

Abstract
Background & Aims
Hepatic production and release of endothelin 1 plays a central role in experimental hepatopulmonary syndrome after common bile duct ligation by stimulating pulmonary endothelial nitric oxide production. In thioacetamide-induced nonbiliary cirrhosis, hepatic endothelin 1 production and release do not occur, and hepatopulmonary syndrome does not develop. However, the source and regulation of hepatic endothelin 1 after common bile duct ligation are not fully characterized. We evaluated the sources of hepatic endothelin 1 production after common bile duct ligation in relation to thioacetamide cirrhosis and assessed whether transforming growth factor β1 regulates endothelin 1 production.
Methods
Hepatopulmonary syndrome and hepatic and plasma endothelin 1 levels were evaluated after common bile duct ligation or thioacetamide administration. Cellular sources of endothelin 1 were assessed by immunohistochemistry and laser capture microdissection of cholangiocytes. Transforming growth factor β1 expression and signaling were assessed by using immunohistochemistry and Western blotting and by evaluating normal rat cholangiocytes.
Results
Hepatic and plasma endothelin 1 levels increased and hepatopulmonary syndrome developed only after common bile duct ligation. Hepatic endothelin 1 and transforming growth factor β1 levels increased over a similar time frame, and cholangiocytes were a major source of each peptide. Transforming growth factor β1 signaling in cholangiocytes in vivo was evident by increased phosphorylation and nuclear localization of Smad2, and hepatic endothelin 1 levels correlated directly with liver transforming growth factor β1 and phosphorylated Smad2 levels. Transforming growth factor β1 also stimulated endothelin 1 promoter activity, expression, and production in normal rat cholangiocytes.
Conclusions
Cholangiocytes are a major source of hepatic endothelin 1 production during the development of hepatopulmonary syndrome after common bile duct ligation, but not in thioacetamide-induced cirrhosis. Transforming growth factor β1 stimulates cholangiocyte endothelin 1 expression and production. Cholangiocyte-derived endothelin 1 may be an important endocrine mediator of experimental hepatopulmonary syndrome.
The hepatopulmonary syndrome (HPS) causes impaired oxygenation due to vasodilatation in the pulmonary microcirculation in patients with cirrhosis.1,2 HPS significantly increases mortality in affected patients, and no effective medical treatments are currently available.3 The pathogenesis of HPS remains an area of active investigation.
Chronic common bile duct ligation (CBDL) in the rat is the only recognized model system for the study of HPS.4 In this model, biliary cirrhosis is associated with increased pulmonary endothelial nitric oxide synthase (eNOS) levels, intrapulmonary vasodilatation, and gas exchange abnormalities analogous to human HPS.4,5 Increased hepatic production and plasma levels of endothelin (ET)-1 accompanied by pulmonary microvascular endothelial ETB receptor overexpression occur at the onset of HPS after CBDL and stimulate eNOS-derived nitric oxide production in the microvascular endothelium.6,7 Chronic intravenous ET-1 infusion in animals that develop portal hypertension and increased pulmonary microvascular ETB levels without increased hepatic ET-1 production after partial portal vein ligation also increases pulmonary eNOS levels and triggers HPS.8 Similarly, exogenous ET-1 directly increases eNOS expression and activity in an ETB receptor–dependent manner in pulmonary artery endothelial cells.9 In contrast, hepatic and plasma ET-1 levels do not increase and HPS does not develop in thioacetamide (TAA)-induced nonbiliary cirrhosis despite an increase in pulmonary microvascular ETB receptor expression.10 Together, these observations indicate that hepatic ET-1 production and release is a critical event that drives pulmonary vascular eNOS-derived NO production during the onset of experimental HPS.
The source and regulation of hepatic ET-1 production in experimental HPS have not been clearly defined. CBDL is unique among cirrhosis models in that marked progressive cholangiocyte proliferation occurs over time and could be an important source of ET-1 production. Both human and rodent cholangiocytes have been found to contain ET-1 in cirrhosis, although detailed analysis after CBDL has not been performed.6,11,12 Proliferating biliary epithelium has also been recognized to contribute to the development of fibrosis after CBDL by producing a number of profibrogenic cytokines.13 Among these, transforming growth factor (TGF)-β1 seems to be important, and TGF-β1 can modulate ET-1 production in several hepatic cell types14,15 and in renal ductular epithelium.16 TGF-β1 signaling involves binding to a heteromeric complex of 2 cell-surface receptors: type I and type II. Binding triggers phosphorylation of Smad2 and Smad3, followed by complex formation with Smad4 and translocation to the nucleus.17 Recently, a Smad-binding element has been identified in the ET-1 promoter that is critical for TGF-β1 induction of gene expression.18 One hypothesis based on these observations is that proliferating biliary epithelial cells may be a major source of hepatic ET-1 production and that TGF-β1 might drive ET-1 expression in these cells after CBDL.
The aim of this study was to define whether cholangiocytes are an important source of hepatic ET-1 production after CBDL and to investigate whether TGF-β1 may influence ET-1 expression in these cells and contribute to the development of HPS. To address this aim, we assessed the cellular localization and expression of hepatic ET-1, TGF-β1, and phospho-Smad2 (p-Smad2) after CBDL in comparison to TAA administration, particularly in biliary epithelial cells. We also directly evaluated whether TGF-β1 modulates ET-1 expression in normal rat cholangiocytes (NRCs). Our findings show that cholangiocytes are a major source of both ET-1 and TGF-β1 production after CBDL and that there is a temporal correlation in their expression in these cells. In addition, we found evidence of TGF-β1 activation in cholangiocytes on the basis of analysis of p-Smad2 levels and localization. Finally, we showed that TGF-β1 directly activates ET-1 expression in NRCs.
Materials and Methods
Animals
Male Sprague–Dawley rats (200–250 g; Charles River, Wilmington, MA) were used in all experiments. CBDL was performed as previously described.19,20 Normal control animals underwent mobilization of the common bile duct without ligation. Some rats were intraperitoneally injected with TAA 200 mg/kg body weight (Sigma-Aldrich, St Louis, MO) or saline 3 times each week for 2 or 8 weeks as previously described.10 Five to 8 animals from each group (control; 1-, 2-, and 3-week CBDL; and 2- and 8-week TAA administration) were used. Plasma, tissue, and physiological measurements were similar in sham CBDL and saline-treated controls, and measurements were pooled for analysis. All animals had hepatic biochemical and histological analysis and measurements of portal venous pressure and spleen weight.4,6,8,21 Blood and liver tissues were obtained from each animal. The study was approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and conformed to National Institutes of Health guidelines on the use of laboratory animals.
Liver Histology and Immunohistochemistry
Liver samples were fixed in 10% neutral buffered formalin solution. Paraffin-embedded tissues were sectioned at 5 μm. Sections for histology were stained with the Masson trichrome stain.22 After preparation and blocking, sections were incubated with ET-1 (Peninsula, San Carlos, CA), cytokeratin 19 (CK19; Novocastra, