

Abstract
OBJECTIVE
Apolipoprotein E (ApoE) regulates plasma lipid levels via modulation of lipolysis and serving as ligand for receptor-mediated clearance of triglyceride (TG)-rich lipoproteins. This study tested the impact of modulating lipid delivery to tissues on insulin responsiveness and diet-induced obesity.
RESEARCH DESIGN AND METHODS
ApoE+/+ and apoE−/− mice were placed on high-fat–high-sucrose diabetogenic diet or control diet for 24 weeks. Plasma TG clearance, glucose tolerance, and tissue uptake of dietary fat and glucose were assessed.
RESULTS
Plasma TG clearance and lipid uptake by adipose tissue were impaired, whereas glucose tolerance was improved in control diet–fed apoE−/− mice compared with apoE+/+ mice after an oral lipid load. Fat mass was reduced in apoE−/− mice compared with apoE+/+ mice under both dietary conditions. The apoE−/− mice exhibited lower body weight and insulin levels than apoE+/+ mice when fed the diabetogenic diet. Glucose tolerance and uptake by muscle and brown adipose tissue (BAT) was also improved in mice lacking apoE when fed the diabetogenic diet. Indirect calorimetry studies detected no difference in energy expenditure and respiratory quotient between apoE+/+ and apoE−/− mice on control diet. Energy expenditure and uncoupling protein-1 expression in BAT were slightly but not significantly increased in apoE−/− mice on diabetogenic diet.
CONCLUSIONS
These results demonstrated that decreased lipid delivery to insulin-sensitive tissues improves insulin sensitivity and ameliorates diet-induced obesity.
Insulin resistance, characterized by reduced responsiveness of peripheral tissues to insulin, plays a major role in the development of type 2 diabetes (1,2). Although glucose homeostasis still takes the center stage in the definition of insulin resistance, more emphasis has been attributed recently to aberrant lipid metabolism as a contributing factor of this metabolic disorder. The concept of “lipotoxicity” describes how abnormalities in fatty acid metabolism, resulting in inappropriate storage of lipids in muscle and liver, ultimately lead to the pathogenesis of insulin resistance (3,4). Ectopic lipid accumulation in skeletal muscle and liver may be a result of increased delivery of fatty acid when energy intake exceeds adipose tissue storage capacity (4). On reaching insulin-sensitive tissues, excessive intracellular fatty acid decreased insulin-mediated glucose transport (5-7) through modulation of the insulin-signaling cascade, leading to decreased GLUT-4 translocation to the plasma membrane and reduced intracellular glucose concentrations (8). In contrast, decreasing fatty acid uptake into skeletal muscle with deletion of fatty acid transporters improved insulin sensitivity and protected mice from developing diet-induced insulin resistance (9,10).
Dietary and endogenously synthesized lipid nutrients are also delivered to peripheral tissues via triglyceride (TG)-rich lipoproteins (TGRLs) such as chylomicrons, VLDL, and their remnant lipoproteins. The lipids associated with TGRLs are taken up by cells and tissues via two mechanisms: one requiring lipoprotein lipase (LpL)–catalyzed hydrolysis of the TGs to fatty acid at the cell surface followed by the subsequent cellular uptake of nonesterified fatty acid and the second involving whole–lipoprotein particle uptake via receptor-mediated pathway(s). Perturbations in one or both of these pathways have been shown to significantly influence sensitivity to diet-induced obesity and insulin resistance in several genetically modified mouse models. For example, the absence of apolipoprotein (apo) CIII, a natural inhibitor of LpL, enhances LpL-dependent uptake of TGRL-derived fatty acids by adipose tissue, resulting in elevated susceptibility to diet-induced obesity and insulin resistance (11). In contrast, mice overexpressing apoCI are protected from obesity and insulin resistance (12), apparently through a direct interaction of apoCI with nonesterified fatty acids to prevent their cellular uptake (13). Although apoCI also inhibits TRGL binding to hepatic lipoprotein receptors (14,15), whether receptor-mediated TRGL uptake by insulin-sensitive high-energy–metabolism tissue plays a role in modulating susceptibility to obesity and diabetes is less clear from these studies. The VLDL receptor–deficient mice have also been shown to be less susceptible to diet-induced obesity and insulin resistance (16). Defective tissue lipid uptake in this latter model was attributed to reduced LpL activity (17).
The current study took advantage of the apoE-deficient (apoE−/−) mice, with well-established defects in receptor-mediated lipoprotein uptake (18,19), to investigate whether the suppression of whole particle lipoprotein uptake by high energy metabolism tissues affects insulin responsiveness and diet-induced obesity.
RESEARCH DESIGN AND METHODS
Male apoE −/− and apoE +/+ C57BL/6 mice (The Jackson Laboratories, Bar Harbor, ME) were housed in a pathogen-free environment with all animal care and experimental procedures conforming to institutional guidelines for animal experiments. Mice were initially fed the D12328 control diet from Research Diets (New Brunswick, NJ) containing 16 kcal% protein, 73 kcal% corn starch, and 11 kcal% fat. Subsequently, apoE−/− and apoE+/+ mice at 3 months of age were randomly divided into four groups (n = 8) and placed on either the control diet or the D12331 diabetogenic diet from Research Diets composed of 16.4 kcal% protein, 26 kcal% sucrose, and 58 kcal% fat (20). Whole-body fat and lean mass were measured in conscious mice using 1H magnetic resonance spectroscopy (EchoMRI-100; Echomedical Systems, Houston, TX).
Biochemical assays
Intraperitoneal glucose tolerance test (ipGTT) was performed by injection of glucose (2 g/kg) after 6 h or overnight fast. Blood samples were collected immediately before and 15, 30, 60, and 120 min after injection. Plasma TG clearance was determined by gastric gavage of olive oil (5 μl/g body wt) after a 16-h overnight fast. Blood samples were collected immediately before and at 1, 2, and 3 h after oral bolus load. Blood glucose was determined with an Accu Check Active Glucometer (Roche Applied Science, Penzberg, Germany). Plasma insulin levels were measured with the Ultra Sensitive Rat Insulin ELISA kit (Crystal Chem, Chicago, IL) using rat insulin as the standard. Plasma TGs and cholesterol levels were measured by enzymatic assay kits (Thermo Electron, Waltham, MA). Samples were analyzed individually except for lipoprotein separation in which pooled samples (0.25 ml) from five animals per group were subjected to fast-performance liquid chromatography (FPLC) gel filtration on two Superose 6 columns connected in series.
Measurement of 2-deoxy-d-[1,2-3H]glucose uptake by target tissues
Mice were fasted overnight and then gavaged with olive oil (5 μl/g body wt). After 45 min, 2-deoxy-d-[1,2-3H]glucose (NEN Life Science Products, Boston, MA) was administered as an intravenous bolus of 0.5 μCi/g body wt. Plasma samples were collected to measure glucose levels and radioactivity determinations for calculation of specific 3H radioactivity per unit of glucose in plasma. Animals were killed after 15 min and extensively perfused with ice-cold saline, and tissues were excised, weighed, and solubilized with NCSR-II Tissue Solubilizer (Amersham, Arlington Heights, IL) for radioactivity determinations in a liquid scintillation counter. Data are reported as total amount of glucose, based on the amount of radioactivity, taken up per milligram tissue. The phosphorylation of 2-deoxy[3H]glucose was not assessed in this study because the contribution of glucose kinase is expected to be minimal during the 15-min measurement period.
Determination of energy balance
ApoE −/− and apoE +/+ mice were acclimated to respiratory chambers for 4 days before measurements. Energy expenditure, respiratory quotient, food intake, fluid intake, and locomotor activity were measured simultaneously over a 24-h period using a customized 32-cage, Indirect Calorimetry System combined with Drinking and Feeding Monitor and TSE ActiMot system (TSE-Systems, Germany). The volume of oxygen consumed (Vo2) and carbon dioxide produced (Vco2) each hour was measured using a paramagnetic oxygen sensor and a spectrophotometric CO2 sensor over a 24-h period. Respiratory quotient was calculated as the ratio of Vco2 produced to Vo2 consumed. Energy expenditure was calculated as the product of calorific value of O2 and Vo2 per kilogram body weight, where the calorific value of O2 = 3.815 + (1.232) (respiratory quotient). Total calories expended were calculated to determine daily fuel use. The proportion of protein, fat, and carbohydrate that was used during that 24-h period was calculated under the assumption that protein use is equivalent to protein intake for adult animals (21,22). The percent of daily fuel use derived from carbohydrate and fat was determined using formulae and constants derived by Elia and Livesey (23). Daily caloric intake was calculated by multiplying the mass of daily food intake in grams by the physiological fuel value of the diet in kilocalories per gram. Complete energy balance measurements were taken at the beginning and at 4 and 16 weeks after initiation of the dietary treatment.
Immunoblot analysis
An aliquot of 25 μl from each FPLC fraction was separated by SDS-PAGE, transferred to polyvinylidene fluoride paper, and blotted with antibodies against apoAI (Novus Biologicals, Littleton, CO) and apoE (Santa Cruz Biotechnology, Santa Cruz, CA). Expression of uncoupling protein-1 (UCP-1) in brown adipose tissue (BAT) was analyzed by immunoblotting tissue homogenates in radioimmunoprecipitation assay buffer with UCP-1–specific antibodies (Calbiochem, San Diego, CA). Immunoreactive proteins were detected by incubating the blots with fluorescently labeled species-specific secondary antibodies (Molecular Probes, Eugene, OR) and visualized by the Infrared Imaging System Odyssey (Li-Cor Biosciences, Lincoln, NE).
Statistical analysis
Numerical data are expressed as means ± SE. An unpaired two-tailed Student’s t or one-way ANOVA test with Bonferroni’s multiple comparison test was used for statistical analysis of one-group variables. P values <0.05 were considered significant.
RESULTS
The apoE−/− mice had markedly increased fasting plasma cholesterol levels compared with apoE+/+ mice when fed either control or diabetogenic diets (Table 1). Fasting plasma TG levels were markedly higher in apoE−/− mice compared with apoE+/+ mice, regardless of the diet (Table 1). Diabetogenic diet increased fasting TG levels significantly in both groups of mice but to a larger extent in apoE−/− mice. Although we found similar TG levels in nonfasted apoE+/+ and apoE−/− mice under control dietary conditions, the nonfasting TG levels were higher in apoE−/− mice compared with apoE+/+ mice when they were fed diabetogenic diet. Fractionation of plasma lipoproteins by FPLC showed markedly increased cholesterol levels in the VLDL and IDL/LDL fractions of apoE−/− mice compared with apoE+/+ mice (Fig. 1A and B) as described previously (24). Feeding the diabetogenic diet resulted in a marked increase in HDL cholesterol in both apoE−/− and apoE+/+ mice compared with control diet–fed groups (Fig. 1A and B). In addition, significant increase in cholesterol level was observed in fractions 20–26 of diabetogenic diet–fed apoE+/+ mice (Fig. 1A). Because of the expected overlaps in particle sizes between IDL/LDL and HDL1 lipoproteins, immunoblot analysis was performed with antibodies against apoAI and apoE, two proteins present in HDL1 but not in LDL. Results confirmed that the cholesterol increase in these fractions was due primarily to increase levels of HDL1 (Fig. 1C). The diabetogenic diet–induced elevation of HDL1 level was not apparent in apoE−/− mice nor in mice fed control diet (Fig. 1B and C). The increased VLDL and decreased HDL1 concentrations in apoE−/− mice are consistent with the well-established defects in receptor-mediated lipid transport pathways associated with apoE deficiency. These results also established that apoE−/− mice are appropriate models for investigating whether suppression of receptor-mediated lipid transport to high–energy metabolism tissues affects insulin responsiveness and diet-induced obesity.
TABLE 1.
Metabolic characteristics of apoE−/− and apoE+/+ mice
| Control diet |
Diabetogenic diet |
|||
|---|---|---|---|---|
| ApoE−/− | ApoE+/+ | ApoE−/− | ApoE+/+ | |
| Fasted state (6 h) | ||||
| TG (mg/dl) | 73.2 ± 21*† | 44 ± 18*†‡ | 101 ± 24*†‡ | 60 ± 11*†‡ |
| Cholesterol (mg/dl) | 550 ± 137* | 77 ± 39*† | 743 ± 224* | 235 ± 50*† |
| Glucose (mg/dl) | 132 ± 17*†‡ | 155 ± 16*†‡ | 188 ± 40† | 211 ± 35† |
| Insulin (ng/ml) | 0.7 ± 0.2†‡ | 0.8 ± 0.2‡ | 4.8 ± 2.9*† | 7.6 ± 1.8* |
| Fed state | ||||
| TG (mg/dl) | 101 ± 32 | 102 ± 31‡ | 138 ± 27*‡ | 108 ± 20*‡ |
| Cholesterol (mg/dl) | 564 ± 138*† | 104 ± 30*† | 813 ± 217*† | 237 ± 49*† |
| Glucose (mg/dl) | 150 ± 12*†‡ | 182 ± 28*‡ | 195 ± 25† | 186 ± 19 |
| Insulin (ng/ml) | 1.1 ± 0.2*†‡ | 1.5 ± 0.2*†‡ | 5.9 ± 3.3*† | 10.5 ± 4.3*† |
Data are means ± SD from blood samples collected from eight mice per group at week 12 of the study.
P < 0.05 apoE−/− vs. apoE+/+ mice;
P < 0.05 control vs. diabetogenic diet within genotype;
P < 0.05 fasted vs. fed state within genotype.
FIG. 1.
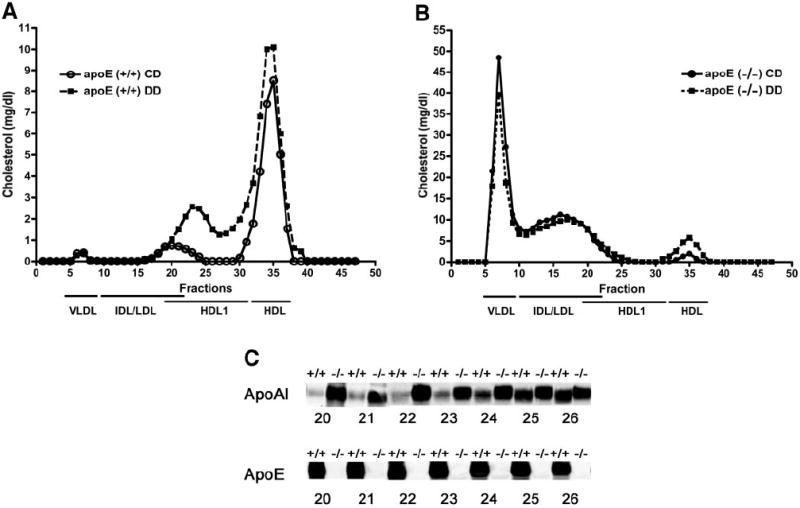
Analysis of plasma lipoproteins in apoE−/− and apoE+/+ mice. Pooled plasma (0.25 ml) from five apoE+/+ (A) and five apoE−/− (B) mice maintained on control diet (○) or diabetogenic diet (■) was applied to two FPLC Superose columns, and 0.5-ml fractions were collected for cholesterol determinations. According to calibration with standard lipoproteins, the peak fractions 7–9 represent VLDL, fractions 15–22 are mainly IDL/LDL particles, and fractions 20–26 contain mainly large HDL1. The second peak (fractions 30–37) is mainly HDL. C: Immunoblot analysis of apoAI and apoE in fractions 20–26 from apoE+/+ and apoE−/− mice.
The control diet–fed apoE−/− mice exhibited lower fasting glucose but not insulin levels compared with apoE+/+ mice (Table 1). Importantly, both glucose and insulin levels were significantly lower in apoE−/− mice than in apoE+/+ mice during the fed state when the animals were maintained on the control diet (Table 1), suggesting that apoE−/− mice on control diet were more insulin sensitive than apoE+/+ mice. Diabetogenic diet feeding increased insulin levels in both groups of mice under both fasting and fed states, but the increases were significantly less in apoE−/− mice compared with apoE+/+ mice (6.86- vs. 9.5-fold in fasted state and 5.36- vs. 7-fold in fed state, respectively). Although glucose levels were similar in both groups of diabetogenic diet–fed mice, the markedly lower fasting and fed insulin levels in apoE−/− mice are indicative of their reduced sensitivity to diet-induced insulin resistance. These latter findings were confirmed by ipGTT after a 6-h fasting period. In these experiments, the response to glucose was found to be significantly improved in apoE−/− mice compared with apoE+/+ mice regardless of the diet (Fig. 2A).
FIG. 2.
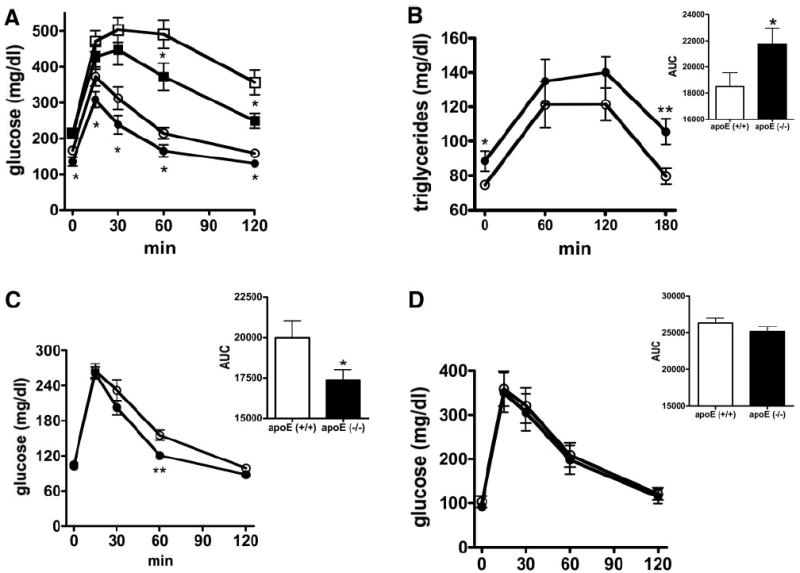
Glucose and lipid tolerance tests in apoE−/− and apoE+/+ mice. The apoE−/− (filled symbols) and apoE+/+ (open symbols) mice were maintained on control (n = 20; circles) or diabetogenic (n = 8; squares) diets. ipGTT was performed after a 6-h fasting period (A). Alternatively, the mice were fasted overnight (B–D) before determination of plasma TG levels in response to oral fat load (B) or ipGTT in the presence (C) or absence (D) of an oral lipid load. Calculations of area under curve (AUC) of the data presented in B–D are included in the insets to each figure. Data are expressed as means ± SE. *P < 0.05; **P < 0.005.
