

Abstract
The first enantioselective synthesis of a member of the chlorosulfolipid family of natural products is reported. All of the polar substituents of malhamensilipin A are introduced with high stereoselectivity, and the unique E-chlorovinyl sulfate is created by a chemo-, regio-, and stereoselective E2 elimination of HCl in a reaction that likely has a counterpart in the biosynthesis of this fascinating natural product.
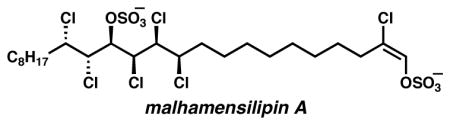
The chlorosulfolipids (1–5, Figure 1)1–3 are a fascinating class of natural products that, after decades of inattention from organic chemists, have recently garnered interest as targets of synthesis4,5 and have inspired methodology for stereoselective chlorination.6–8 In early 2009, the Carreira group published the synthesis of mussel toxin chlorosulfolipid 3, representing the first synthesis in this class.4 Shortly thereafter, our laboratories disclosed the NMR-based relative and absolute stereochemical elucidation of algae-derived lipid danicalipin A 1, which was supported by a concise synthesis.5 Recently, we reported the structural revision and stereochemical determination of the protein tyrosine kinase inhibitor malhamensilipin A (2), a chlorosulfolipid related to danicalipin A that is produced by the algae Poterioochromonas malhamensis.2b In this communication, we describe a stereocontrolled and concise synthesis of malhamensilipin A, which constitutes the first enantioselective synthesis of a chlorosulfolipid. Our synthesis features a selective, base-mediated elimination of HCl from a penultimate intermediate bearing seven chlorine atoms and two sulfate groups as the final step to introduce the unique E-chlorovinyl sulfate functional group.
Figure 1.

Representative chlorosulfolipids from freshwater algae (1 and 2) and from toxic Adriatic mussels (3–5).
In our work on the stereochemical elucidation of the algae-derived chlorosulfolipids,2b,5 we were surprised to find that the stereochemistry among lipids 1, 2, and 3, which are presumably derived from similar biosynthetic pathways, was not fully conserved. Initially, we planned to adapt the strategy used in our synthesis of danicalipin A for malhamensilipin A; however, we quickly encountered a problem. Whereas in our efforts toward 1 regioselective and stereospecific dichlorination of the γ,δ-alkene in E,E-diene 6 proceeded smoothly (Eq. 1) and enabled a subsequent diastereoselective dihydroxylation, we were surprised to discover that the dichlorination of related (E,Z)-α,β,γ,δ-unsaturated esters generated equimolar mixtures of diastereomers, in a reaction that would prove unsuitable as the foundation of a stereocontrolled synthesis. We also knew from our work on the diastereoselective dichlorination of Z-allylic alcohol derivatives6a that the dichlorination of these substrates (8, Eq. 2) tends to be unselective in most cases, but can favor the formation of the undesired syn, syn dichloroalcohol products when the hydroxyl group is acylated; therefore, the possibility of using the stereochemistry of the C14 hydroxyl-bearing stereogenic center to introduce the C15 and C16 chlorides (malhamensilipin A numbering) appeared unlikely (Eq. 3). Nonetheless, we felt that the opportunity to begin from compounds of type 11, wherein the diol could be introduced enantioselectively via Sharpless asymmetric dihydroxylation,9 was worthy of study for the ultimate ability to complete the first enantiocontrolled synthesis of a chlorosulfolipid. We hoped that in some context, the stereogenicity of either C13 or C14 might be relayed in a dichlorination reaction of a C15–C16 alkene. Realization of this strategy was key to our stereocontrolled synthesis of malhamensilipin A, as described below.
