic alcohols.1-5 The first intermolecular reductive coupling of alkyne and aldehyde was reported in 2000,2 utilizing a Ni(0) catalyst, tributylphosphine ligand, and triethylborane as the stoichiometric reducing agent. Ni(0) can also catalyze the coupling reaction using N-heterocyclic carbene (NHC) ligand and triethylsilane reducing agent, as reported by Montgomery.3 The reductive coupling reaction was later applied to various groups of substrates. In addition to the reaction with aldehydes, alkynes were shown to undergo catalytic reductive coupling reactions with epoxides6 and imines.7 The coupling reactions with allenes8 and alkenes9 were also achieved recently using nickel catalysts.
One of the major features of these intermolecular coupling reactions is ability to control regioselectivity. For many combinations of alkynes and aldehydes the regioselectivity is very high (> 95:5), and in some of these, either the PR3/BEt3 or the N-heterocyclic carbene (NHC)/Et3SiH variant can be used (Scheme 1). However, the origin of the steric and electronic control of regioselectivity is yet to be explained. Of particular interest to us was that in both the PR3/BEt3 and NHC/Et3SiH systems, conjugated 1,3-enynes have enhanced reactivities compared to nearly all other alkynes and strong directing effects leading to products with alkenyl groups distal to the forming C–C bond.4 For example, coupling with enyne t-Bu–C≡C–CH=CH2 using phosphine ligand and BEt3 reductant leads to C–C bond formation at the very hindered tert-butyl position exclusively (Scheme 1).4a By contrast, simple alkynes with tert-alkyl substituents (t-Bu–C≡C–alkyl and t-Bu–C≡C–Ar) do not undergo reductive coupling under similar conditions. Similar alkene-directing effects were also observed in Ni-catalyzed reductive coupling reactions with conjugated enynes and epoxides,6b and in Rh- and Zr-catalyzed coupling reactions with 1,3-enynes.10 The dramatic effects of conjugated enynes on reactivity and regioselectivity are not consistent with a purely steric and/or electronic phenomenon. Tethered alkenyl groups in enynes and dienes are also known to increase the reactivity and regioselectivity of transition metal-catalyzed coupling reactions.11 The directing effects of tethered alkenyl groups are usually explained by coordination with the metal center during the rate- and regioselectivity-determining step, since the length of tether has dramatic effects on the reactivity and regioselectivity of enynes. While 1,6-enynes show strong directing effects in Ni-catalyzed ligand-free reductive couplings, those enynes with a shorter one- or two-atom tethers are not reactive under the same conditions, presumably due to the large strain energy required for the alkenyl group to coordinate with metal.5b In contrast, conjugated 1,3-enynes have strong directing effects in Ni-catalyzed reductive coupling reactions, even though coordination of the alkenyl group with the metal is expected intuitively to require large distortions of the enyne in the transition state.
Scheme 1.

Ni-catalyzed reductive coupling reactions.
We report a theoretical study on the origins of regioselectivity in Ni-catalyzed reductive coupling reactions, and the mechanism of directing groups in the reactions with conjugated enynes. Directing effects of other conjugated alkynes are also predicted. The directing effects are attributed to a unique 1,4-attack transition state involving strong coordination of the double bond of enyne with metal. Distortion/interaction analysis on transition state structures suggest that the high reactivity and regioselectivity of 1,3-enynes result from not only the π-metal stabilizing interaction but also smaller alkyne distortion in the 1,4-attack transition state.
Computational Method
All minima and transition state geometries were optimized with the B3LYP functional implemented in Gaussian 03.12 The LANL2DZ basis set was used for nickel, and 6-31G(d) for other atoms. All relative energies presented in this paper are Gibbs free energies in kilocalories per mole, with respect to the most stable pre-reaction complex, alkyne(diphosphine)nickel(0). Zero point vibrational energies and thermodynamic corrections at 298K were calculated at the same level as geometry optimization and were included in the Gibbs free energies reported.
Regioselectivity of Simple Terminal Alkynes
The mechanism of nickel-catalyzed reductive alkyne–aldehyde coupling reactions using a phosphine ligand and organoborane reductant has been studied previously by density functional theory.13,14 The rate- and regioselectivity-determining step is the oxidative addition of the Ni-alkyne-aldehyde complex to form the oxametallacyclopentene intermediate 3 (Table 1).15 The oxidative addition of simple alkyne and aldehyde occurs via a planar five-membered cyclic transition state. Lewis acids promote the oxidative addition by coordinating with the aldehyde oxygen in the oxidative addition transition state; in contrast, organoborane reductants are not involved in the oxidative addition step, due to relatively low Lewis acidity. The overall activation barrier is determined by the energy difference between the oxidative addition transition state TS1 and the most stable pre-reaction π-complex, alkyne(diphosphine)nickel 5. In the oxidative addition transition state, unsymmetrically substituted alkynes may adopt two different orientations, leading to two regioisomeric products 4-A and 4-B. The regioselectivity between 4-A and 4-B is determined by the relative activation barriers of the two pathways (ΔΔG‡). The activation energies and predicted regioselectivities of the coupling with acetaldehyde and several terminal and internal alkynes were calculated at the B3LYP/LANL2DZ-6-31G(d) level and are given in Table 1. The smaller PMe3 ligand was used in the calculations, since the size of ligand has little effect on regioselectivity (see below).
Table 1.
Activation energies for Ni-catalyzed reductive couplings of alkynes and acetaldehyde.a
 | |||||||
|---|---|---|---|---|---|---|---|
| alkyne | R1 | R2 | ΔG‡ (kcal/mol) | ΔΔG‡ (kcal/mol) |
major product | exp. selectivity1-3 (4-A:4-B) |
|
| TS1-A | TS1-B | ||||||
| 1a | H | H | 27.9 | - | 4a | - | |
| 1b | Me | H | 27.5 | 30.0 | 2.5 | 4b-A | 96:4 b,c |
| 1c | t-Bu | H | 29.1 | 35.4 | 6.3 | 4c-A | - |
| 1d | Ph | H | 28.2 | 33.8 | 5.6 | 4d-A | > 98:2 b,d |
| 1e | NH2 | H | 25.2 | 29.2 | 4.0 | 4e-A | - |
| 1f | CN | H | 29.5 | 35.3 | 5.8 | 4f-A | - |
| 1g | CH=O | H | 28.9 | 33.5 | 4.6 | 4g-A | - |
| 1h | COMe | H | 28.1 | 33.8 | 5.7 | 4h-A | - |
| 1i | CO2Me | H | 27.5 | 33.1 | 5.6 | 4i-A | - |
| 1j | CF3 | H | 28.8 | 34.7 | 5.9 | 4j-A | - |
| 1k | CH=CH2 | H | 19.8 | 32.5 | 12.7 | 4k-A | > 98:2 e |
| 1l | CMe=CH2 | H | 21.3 | 33.4 | 12.1 | 4l-A | - |
| 1m | CMe=CMe2 | H | 25.1 | 34.3 | 9.2 | 4m-A | - |
| 1n | C≡CH | H | 21.0 | 35.4 | 14.4 | 4n-A | - |
| 1o | Ph | Me | 30.8 | 34.0 | 3.2 | 4o-A | 92:8 b,f |
| 1p | CH=CH2 | Ph | 20.0 | 33.7 | 13.7 | 4p-A | 95:5 g |
| 1q | t-Bu | Me | 36.9 | 38.1 | 1.2 | 4q-A | no rxn |
| 1r | CH=CH2 | t-Bu | 19.3 | 39.5 | 20.2 | 4r-A | > 95:5 g |
| 1s | COMe | Me | 32.6 | 36.6 | 4.1 | 4s-A | - |
| 1t | COMe | t-Bu | 30.6 | 41.5 | 11.0 | 4t-A | - |
| 1u | CO2Me | Me | 32.5 | 34.8 | 2.4 | 4u-A | - |
B3LYP/LANL2DZ-6-31G(d) free energies with respect to alkyne (diphosphine)nickel complex 5. PMe3 was used as ligand in the calculations.
P(n-Bu)3 used as ligand. BEt3 used as reductant.
1-Octyne used in place of propyne. Benzaldehyde used in place of acetaldehyde.
Octanal used in place of acetaldehyde.
N-heterocyclic carbene used as ligand. Et3SiH used as reductant. HC≡C–CH=CH–C6H13 used in place of HC≡C–CH=CH2.
Butyraldehyde used in place of acetaldehyde.
Tricyclopentylphosphine (PCyp3) used as ligand. BEt3 used as reductant. Isobutyraldehyde used in place of acetaldehyde.
All predicted regioselectivities are in agreement with experimental data, where available. For simple alkyl and aryl alkynes (1a-d), the regioselectivities are mainly controlled by steric effects. For all terminal alkynes, substituents prefer to be distal to the forming C–C bond, and bulkier substituents lead to greater regioselectivity. This suggests that the steric repulsions in the oxidative addition are greater in the forming C–C bonds than in the forming Ni–C bonds. The steric effects can be observed by comparing the lengths of the forming Ni–C and C–C bonds: the forming C–C bond lengths are more sensitive to the size of the alkyne substituent than the forming Ni–C bond lengths (Table 2). When the substituents are proximal to the metal (pathway A), the Ni–C bonds in TS1b-A, TS1c-A, and TS1d-A are only slightly lengthened by 0.02, 0.03, and 0.02 Å, respectively, compared to the unsubstituted TS1a. When the substituents are proximal to the forming C–C bond (pathway B), the forming C–C bonds in TS1b-B, TS1c-B, and TS1d-B are lengthened 0.05, 0.07, and 0.05 Å, respectively. The greater changes of bond lengths in pathway B suggest that the repulsion of the alkyne substituents with aldehyde is much larger than the repulsion with the nickel and phosphine ligand in pathway A.
Table 2.
The forming Ni–C and C–C bond lengths in the oxidative addition transition states TS1. Changes of bond lengths compared to TS1a are given in parentheses.
| TS | R1 | R2 | pathway | r(Ni–C) (Å) | r(C–C) (Å) |
|---|---|---|---|---|---|
| TS1a | H | H | - | 1.81 (0) | 1.67 (0) |
| TS1b-A | Me | H | A | 1.83 (0.02) | 1.64 (−0.03) |
| TS1b-B | Me | H | B | 1.81 (0.00) | 1.72 (0.05) |
| TS1c-A | t-Bu | H | A | 1.84 (0.03) | 1.60 (−0.07) |
| TS1c-B | t-Bu | H | B | 1.81 (0.00) | 1.75 (0.08) |
| TS1d-A | Ph | H | A | 1.83 (0.02) | 1.65 (−0.02) |
| TS1d-B | Ph | H | B | 1.81 (0.00) | 1.72 (0.05) |
Electron-rich and electron-deficient alkynes are predicted to have similar regioselectivities (alkynes 1e and 1f-j, respectively). The regioselectivities of polarized alkynes are also controlled by steric effects. The lack of dependence on donor or acceptor character of the substituent suggests that electronic interactions have little effect on regioselectivity. Electron-deficient alkynes 1f-j have slightly higher activation barriers than electron-rich alkynes 1b and 1e, due to the stronger binding of nickel with electron-deficient alkynes in the catalyst resting complex 5.
Regioselectivity of Simple Internal Alkyne Reactions
The steric effects of alkyne substituents on regioselectivity are additive. For alkyl or aryl substituted internal alkynes, bulkier groups prefer to be distal to the forming C–C bond. C–C bond formation distal to the large phenyl group of 1-propynylbenzene (R1 = Ph, R2 = Me, 1o) is favored by 3.2 kcal/mol, in agreement with the experimental regioselectivity. Large alkyne substituents increase the activation barriers for both pathways. The experimentally unreactive 4,4-dimethylpent-2-yne (R1 = t-Bu, R2 = Me, 1q) is predicted to have a high activation energy of 36.9 kcal/mol.
Regioselectivity of Conjugated 1,3-Enyne Reactions – Alkene Directing Effects by 1,4-addition
Conjugated 1,3-enynes have dramatic effects on both reactivity and regioselectivity. The vinyl substituent of 1,3-enynes strongly prefers pathway A (TS1-A) by 12.7, 13.7, and 20.2 kcal/mol in the coupling with enynes 1k, 1p, and 1r (R2 = H, Ph, and t-Bu), respectively. This suggests that the C–C bond is exclusively formed distal to the alkenyl group on the enyne. The activation barriers (ΔG‡ = ∼20 kcal/mol) of these reactions are much lower than the coupling with simple alkynes, leading to greater reactivities of 1,3-enynes. These results are in good agreement with experimental reactivities and regioselectivities of enynes.
Figure 1 illustrates the structures of the rate-determining oxidative addition transition states and the metallacycle intermediates in the three possible alkyne–aldehyde coupling pathways for the reaction with 1,3-butenyne 1k and acetaldehyde. Three alternative pathways involving coupling at the double bond of the enyne were calculated, and all are predicted to have higher activation energies than the coupling at the triple bond (Figure 2). The most favorable transition state TS1k-A involves attack of nickel and aldehyde to the 1 and 4-positions of the enyne. The 1,4-attack requires only 19.8 kcal/mol activation energy, much lower than the 1,2-attack in pathway A′ which leads to the same regioisomeric product (ΔG‡ = 28.2 kcal/mol). When the vinyl group is proximal to the forming C–C bond (pathway B), only 1,2-attack is possible. The activation energy for pathway B is 32.5 kcal/mol, also much higher than that for pathway A.
Figure 1.
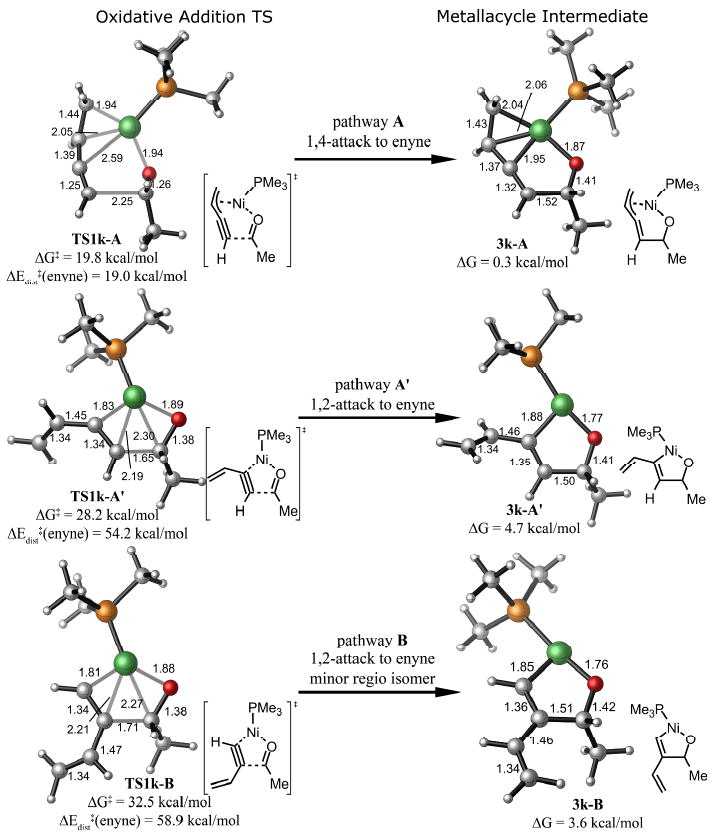
Oxidative addition transition states for the reaction with 1,3-butenyne 1k and acetaldehyde.