The highly efficient glucose tolerance mechanism in the lipid transport–defective apoE−/− mice suggested that the delayed uptake of lipids into tissues during the postprandial state may reduce intracellular lipid accumulation and thereby lead to increased insulin sensitivity. This hypothesis was examined directly by performing ipGTT with or without a simultaneously administered oral fat tolerance test (oFTT) in mice on control diet after an overnight fast. This experimental design allowed us to compare glucose disposal in a truly fasted state with or without an acute influx of lipids. We found that whereas plasma TG clearance after an oral lipid load was significantly impaired in apoE−/− mice (Fig. 2B), glucose tolerance was notably improved in apoE−/− mice compared with apoE+/+ mice when an oFTT was conducted simultaneously (Fig. 2C). In contrast, no difference in glucose response between apoE−/− and apoE+/+ mice was observed when ipGTT was performed after an overnight fast without concomitant influx of dietary lipids (Fig. 2D). The latter results contrasted with results obtained after a 6-h fasting period (Fig. 2A). The difference is likely due to ongoing lipid uptake and metabolism in apoE+/+ mice 6 h after feeding, which was not apparent in apoE−/− mice with defective receptor-mediated lipid uptake.
The impact of defective lipid uptake on glucose disposal by various tissues was investigated by intravenous GTT using deoxy[3H]glucose as a tracer after its infusion with lipids into apoE+/+ and apoE−/− mice. Based on our previous observations that circulating glucose levels peaked at 2 min and were eliminated thereafter by first-order kinetics after intravenous administration of glucose with plasma glucose levels reducing to 50% levels after 15 min (25), we examined the disposition of the deoxy[3H]glucose in various tissues 15 min after intravenous injection as an indication of the rate of glucose uptake by each tissue. We observed a significant increase in glucose uptake by BATs and skeletal muscle but not the heart of apoE−/− mice compared with apoE+/+ mice on diabetogenic diet (Fig. 3A).
FIG. 3.
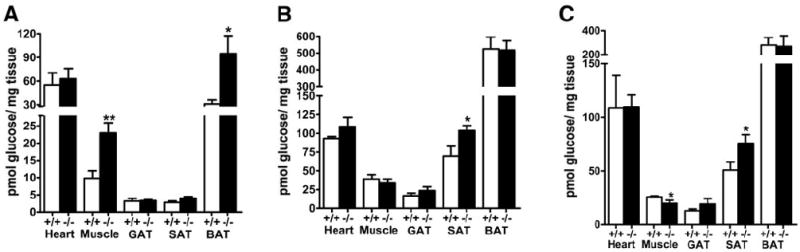
Tissue uptake of deoxy-[3H]glucose by apoE−/− and apoE+/+ mice. A: 2-deoxy-[3H]glucose uptake by heart, skeletal muscle, gonadal (GAT), SAT, and BAT of diabetogenic diet–fed apoE+/+ (□) and apoE−/− (■) mice 15 min after intraperitoneal injection of the radiolabeled tracer. B and C: Tissue uptake of deoxy-[3H]glucose by control diet–fed apoE+/+ (□) and apoE−/− (■) mice with or without an oral fat load, respectively. Data are expressed as means ± SE for n = 5 mice per group. *P < 0.05; **P < 0.005.
Additional experiments were also conducted to assess the impact of apoE-mediated lipid uptake on glucose metabolism in various tissues by analyzing tissue distribution of deoxy[3H]glucose with and without a concomitant oral lipid load in apoE+/+ and apoE−/− mice maintained on control low-fat diet. When deoxy[3H]glucose was administered in the presence of an oral fat load, a significant increase in deoxy[3H]glucose uptake by the subcutaneous white adipose tissue (SAT) was observed in control diet–fed apoE−/− mice compared with that in apoE+/+ mice (Fig. 3B). The uptake of deoxy[3H]glucose by other tissues was similar between both groups under these conditions. The adipose-specific difference in deoxyglucose uptake between apoE+/+ and apoE−/− mice with a fatty meal may explain the slight but significant improvement of glucose tolerance in the apoE−/− mice when glucose was administered together with a fat load. Interestingly, whereas the difference in SAT uptake of deoxy[3H]glucose between control diet–fed apoE+/+ and apoE−/− mice was sustained in the absence of an oral lipid load, significantly less deoxyglucose uptake was observed in the skeletal muscle of apoE−/− mice compared with that in apoE+/+ mice in the absence of an oral fat load (Fig. 3C). The latter observation is consistent with data showing no difference in glucose tolerance between chow-fed apoE+/+ and apoE−/− mice in the absence of an oral lipid load.
The decrease in postprandial fat clearance in apoE−/− mice compared with apoE+/+ mice suggested that the apoE−/− mice may also be resistant to diet-induced adiposity. Therefore, body weight and fat mass of apoE−/− and apoE+/+ mice were analyzed after feeding them with either control or diabetogenic diet. Despite similar body weights between the two groups of animals during regular control diet feeding, the apoE−/− mice had significantly lower whole-body fat mass compared with that observed in apoE+/+ mice (Fig. 4A). The apoE−/− mice also gained significantly less whole-body fat mass after diabetogenic diet feeding compared with apoE+/+ mice, resulting in significantly reduced total body weights after 16 weeks (Fig. 4A and B). The differences in fat mass and body weight gain between apoE+/+ and apoE−/− mice were not related to major differences in energy use, because both groups of mice showed similar energy expenditure (base on lean body mass) when fed either the basal or diabetogenic diet (Fig. 5A and B). Caloric intake and respiratory quotient were also similar between apoE+/+ and apoE−/− mice on both diets (Fig. 5C and D; Supplemental Figure S1, which is detailed in the online appendix [available at http://dx.doi.org/10.2337/db07-0403]). Interestingly, a slight increase in UCP-1 expression in BAT of apoE−/− mice was consistently observed, but the increase did not reach statistical significance (Fig. 5E).