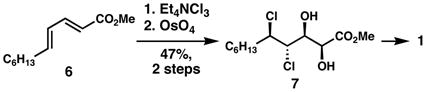 |
[Eq. 1] 5 |
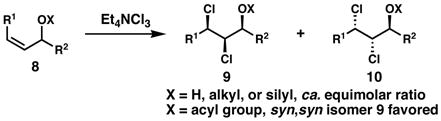 |
[Eq. 2] 6a |
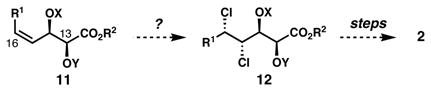 |
[Eq. 3] |
We began our studies with unsaturated ester 13,10 which underwent a slow dihydroxylation (>95% ee) under the modified Sharpless dihydroxylation conditions reported by Bittman for a similar enyne ester substrate (Scheme 1).11 Semireduction of the alkyne using Lindlar’s catalyst was unreliable, but P-2 Ni/ethylenediamine cleanly afforded Z-allylic alcohol 14.12 A variety of derivatives of this diol were studied for diastereoselectivity under alkene dichlorination conditions;13,14 dichlorination of 15, readily prepared by selective nosylation of the α-hydroxyl group,5,15 was highly selective (>10:1 dr) for the stereoisomer required for a synthesis of malhamensilipin A. At this time, we do not have a compelling explanation for the selectivity ostensibly imparted on the reaction by the remote nosyl ester; serendipitously, this group was required for the subsequent base-mediated cis-epoxide formation (15 → 16).15 Reduction of the ester to the aldehyde (17) preceded a Wittig reaction with phosphorane 18,5 which proceeded with only modest stereoselectivity (2:1 Z:E) in the generation of 19. We had previously developed a chloride-induced vinyl epoxide ring-opening reaction that favors inversion of stereochemistry5 in the face of potentially complicating participation of the remote chlorides;4 when applied to the stereoisomeric mixture of Wittig products, this method delivered Z-allylic chloride 20 in 32% yield from aldehyde 17. Stereoselective dichlorination of the remaining alkene completed the stereochemical array of malhamensilipin A. The stereocontrol in this step is consistent with that observed by the Carreira group in their synthesis of 1.4 At this stage, comparison of the 1H NMR signals attributed to the C11–C16 region of 21 with those from a desulfated derivative of malhamensilipin A2b revealed a nearly perfect match with respect to chemical shift and 1H–1H coupling constants, providing strong evidence that we had obtained the natural stereochemical relationships.
Scheme 1.

Synthesis of Malhamensilipin A
To complete the synthesis, introduction of the C14 sulfate and the E-chlorovinyl sulfate was required. To this end, silyl ether 21 was treated directly with chlorosulfonic acid followed by saturated aqueous NaHCO3 to generate bis-sulfate 22. In a reaction that we had studied in very simple model systems,2b and that caused us some degree of trepidation, we subjected heptachloride bis-sulfate 22 to an excess of LDA in THF. Examination of the 1H NMR spectrum of the crude reaction mixture revealed the formation of a single alkene corresponding to malhamensilipin A; an undesired side product with no alkene was produced in equimolar quantities.16 Given the presence of β,β-dichlorinated terminal sulfates in the related lipids of O. danica (see 1), it is plausible that this reaction has its counterpart in the biosynthesis of malhamensilipin A. That such a strong base was required to effect elimination suggests that the chlorovinyl sulfate is not an artifact of isolation. Malhamensilipin A (2) of approximately 90% purity could be isolated in 20–30% yield,17 and demonstrated spectral data that matched those obtained from a natural sample, thereby confirming the recent structural revision.2b
The enantioselective synthesis of malhamensilipin A was completed in 11 steps (longest linear sequence) from known enyne 13 with high stereocontrol in the introduction of all of the polar substituents. The selectivity imposed on the dichlorination of alkene 15 by the remote nosyl ester and the regio- and diastereocontrol of the final elimination reaction are novel aspects of this work; experiments to understand these processes are ongoing and will be described in a more detailed disclosure.
Supplementary Material
Acknowledgments
We thank the School of Physical Sciences, UC Irvine for startup funds and a Faculty Research Grant to C.D.V. G.M.S. was the recipient of a UCI GAANN Fellowship and D.K.B. was supported by an Eli Lilly Graduate Fellowship. A.R.P. and W.H.G. were supported by the NIH (NS053398). We thank Ariane Jansma for assistance with NMR experiments and Dr. John Greaves for mass spectrometric assistance. Prof. Robert Bittman and Dr. Hoe-Sup Byun (Queens College, CUNY) are acknowledged for advice regarding the dihydroxylation of 13.