Figure 2.

Oxidative addition transition states in alternative pathways for the reaction with 1,3-butenyne 1k and acetaldehyde.
The transition state structure of the 1,4-addition TS1k-A is very different from the five-membered cyclic oxidative addition transition states in the 1,2-pathway (TS1k-A′). The lengths of the forming C–C and Ni–C bonds in TS1k-A are 2.25 and 2.59 Å, respectively, much longer than in the 1,2-attack (1.65 and 1.83 Å, respectively). This suggests a much earlier transition state in the 1,4-attack. In TS1k-A, Ni is strongly bonded to the vinyl group. The distances between Ni and the two vinyl carbons are only 1.94 and 2.05 Å, respectively.
The oxidative addition via the 1,4-addition TS1k-A leads to an η3-allyl/metal complex 3k-A. Similar η3-allyl/metal complexes have been reported for other group 10 transition metals.16,17 In contrast, higher energy five-membered oxametallacyclopentene intermediates 3k-A′ and 3k-B are formed via the 1,2-pathways. It is noted that the η3-allyl/metal interaction only stabilizes intermediate 3k-A by 4.4 kcal/mol compared to complex 3k-A′, while the energy difference in the oxidative addition transition state is 8.4 kcal/mol. This also suggests that the η3-allyl/metal interaction is not the only factor that stabilizes the 1,4-addition transition state.
In addition to the alkyne–aldehyde coupling pathways shown in Figure 1, the alkene–aldehyde coupling pathways were also calculated for the reaction with 1,3-butenyne 1k (Figure 2). Both 1,4- and 1,2-addition transition states were located for the alkene–aldehyde couplings. The 1,4-addition TS1k-C is also much more stable than the 1,2-addition TS1k-C′ and TS1k-D. However, TS1k-C is still 4.1 kcal/mol higher in energy than the coupling at the triple bond (TS1k-A). This agrees with experimental observation that the coupling occurs at the the acetylenic terminus of the enyne.18
Steric Effects on the Regioselectivity of Conjugated 1,3-Enynes
The regioselectivities of various 1,3-enynes with substitution at the terminal alkyne position, and at internal and terminal alkene positions were investigated (alkynes 1k-m, 1p, 1r in Table 1). All substituted 1,3-enynes show strong and dominant directing effects of alkenyl groups. The favorable 1,4-attack pathway dominates the regioselectivity and greatly increases reactivity of all 1,3-enynes. This favor bulky phenyl and tert-butyl groups to be adjacent to the forming C–C bond in enynes 1p and 1r. Vinyl tert-butyl acetylene (1r), which is very reactive experimentally, has an activation barrier of 19.3 kcal/mol, much lower than the unreactive methyl tert-butyl acetylene (1o, ΔG‡ = 36.9 kcal/mol).
The size of the substituent at the terminal alkyne position has little effects on the barrier of the 1,4-addition pathway A. Enynes 1k (R2 = H), 1p (R2 = Ph), and 1r (R2 = t-Bu) have very similar activation barriers for the favored pathway A (19.8, 20.0, and 19.3 kcal/mol, respectively). As mentioned before, the 1,4-addition occurs via an early transition state with a long forming C–C bond. Thus, steric repulsions in the forming C–C bond are relatively small. Also, alkyl or aryl substituents may stabilize the partial radical character of the terminal alkyne carbon in the 1,4-addition transition state. In contrast, bulky terminal alkyne substituents increase the repulsion with the ligand in the disfavored 1,2-addition pathway B, and thus lead to higher activation barriers for pathway B and higher regioselectivity.
Substitution by a methyl group at the internal alkene position of the 1,3-enyne slightly increases the activation barriers of both 1,4- and 1,2-pathways (Table 1, alkyne 1l, R1 = CMe=CH2, R2 = H). Thus, the effects of internal alkene substitution on regioselectivity are small. The regioselectivity of the reaction of enyne 1l is very similar to that of butenyne 1k (R1 = CH=CH2, R2 = H).
Substitution by two methyl groups at the terminal alkene position increases the activation barrier of the 1,4-addition pathway A. The activation barrier of the reaction of acetaldehyde with enyne 1m (R1 = CMe=CMe2) is 3.8 kcal/mol higher than that with enyne 1l (R1 = CMe=CH2). Pathway A is still strongly favored for enyne 1m (ΔΔG‡ = 9.2 kcal/mol), but the regioselectivity is lower than with enyne 1l, which ΔΔG‡ is 12.1 kcal/mol.