FIG. 4.
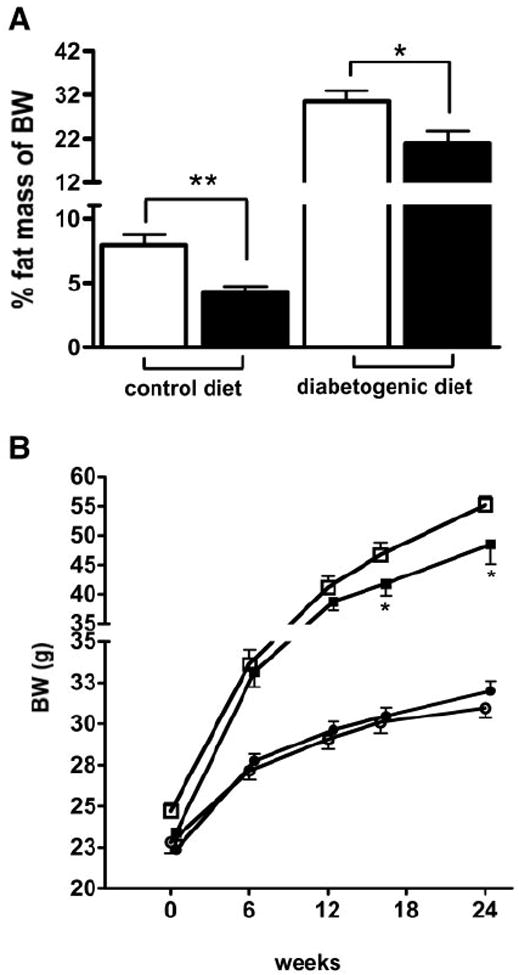
Whole-body fat, as percentage of whole-body weight (BW) at week 16 of the study (A) and body weight distribution (B) in apoE−/− (filled bars) and apoE+/+ (open bars) mice on control (circles) and diabetogenic diet (squares). Data are expressed as means ± SE for n = 8 mice per group. *P < 0.05; **P < 0.005.
FIG. 5.
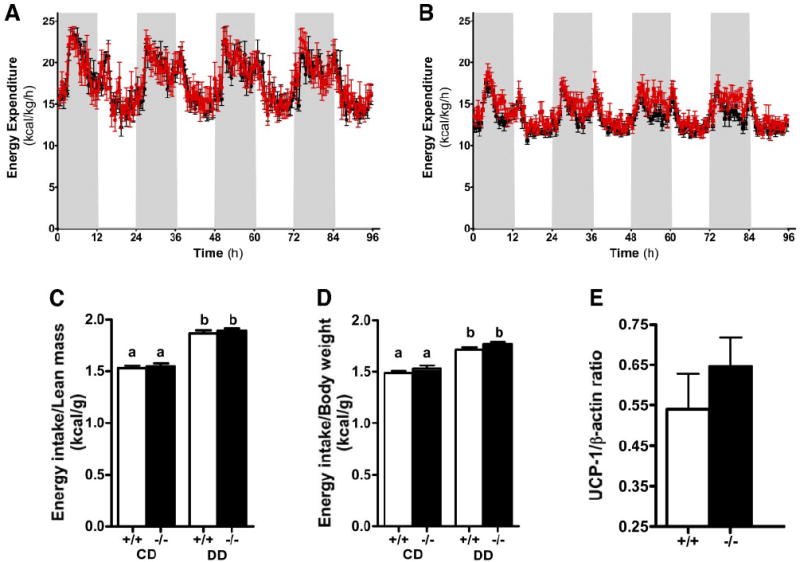
Energy expenditure in apoE−/− (red tracings) and apoE+/+ (black tracings) mice on control diet (CD) (A) and diabetogenic diet (DD) (B) per kilogram lean body mass during dark (gray shaded areas) and light (white shaded areas) phases at week 16 of the study. C and D: Energy intake per gram lean body mass and gram body weight, respectively, of apoE+/+ and apoE−/− mice fed control diet or diabetogenic diet. E: UCP-1 protein levels normalized to β-actin levels in BATs of apoE+/+ and apoE−/− mice maintained on the diabetogenic diet at week 16 of the study. a and b denote differences between the bars at P < 0.05.
DISCUSSION
Defective receptor-mediated plasma lipoprotein clearance is a well-established hallmark of apoE deficiency in mice (26). In addition, the apoE−/− mice have also been reported to have altered immune functions (27) with an exaggerated inflammatory response to infection (28) and an age-dependent neurodegeneration of the central nervous system (29). The present study reveals that apoE−/− mice are more insulin sensitive in skeletal muscle and BAT and have reduced adiposity and insulin resistance compared with apoE+/+ mice when they are challenged with a diet rich in fat and sucrose. The increased insulin sensitivity and resistance to diet-induced obesity in the apoE−/− mice is probably unrelated to its altered immune functions because increased inflammatory response is generally associated with diet-induced obesity and insulin resistance (30,31). The resistance to diet-induced obesity and improved insulin sensitivity of apoE−/− mice are also unlikely due to the altered functions in the central nervous system, because 3-month-old mice were used in the current study and neurological functions were reported to be normal in 8-month-old apoE−/− mice (32) with neurodegeneration observed only in older apoE−/− mice (29). The normal food intake and locomotive activities observed in our apoE−/− mice are consistent with this interpretation. Therefore, the obesity resistance and improved insulin sensitivity in apoE−/− mice reported in the current study is most likely due to reduced lipid transport to insulin-sensitive tissues, ameliorating diet-induced obesity and insulin resistance in these animals.