Footnotes
Supporting Information Available: Experimental details, characterization data, and copies of NMR spectra are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Elovson J, Vagelos PR. Proc Natl Acad Sci USA. 1969;62:957–963. doi: 10.1073/pnas.62.3.957. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haines TH, Pousada M, Stern B, Mayers GL. Biochem J. 1969;113:565–566. doi: 10.1042/bj1130565. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Elovson J, Vagelos PR. Biochemistry. 1970;9:3110–3126. doi: 10.1021/bi00818a002. [DOI] [PubMed] [Google Scholar]; (d) Haines TH. In: Lipids and Biomembranes of Eukaryotic Microorganisms. Erwin JA, editor. Academic Press; New York: 1973. pp. 197–232. [Google Scholar]; (e) Haines TH. Annu Rev Microbiol. 1973;27:403–412. doi: 10.1146/annurev.mi.27.100173.002155. [DOI] [PubMed] [Google Scholar]
- 2.(a) Chen JL, Proteau PJ, Roberts MA, Gerwick WH, Slate DL, Lee RH. J Nat Prod. 1994;57:524–527. doi: 10.1021/np50106a015. [DOI] [PubMed] [Google Scholar]; (b) Pereira AR, Byrum T, Shibuya GM, Vanderwal CD, Gerwick WH. J Nat Prod. doi: 10.1021/np900672h. Published online 25 January 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Ciminiello P, Fattorusso E, Forino M, Magno S, Di Rosa M, Ianaro A, Poletti R. J Org Chem. 2001;66:578–582. doi: 10.1021/jo001437s. [DOI] [PubMed] [Google Scholar]; (b) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Di Rosa M, Ianaro A, Poletti R. J Am Chem Soc. 2002;124:13114–13120. doi: 10.1021/ja0207347. [DOI] [PubMed] [Google Scholar]; (c) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Magno S. Pure Appl Chem. 2003;75:325–336. [Google Scholar]; (d) Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Magno S, Di Meglio P, Ianaro A, Poletti R. Tetrahedron. 2004;60:7093–7098. [Google Scholar]; (e) Ciminiello P, Fattorusso E. Eur J Org Chem. 2004:2533–2551. [Google Scholar]
- 4.Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]
- 5.Bedke DK, Shibuya GM, Pereira A, Gerwick WH, Haines TH, Vanderwal CD. J Am Chem Soc. 2009;131:7570–7572. doi: 10.1021/ja902138w. [DOI] [PubMed] [Google Scholar]
- 6.(a) Shibuya GM, Kanady JS, Vanderwal CD. J Am Chem Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]; (b) Kanady JS, Nguyen JD, Ziller JW, Vanderwal CD. J Org Chem. 2009;74:2175–2178. doi: 10.1021/jo802390e. [DOI] [PubMed] [Google Scholar]
- 7.Yoshimitsu T, Fukumoto N, Tanaka T. J Org Chem. 2009;74:696–702. doi: 10.1021/jo802093d. [DOI] [PubMed] [Google Scholar]
- 8.Nilewski C, Geisser RW, Ebert MO, Carreira EM. J Am Chem Soc. 2009;131:15866–15876. doi: 10.1021/ja906461h. [DOI] [PubMed] [Google Scholar]
- 9.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 10.Ester 13 is known: Dixon DJ, Lucas AC. Synlett. 2004:1092–1094.
- 11.He L, Byun HS, Bittman R. J Org Chem. 2000;65:7627–7633. doi: 10.1021/jo001226n. [DOI] [PubMed] [Google Scholar]
- 12.Brown CA, Ahuja VK. J Chem Soc Chem Commun. 1973:553–554. [Google Scholar]
- 13.Et4NCl3 is our reagent of choice for alkene dichlorination: Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Angew Chem Int Ed. 1997;36:2342–2344.
- 14.Please see the Supporting Information for the results of our dichlorination studies on compounds related to diol 14.
- 15.Fleming PR, Sharpless KB. J Org Chem. 1991;56:2869–2875. [Google Scholar]
- 16.Attempts to isolate and characterize this side product were unsuccessful. Given the lack of alkene-derived signals associated with this product, a C1–C2 alkyne (an alkynyl sulfate) formed via double elimination is a plausible structure. MS analysis of the crude reaction mixture reveals a product with such a mass, but efforts to further characterize it as part of the mixture by 13C NMR and IR did not provide unambiguous evidence in support of this assignment.
- 17.Purification via normal and reverse phase flash chromatography, HPLC, preparative TLC, extractions, and triturations were attempted; separation of 2 from starting material and the major side product, all of which contain two sulfates, proved exceptionally challenging.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.