Distortion/Interaction Analysis on the 1,2- and 1,4-Transition States with Enynes
To understand the factors that stabilize the 1,4-addition transition state with conjugated enynes, a distortion/interaction analysis of the oxidative addition transition states for the coupling of 1,3-butenyne and acetaldehyde was performed on both TS1k-A (1,4-attack) and TS1k-A′ (1,2-attack).19 The distortion energy of the enyne and the interaction energy between the enyne and the Ni-aldehyde complex along the reaction coordinate are shown in Figure 3. The distortion energy of enyne (Edist(enyne)) is defined as the energy difference between the ground state of enyne and the distorted enyne in the transition state geometry. The interaction energy of enyne with nickel and aldehyde (Eint) is defined as the energy difference between the transition state and the separated enyne and nickel/aldehyde complex at the transition state geometry. For all points along the reaction coordinate, the enyne distortion energy is lower, and the interaction energy is stronger in the 1,4-attack than in the 1,2-attack. This suggests that the origin of the alkene directing effects is the stabilizing π-metal interaction as well as the smaller distortion of enyne in the 1,4-attack pathway A. The stronger interaction energy in the 1,4-attack is obviously due to the π-coordination of the vinyl group with metal. The smaller enyne distortion in the 1,4-attack is surprising, but the alkenyl group is actually less bent in the 1,4-attack, since it is closer to the metal. Since the 1,2-attack transition state is much later than the 1,4-attack, much greater enyne distortion and smaller interaction energy is observed in the disfavored TS1k-A′ than in TS1k-A (Table 3).
Figure 3.

Distortion energy of enyne (Edist(enyne)) and interaction energy of enyne with nickel and aldehyde (Eint) as a function of the forming C–C bond length in the reaction of 1,3-butenyne 1k and acetaldehyde.
Table 3.
Distortion/interaction analysis for the oxidative addition transition states. Energies are in kcal/mol.
| entry | Structure | R1 | R2 | ΔG‡ | ΔEdist‡ (alkyne)a |
ΔEint‡ |
|---|---|---|---|---|---|---|
| 1 | TS1a | H | H | 27.9 | 51.6 | −89.0 |
| 2 | TS1b-A | Me | H | 26.2 | 57.3 | −91.6 |
| 3 | TS1b-B | Me | H | 28.7 | 58.5 | −88.2 |
| 4 | TS1k-A | CH=CH2 | H | 19.8 | 19.0 | −69.3 |
| 5 | TS1k-A′ | CH=CH2 | H | 28.2 | 54.2 | −89.5 |
| 6 | TS1k-B | CH=CH2 | H | 32.5 | 58.9 | −88.6 |
Energy to distort alkynes to transition state geometry.
Energy of interaction between the distorted alkyne and the Ni(PR3)-aldehyde complex in the transition state.
The 1,2-addition pathway B of the reaction with 1,3-butenyne leads to the minor regioisomeric product; in this pathway, the alkyne distortion is even greater than in the other 1,2-addition pathway A′ (ΔEdist‡(alkyne) = 54.2 and 58.9 kcal/mol for TS1k-A′ and TS1k-B, respectively). In pathway B, the steric repulsion of the vinyl group in the forming C–C bond is stronger than the repulsion of the vinyl group with the metal and the ligand in pathway A. This leads to the greater alkyne distortions in pathway B.
Distortion/Interaction Analysis on the Transition States with Unconjugated Alkynes
A distortion/interaction analysis was also performed on the transition states of the couplings with simple acetylene and propyne (Table 3, entries 1-3). Transition state distortion energies of unconjugated alkynes parallel the size of the substituents on alkyne. The distortion energy of propyne is 5 kcal/mol greater than that of simple acetylene. The distortion energy of propyne is smaller in the favored pathway A in which smaller steric repulsion is expected. For simple unconjugated alkynes, larger steric repulsions also lead to weaker interaction energy, since the forming bond lengths are longer for sterically hindered alkynes.
Directing Effects of Other Conjugated Alkynes
Possible directing effects with other unsaturated substituents were investigated. Favorable 1,4-attack is also predicted for the reaction with 1,3-butadiyne (R1 = C≡CH, 1n). The activation energy of the 1,2-attack is 8.0 kcal/mol higher than that for the 1,4-attack. This predicts that 1,3-diynes may have similar reactivity and regioselectivity as 1,3-enynes.20 While vinyl and ethynyl substituents promote 1,4-attack leading to an η3-allyl/metal complex, phenyl and most unsaturated heteroatom substituted groups do not. For alkynes 1d (R1 = Ph), 1f (R1 = CN), 1g (R1 = CHO), 1h (R1 = COMe), and 1i (R1 = COOMe), the 1,2-attack transition states are more stable than the 1,4-transition states by 12.6, 9.7, 1.4, 3.9, and 16.2 kcal/mol, respectively.