Two independent research groups have previously shown that apoE−/− mice have lower body fat and reduced adipocyte size compared with apoE+/+ mice (33,34). However, their results in diet-induced obesity experiments were inconsistent. Chiba et al. (34) found that apoE−/− mice in the ob/ob background failed to gain weight compared with apoE+/+ ob/ob mice on a high-fat diet, with most of the difference in body weight attributed to the difference in fat mass. These investigators used food intake restriction analysis to show that food intake did not play a role in the observed difference in body weight. In contrast, Schreyer et al. (33) reported no difference in body weight gain between apoE−/− and apoE+/+ mice fed a high-fat–high-sucrose diet, but their study showed that food intake was significantly increased in apoE−/− mice compared with apoE+/+ mice. Of note, they also detected a substantial decrease in adiposity despite the increased food intake in apoE−/− mice compared with apoE+/+ mice fed a high-fat–high-sucrose diet. Our study confirmed earlier findings that apoE−/− mice have a significant lower body fat percentage compared with apoE+/+ mice. In contrast to the results published by Schreyer et al. (33), we detected significant lower body weights in apoE−/− compared with apoE+/+ mice after 16 weeks of feeding diabetogenic diet with no differences in caloric intake between the two groups. Our findings indicate that the observed difference in body weight was largely due to a difference in fat mass (Fig. 4A).
The reduced body fat percentage in apoE−/− mice may be explained by impaired TG clearance from circulation and decreased lipid uptake by adipose tissue after an oral lipid load (Figs. 2B and 3). Three distinct mechanisms may play a role in the observed impaired lipid clearance from circulation and delivery to target tissues. First, the absence of apoE impaired receptor-mediated clearance of TGRLs from circulation (26). Second, apoE−/− mice were reported to be resistant to heparin-induced lipolysis (35), and LpL activity in these animals was moderately reduced (35). Our data showing small but statistically significant delay in postprandial TG clearance and elevated plasma TG levels in apoE−/− mice compared with those observed in apoE+/+ mice are consistent with the earlier reports. The moderately decreased TGRL lipolysis and clearance mechanism may decrease lipid uptake by the fat tissues. Third, the impaired hepatic VLDL secretion in apoE−/− mice (36,37) may also result in reduced amount of lipids transported from the liver to target tissues. Data presented in the online appendix (Supplemental Fig. S2) confirming previously reported results of TG accumulation in liver of apoE−/− mice (38,39) are supportive of this possibility. All three of these mechanisms may be working together in conferring the resistance to diet-induced obesity in the apoE−/− mice. Finally, fat absorption has also been reported to be reduced in apoE−/− mice (40), although this observation was not consistently observed in other studies (33,41). Regardless of whether fat absorption is impaired in apoE−/− mice, their elevated plasma TG and lower glucose and insulin levels in both fasted and fed states, even under basal dietary conditions, indicated that abnormalities in plasma lipid transport are independent variables in modulating sensitivity to diet-induced obesity and insulin resistance.
Recent evidence suggested that resistance to insulin-stimulated glucose metabolism is not a primary event in obesity but is secondary to lipid accumulation in target tissues resulting from full responsiveness to insulin-stimulated lipogenic activity (3,4). Our results demonstrating similar glucose tolerance between overnight-fasted apoE+/+ and apoE−/− mice despite their difference in adiposity, lower glucose and insulin levels in apoE−/− mice compared with apoE+/+ mice during the fed state, and better glucose tolerance in apoE−/− mice than in apoE+/+ mice after an oral lipid load are consistent with this hypothesis. Based on a novel concept that resistance to insulin-stimulated glucose metabolism might be a protective mechanism that reduces lipid-induced damage to tissue by excluding glucose from cells, and therefore decreasing glucose-derived lipogenesis (42-45), we interpreted the observed deterioration of glucose tolerance after an oral lipid load in the apoE+/+ mice as an acute measure to prevent excessive intracellular fat storage. In contrast, in the absence of apoE, lipid uptake may be impaired after an oral fat load, resulting in reduced lipid accumulation in tissues, and consequently, changes in insulin-stimulated glucose metabolism may not be necessary to prevent lipid-induced damage. The same decrease of lipid delivery in the absence of apoE may well be the mechanism responsible for the observed reduction in diet-induced adiposity and insulin resistance of apoE−/− mice.
Results reported in the current study also showed no detectable difference in food intake between apoE−/− and apoE+/+ mice fed control or diabetogenic diet. Nevertheless, total body fat was reduced in apoE−/− mice, and body weight gain was lower in apoE−/− mice fed a diabetogenic diet. These findings suggested that a potential increase in energy expenditure may account for the surplus in energy intake that is not stored in adipose tissue of apoE−/− mice. However, indirect calorimetry analysis of apoE−/− and apoE+/+ mice showed that energy expenditure was similar between apoE−/− and apoE+/+ mice on control diet throughout the study. Whereas the apoE−/− mice exhibited a slight increase in energy expenditure during the dark phase compared with apoE+/+ mice after 16 weeks on diabetogenic diet, this difference did not reach statistical significance. We hypothesize that a difference in energy metabolism that is below the detection limit of the generally insensitive 24-h indirect calorimetry, but nevertheless relevant for chronic body composition development, is responsible for the differences in energy balance, which are leading to different levels of adiposity. A trend toward increased UCP-1 expression in BAT of apoE−/− mice compared with that in apoE+/+ mice is consistent with this possibility. Additionally, the prolonged retention of dietary lipids in circulation may also contribute significantly to the decreased adiposity in apoE−/− mice compared with that observed in apoE+/+ mice.