While 1,2-attack is favored for butynone (R1 = COMe, R2 = H, 1h) by 3.9 kcal/mol, 1,4-attack is slightly favored by 1.8 kcal/mol for pent-3-yn-2-one (R1 = COMe, R2 = Me, 1s), and is favored by 10.9 kcal/mol for 5,5-dimethylhex-3-yn-2-one (R1 = COMe, R2 = t-Bu, 1t). For the weakly directing acetyl group,21 bulky substitution at the terminal alkyne position promotes the 1,4-attack pathway. Since the 1,4-attack is less sensitive to the steric repulsions at the forming C–C bond (see discussions above), the activation barriers for the 1,4-attack remain similar when substituting terminal alkyne hydrogen with methyl or tert-butyl group, while the barriers for 1,2-attack rise substantially due to increased steric repulsions.
Effects of Ligands on Regioselectivities
Various phosphine ligands have been used in the Ni-catalyzed reductive coupling reactions. The activation barriers for both pathways A and B were calculated for the reaction of propyne with acetaldehyde using various phosphine ligands. The results are shown in Table 4. Larger phosphine ligands, including PEt3, P(n-Bu)3, and PPh3 are predicted to be more reactive than PMe3. The catalyst resting state propyne(PMe3)2Ni(0) complex is less crowded and thus more stable than complexes with other phosphine ligands, leading to the higher activation barrier for the smaller PMe3 ligand. The differences of activation energies between pathways A and B are similar (2.5-3.2 kcal/mol) for all phosphine ligands studied (Table 4). This suggests that the effects of ligands on regioselectivities are small. Thus, the small PMe3 ligand was used when calculating regioselectivities of other alkynes to save computation time.
Table 4.
Activation free energies for Ni-catalyzed reductive coupling reactions between propyne and acetaldehyde.a
 | |||||
|---|---|---|---|---|---|
| entry | R | ΔG‡ (kcal/mol) | ΔΔG‡ (kcal/mol) |
major product | |
| TS1-A | TS1-B | ||||
| 1 | PMe3 | 27.5 | 30.0 | 2.5 | 4b-A |
| 2 | PEt3 | 24.0 | 26.8 | 2.8 | 4b-A |
| 3 | P(n-Bu)3 | 23.7 | 26.9 | 3.2 | 4b-A |
| 4 | PPh3 | 21.8 | 24.7 | 2.9 | 4b-A |
B3LYP/LANL2DZ-6-31G(d) free energies with respect to propyne(diphosphine)nickel complex 5b.
Effects of Aldehyde Substituents on Regioselectivities
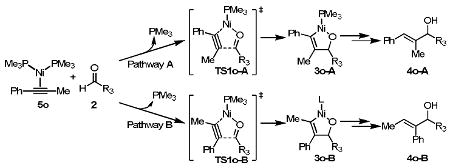
In this theoretical study, acetaldehyde was used as the aldehyde component. Regioselectivities were also computed with butyraldehyde, isobutyraldehyde, and benzaldehyde in the reaction with 1-propynylbenzene. The predicted regioselectivities are shown Table 5. Acetaldehyde, n-butyraldehyde, and isobutyraldehyde have similar activation energies and regioselectivities. The activation barrier for benzaldehyde is 3.5 kcal/mol higher than that for acetaldehyde, but the regioselectivities of the reactions with these aldehydes are very similar (2.8-3.7 kcal/mol). This suggests that the aldehyde substituent does not have strong effects on regioselectivities.
Table 5.
Activation free energies for Ni-catalyzed reductive coupling reactions between 1-propynylbenzene and aldehydes.a
 | |||||
|---|---|---|---|---|---|
| entry | R3 | ΔG‡ (kcal/mol) | ΔΔG‡ (kcal/mol) |
major product | |
| TS1-A | TS1-B | ||||
| 1 | Me | 30.8 | 34.0 | 3.2 | 4o-A |
| 2 | n-Pr | 31.9 | 34.7 | 2.8 | 4o-A |
| 3 | i-Pr | 32.4 | 34.9 | 2.5 | 4o-A |
| 4 | Ph | 34.3 | 38.0 | 3.7 | 4o-A |
B3LYP/LANL2DZ-6-31G(d) free energies with respect to alkyne(diphosphine)nickel complex 5o.
Conclusions
We studied the origins of regioselectivity in nickel-catalyzed reductive coupling reactions. The regioselectivity in coupling reactions of simple alkynes is controlled by steric effects; bulky substituents in simple alkynes prefer to be distal to the forming C–C bond. A very different phenomenon dominates the regioselectivity in coupling reactions of conjugated enynes; the alkenyl group exerts a very strong preference to be distal to the forming C–C bond. These couplings of conjugated enynes (and those of diynes) occur via a 1,4-attack pathway that is stabilized by a π-metal interaction and results in a smaller transition state distortion of the alkyne relative to other possible mechanisms.
Supplementary Material
Acknowledgments
We are grateful to the National Science Foundation for financial support of this research (CHE-0548209). Calculations were performed on the National Science Foundation Terascale Computing System at the NCSA.
Footnotes
Supporting Information Available. Optimized Cartesian coordinates and energies, details of the distortion/interaction study, and complete authorship of reference 13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1 For reviews, see Montgomery J. Acc Chem Res. 2000;33:467. doi: 10.1021/ar990095d.Ikeda S. Angew Chem, Int Ed. 2003;42:5120. doi: 10.1002/anie.200301673.Montgomery J. Angew Chem, Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634.Montgomery J, Sormunen GJ. In: Metal Catalyzed Reductive C-C Bond Formation: A Departure from Preformed Organometallic Reagents. Krische MJ, editor. Springer; Berlin / Heidelberg: 2007. pp. 1–23.Moslin RM, Miller-Moslin K, Jamison TF. Chem Commun. 2007:4441. doi: 10.1039/b707737h.
- 2.Huang WS, Chan J, Jamison TF. Org Lett. 2000;2:4221. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]
- 3.Mahandru GM, Liu G, Montgomery J. J Am Chem Soc. 2004;126:3698. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]
- 4 For more examples of Ni-catalyzed intermolecular alkyne-aldehyde coupling, see: Colby EA, Jamison TF. J Org Chem. 2003;68:1456. doi: 10.1021/jo0264123.Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442. doi: 10.1021/ja034366y.Miller KM, Molinaro C, Jamison TF. Tetrahedron-Asymmetry. 2003;14:3619.Takai K, Sakamoto S, Isshiki T. Org Lett. 2003;5:653. doi: 10.1021/ol0272996.Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org Lett. 2005;7:2937. doi: 10.1021/ol050881k.Moslin RM, Miller KM, Jamison TF. Tetrahedron. 2006;62:7598.Moslin RM, Jamison TF. Org Lett. 2006;8:455. doi: 10.1021/ol052719n.Sa-Ei K, Montgomery J. Org Lett. 2006;8:4441. doi: 10.1021/ol061579u.Chaulagain MR, Sormunen GJ, Montgomery J. J Am Chem Soc. 2007;129:9568. doi: 10.1021/ja072992f.Yang Y, Zhu SF, Zhou CY, Zhou QL. J Am Chem Soc. 2008;130:14052. doi: 10.1021/ja805296k.Trenkle JD, Jamison TF. Angew Chem, Int Ed. 2009;48:5366. doi: 10.1002/anie.200902079.
- 5 For Ni-catalyzed intermolecular coupling reactions with enynes and carbonyl compounds, see: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130. doi: 10.1021/ja0491735.Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342. doi: 10.1021/ja0446799.Miller KM, Jamison TF. Org Lett. 2005;7:3077. doi: 10.1021/ol051075g.Miller KM, Colby EA, Woodin KS, Jamison TF. Adv Synth Catal. 2005;347:1533.; e also see ref. 3
- 6.(a) Molinaro C, Jamison TF. J Am Chem Soc. 2003;125:8076. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]; (b) Sparling BA, Simpson GL, Jamison TF. Tetrahedron. 2009;65:3270. doi: 10.1016/j.tet.2008.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Patel SJ, Jamison TF. Angew Chem, Int Ed. 2003;42:1364. doi: 10.1002/anie.200390349. [DOI] [PubMed] [Google Scholar]; (b) Patel SJ, Jamison TF. Angew Chem, Int Ed. 2004;43:3941. doi: 10.1002/anie.200460044. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ng SS, Jamison TF. J Am Chem Soc. 2005;127:7320. doi: 10.1021/ja0521831. [DOI] [PubMed] [Google Scholar]; (b) Ng SS, Jamison TF. Tetrahedron. 2006;61:11405. [Google Scholar]
- 9.(a) Ogoshi S, Ueta M, Arai T, Kurosawa H. J Am Chem Soc. 2005;127:12810. doi: 10.1021/ja0542486. [DOI] [PubMed] [Google Scholar]; (b) Ng SS, Jamison TF. J Am Chem Soc. 2005;127:14194. doi: 10.1021/ja055363j. [DOI] [PubMed] [Google Scholar]; (c) Ng SS, Ho CY, Schleicher KD, Jamison TF. Pure Appl Chem. 2008;80:929. doi: 10.1351/pac200880050929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10 For examples of Zr-catalyzed coupling reactions with 1,3-enynes, see: Takahashi T, Xi Z, Nishihara Y, Huo S, Kasai K, Aoyagi K, Denisov V, Negishi E. Tetrahedron. 1997;53:9123.Zhou YB, Chen JJ, Zhao CB, Wang EJ, Liu YH, Li YX. J Org Chem. 2009;74:5326. doi: 10.1021/jo900808k. For examples of Rh-catalyzed coupling reactions with 1,3-enynes, see: Jang HY, Huddleston RR, Krische MJ. J Am Chem Soc. 2004;126:4664. doi: 10.1021/ja0316566.Kong JR, Cho CW, Krische MJ. J Am Chem Soc. 2005;127:11269. doi: 10.1021/ja051104i.Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718. doi: 10.1021/ja056474l.Komanduri V, Krische MJ. J Am Chem Soc. 2006;128:16448. doi: 10.1021/ja0673027.Hong YT, Cho CW, Skucas E, Krische MJ. Org Lett. 2007;9:3745. doi: 10.1021/ol7015548.