Overall, our findings demonstrated that lipid delivery to adipose tissue is impaired in the absence of apoE, and as a consequence, diet-induced adiposity and insulin resistance is reduced. Although we cannot rule out a potential role of intracellular apoE in modulating adiposity and tissue insulin sensitivity, nevertheless these results implicated that modulation of TGRL clearance can be a potential treatment strategy to ameliorate diet-induced obesity and insulin resistance. Such an approach must be taken cautiously because impaired TGRL clearance would lead to hyperlipidemia and increased risk of premature coronary artery disease. The selective inhibition of lipid partitioning to insulin-sensitive tissues without incurring hyperlipidemia is necessary for alleviation of diet-induced metabolic disorders. In this regard, the current study revealed that deficiency of the ligand for TGRL transport, namely apoE, is effective in reducing lipid-induced obesity and diabetes but incurring hyperlipidemia. Identifying the receptors on each tissue responsible for mediating apoE-dependent lipid uptake may be valuable in identifying specific targets for reducing diet-induced obesity and diabetes without the adverse effects of hyperlipidemia and premature atherosclerosis.
Supplementary Material
Acknowledgments
D.Y.H. has received National Institutes of Health Grant R01-DK-74932. M.H.T. has received National Institutes of Health Grant R01-DK-69987. S.H. has received Scientist Development Award 0635079N from the American Heart Association.
We thank Jessica Nickel, Joe Drennan, and Lauren Wancata for performing the metabolic assays.
- apo
apolipoprotein
- BAT
brown adipose tissue
- FPLC
fast-performance liquid chromatography
- ipGTT
intraperitoneal glucose tolerance test
- LpL
lipoprotein lipase
- oFTT
oral fat tolerance test
- SAT
subcutaneous white adipose tissue
- TG
triglyceride
- TGRL
triglyceride-rich lipoprotein
- UCP-1
uncoupling protein-1
Footnotes
Additional information for this article can be found in an online appendix at http://dx.doi.org/10.2337/db07-0403.
References
- 1.Kahn BB, Rossetti L. Type 2 diabetes: who is conducting the orchestra? Nat Genet. 1998;20:223–225. doi: 10.1038/3018. [DOI] [PubMed] [Google Scholar]
- 2.Taylor SI. Deconstructing type 2 diabetes. Cell. 1999;97:9–12. doi: 10.1016/s0092-8674(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 3.Unger RH. Minireview. Weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- 4.McGarry JD. Banting Lecture 2001: Dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- 5.Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- 6.Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye JM, Doyle PJ, Iglesias MA, Watson DG, Cooney GJ, Kraegen EW. Peroxisome proliferator–activated receptor (PPAR)-α activation lowers muscle lipids and improves insulin sensitivity in high fat-fed rats: comparison with PPAR-γ activation. Diabetes. 2001;50:411–417. doi: 10.2337/diabetes.50.2.411. [DOI] [PubMed] [Google Scholar]
- 8.Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. doi: 10.1172/JCI5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim JK, Gimeno RE, Higashimori T, Kim HJ, Choi H, Punreddy S, Mozell RL, Tan G, Stricker-Krongrad A, Hirsch DJ, Fillmore JJ, Liu ZX, Dong J, Cline G, Stahl A, Lodish HF, Shulman GI. Inactivation of fatty acid transport protein 1 prevents fat-induced insulin resistance in skeletal muscle. J Clin Invest. 2004;113:756–763. doi: 10.1172/JCI18917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. 2002;109:1381–1389. doi: 10.1172/JCI14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duivenvoorden I, Teusink B, Rensen PC, Romijn JA, Havekes LM, Voshol PJ. Apolipoprotein c3 deficiency results in diet-induced obesity and aggravated insulin resistance in mice. Diabetes. 2005;54:664–671. doi: 10.2337/diabetes.54.3.664. [DOI] [PubMed] [Google Scholar]
- 12.Jong MC, Voshol PJ, Muurling M, Dahlmans VEH, Romijn JA, Pijl H, Havekes LM. Protection from obesity and insulin resistance in mice overexpressing human apolipoprotein C1. Diabetes. 2007;50:2779–2785. doi: 10.2337/diabetes.50.12.2779. [DOI] [PubMed] [Google Scholar]
- 13.Westerterp M, Berbee JFP, Delsing DJM, Jong MC, Gijbels MJJ, Dahlmans VEH, Offerman EH, Romijn JA, Havekes LM, Rensen PCN. Apolipoprotein C-I binds free fatty acids and reduces their intracellular esterification. J Lipid Res. 2007;48:1353–1361. doi: 10.1194/jlr.M700024-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Kowal RC, Herz J, Weisgraber KH, Mahley RW, Brown MS, Goldstein JL. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J Biol Chem. 1990;265:10771–10779. [PubMed] [Google Scholar]
- 15.Jong MC, Dahlmans VE, van Gorp PJ, van Dijk KW, Breuer ML, Hofker MH, Havekes LM. In the absence of the low density lipoprotein receptor, human apolipoprotein CI overexpression in transgenic mice inhibits the hepatic uptake of very low density lipoproteins via a receptor-associated protein-sensitive pathway. J Clin Invest. 1996;98:2259–2267. doi: 10.1172/JCI119036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goudriaan JR, Tacken PJ, Dahlmans VEH, Gijbels MJJ, van Dijk KW, Havekes LM, Jong MC. Protection from obesity in mice lacking the vldl receptor. Arterioscler Thromb Vasc Biol. 2001;21:1488–1493. doi: 10.1161/hq0901.095147. [DOI] [PubMed] [Google Scholar]
- 17.Yagyu H, Lutz EP, Kako Y, Marks S, Hu Y, Choi SY, Bensadoun A, Goldberg IJ. Very low density lipoprotein (VLDL) receptor-deficient mice have reduced lipoprotein lipase activity: possible causes of hypertriglyceridemia and reduced body mass with VLDL receptor deficiency. J Biol Chem. 2002;277:10037–10043. doi: 10.1074/jbc.M109966200. [DOI] [PubMed] [Google Scholar]