- 11 For reviews of alkene-directing effects, see: Johnson JB, Rovis T. Angew Chem, Int Ed. 2008;47:840. doi: 10.1002/anie.200700278. For examples of alkene-directing effects in metal-catalyzed reactions, see: Pd: Trost BM, Lautens M, Chan C, Jebaratnam DJ, Meuller T. J Am Chem Soc. 1991;113:636.Trost BM, Tanoury GJ, Lautens M, Chan C, MacPherson DT. J Am Chem Soc. 1994;116:4255.Krafft ME, Sugiura M, Abboud KA. J Am Chem Soc. 2001;123:9174. doi: 10.1021/ja016446c. Ni:; (e) see ref. 5d, 5h, and 5i. Zn:; (f) Marek I, Beruben D, Normant JF. Tetrahedron Lett. 1995;36:3695. [Google Scholar]; (g) Marek I, Schreiner PR, Normant JF. Org Lett. 1999;1:929. [Google Scholar]
- 12.Frisch MJ, et al. Gaussian 03, Revision D.01. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 13.McCarren PR, Liu P, Cheong PHY, Jamison TF, Houk KN. J Am Chem Soc. 2009;131:6654. doi: 10.1021/ja900701g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14 For a theoretical study on the mechanism of Ni-catalyzed alkyne-enone reductive coupling reactions with ZnMe2 reductant in a ligand-free system, see: Hratchian HP, Chowdhury SK, Gutierrez-Garcia VM, Amarasinghe KKD, Heeg MJ, Schlegel HB, Montgomery J. Organometallics. 2004;23:4636.
- 15 The oxametallacyclopentene intermediate has been observed in the stoichiometric alkyne–aldehyde coupling reaction: Ogoshi S, Arai T, Ohashi M, Kurosawa H. Chem Commun. 2008:1347. doi: 10.1039/b717261c.
- 16 For related η3-allyl/metal complexes, see: Benyunes SA, Brandt L, Fries A, Green M, Mahon MF, Papworth TMT. J Chem Soc, Dalton Trans. 1993:3785.Ogasawara M, Okada A, Watanabe S, Fan L, Uetake K, Nakajima K, Takahashi T. Organometallics. 2007;26:5025.
- 17 It has been proposed that nickel-catalyzed couplings of 1,3-dienes and aldehyde or ketone occur via η3-allylalkoxynickel intermediates, which crystal structures have been reported. See: Ogoshi S, Tonomori K, Oka M, Kurosawa H. J Am Chem Soc. 2006;128:7077. doi: 10.1021/ja060580l. and references therein.
- 18 In contrast, Ru-catalyzed coupling of 1,3-enyne and alcohol occurs at the vinylic terminus of the enyne: Patman RL, Williams VM, Bower JF, Krische MJ. Angew Chem, Int Ed. 2008;47:5220. doi: 10.1002/anie.200801359.
- 19 For previous examples of distortion/interaction analysis on transition metal-catalyzed reactions, see: Legault CY, Garcia Y, Merlic CA, Houk KN. J Am Chem Soc. 2007;129:12664. doi: 10.1021/ja075785o.Jong GT, Bickelhaupt FM. Chemphyschem. 2007;8:1170. doi: 10.1002/cphc.200700092.
- 20 Similar directing effects of 1,3-diynes were observed in transition-metal catalyzed coupling reactions; the alkynyl groups prefer to be distal to the forming C–C bond. For examples, see: Rh: Rourke JP, Batsanov AS, Howard JAK, Marder TB. Chem Commun. 2001:2626.Huddleston RR, Jang HY, Krische MJ. J Am Chem Soc. 2003;125:11488. doi: 10.1021/ja030415v.Cho CW, Krische MJ. Org Lett. 2006;8:3873. doi: 10.1021/ol061485k.Cho CW, Skucas E, Krische MJ. Organometallics. 2007;26:3860. Zr: Hsu DP, Davis WM, Buchwald SL. J Am Chem Soc. 1993;115:10394.
- 21 Acyl groups have been proposed as directing groups in the coupling with enones; an η3-oxaallylnickel moiety was suggested to stabilize the oxidative addition transition state, leading to formation of a C–C bond at the β position of the enone. For examples of Ni-catalyzed couplings with enones, see: Ikeda S, Sato Y. J Am Chem Soc. 1994;116:5975.Herath A, Thompson BB, Montgomery J. J Am Chem Soc. 2007;129:8712. doi: 10.1021/ja073300q.Ho CY, Ohmiya H, Jamison TF. Angew Chem, Int Ed. 2008;47:1893. doi: 10.1002/anie.200705163.Ogoshi S, Haba T, Ohashi M. J Am Chem Soc. 2009;131:10350. doi: 10.1021/ja903510u.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.