- 18.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1998;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 19.Havel RJ, Hamilton RL. Hepatic catabolism of remnant lipoproteins: where the action is. Arterioscler Thromb Vasc Biol. 2004;24:213–215. doi: 10.1161/01.ATV.0000115382.53810.24. [DOI] [PubMed] [Google Scholar]
- 20.Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC, Kuhn CM, Rebuffe-Scrive M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism. 1995;44:645–651. doi: 10.1016/0026-0495(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 21.Rose BS, Flatt WP, Martin RJ, Lewis RD. Whole body composition of rats determined by dual energy X-ray absorptiometry is correlated with chemical analysis. J Nutr. 1998;128:246–250. doi: 10.1093/jn/128.2.246. [DOI] [PubMed] [Google Scholar]
- 22.Flatt J. Use and storage of carbohydrate and fat. Am J Clin Nutr. 1995;61(Suppl 4):952S–959S. doi: 10.1093/ajcn/61.4.952S. [DOI] [PubMed] [Google Scholar]
- 23.Elia M, Livesey G. Energy expenditure and fuel selection in biological systems: the theory and practice of calculations based on indirect calorimetry and tracer methods. World Rev Nutr Diet. 1992;70:68–131. doi: 10.1159/000421672. [DOI] [PubMed] [Google Scholar]
- 24.Zhang SH, Reddick RL, Burkey B, Maeda N. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J Clin Invest. 1994;94:937–945. doi: 10.1172/JCI117460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofmann SM, Dong HJ, Li Z, Cai W, Altomonte J, Thung SN, Zeng F, Fisher EA, Vlassara H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes. 2002;51:2082–2089. doi: 10.2337/diabetes.51.7.2082. [DOI] [PubMed] [Google Scholar]
- 26.Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. 1999;40:1–16. [PubMed] [Google Scholar]
- 27.Laskowitz DT, Lee DM, Schmechel D, Staats HF. Altered immune responses in apolipoprotein E-deficient mice. J Lipid Res. 2000;41:613–620. [PubMed] [Google Scholar]
- 28.Ali K, Middleton M, Pure E, Rader DJ. Apolipoprotein E suppresses the type I inflammatory response in vivo. Circ Res. 2005;97:922–927. doi: 10.1161/01.RES.0000187467.67684.43. [DOI] [PubMed] [Google Scholar]
- 29.Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- 30.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson R, Barnes JC, Bliss TVP, Cain DP, Cambon K, Davies HA, Errington ML, Fellows LA, Gray RA, Hoh T, Stewart M, Large CH, Higgins GA. Behavioural, physiological and morphological analysis of a line of apolipoprotein E knockout mouse. Neuroscience. 1998;85:93–110. doi: 10.1016/s0306-4522(97)00598-8. [DOI] [PubMed] [Google Scholar]
- 33.Schreyer SA, Vick C, Lystig TC, Mystkowski P, LeBoeuf RC. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am J Physiol Endocrinol Metab. 2002;282:E207–E214. doi: 10.1152/ajpendo.2002.282.1.E207. [DOI] [PubMed] [Google Scholar]
- 34.Chiba T, Nakazawa T, Yui K, Kaneko E, Shimokado K. VLDL induces adipocyte differentiation in ApoE-dependent manner. Arterioscler Thromb Vasc Biol. 2003;23:1423–1429. doi: 10.1161/01.ATV.0000085040.58340.36. [DOI] [PubMed] [Google Scholar]
- 35.Zsigmond E, Fuke Y, Li L, Kobayashi K, Chan L. Resistance of chylomicron and VLDL remnants to post-heparin lipolysis in ApoE-deficient mice: the role of apoE in lipoprotein lipase-mediated lipolysis in vivo and in vitro. J Lipid Res. 1998;39:1852–1861. [PubMed] [Google Scholar]
- 36.Kuipers F, Jong MC, Lin Y, Eck M, Havinga R, Bloks V, Verkade HJ, Hofker MH, Moshage H, Berkel TJ, Vonk RJ, Havekes LM. Impaired secretion of very low density lipoprotein-triglycerides by apolipoprotein E-deficient mouse hepatocytes. J Clin Invest. 1997;100:2915–2922. doi: 10.1172/JCI119841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maugeais C, Tietge UJ, Tsukamoto K, Glick JM, Rader DJ. Hepatic apolipoprotein E expression promotes very low density lipoprotein-apolipoprotein B production in vivo in mice. J Lipid Res. 2000;41:1673–1679. [PubMed] [Google Scholar]
- 38.Kuipers F, van Ree JM, Hofker MH, Wolters H, In’t Veld G, Havinga R, Vonk RJ, Princen HMG, Havekes LM. Altered lipid metabolism in apolipoprotein E-deficient mice does not affect cholesterol balance across the liver. Hepatology. 1996;24:241–247. doi: 10.1002/hep.510240138. [DOI] [PubMed] [Google Scholar]
- 39.Mensenkamp AR, Jong MC, van Goor H, van Luyn MJA, Bloks V, Havinga R, Voshol PJ, Hofker MH, van Dijk KW, Havekes LM, Kuipers F. Apolipoprotein E participates in the regulation of very low density lipoprotein-triglyceride secretion by the liver. J Biol Chem. 1999;274:35711–35718. doi: 10.1074/jbc.274.50.35711. [DOI] [PubMed] [Google Scholar]
- 40.Jong MC, Rensen PCN, Dahlmans VEH, van der Boom H, van Berkel TJC, Havekes LM. Apolipoprotein C-III deficiency accelerates triglyceride hydrolysis by lipoprotein lipase in wild-type and apoE knockout mice. J Lipid Res. 2001;42:1578–1585. [PubMed] [Google Scholar]
- 41.Chang S, Zhang SH, Maeda N, Borensztajn J. Hepatic clearance of chylomicron remnants in mice lacking apolipoprotein E. Biochim Biophys Acta. 1994;1215:205–208. doi: 10.1016/0005-2760(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 42.Utsugi T, Ohno T, Ohyama Y, Uchiyama T, Saito Y, Matsumura Y, Aizawa H, Itoh H, Kurabayashi M, Kawazu S, Tomono S, Oka Y, Suga T, Kuro-o M, Nabeshima Y, Nagai R. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism. 2000;49:1118–1123. doi: 10.1053/meta.2000.8606. [DOI] [PubMed] [Google Scholar]
- 43.Unger RH. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol Metab. 2003;14:398–403. doi: 10.1016/j.tem.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 44.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Unger RH. Klotho-induced insulin resistance: a blessing in disguise? Nat Med. 2006;12:56–57. doi: 10.1038/nm0106-56. o:p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.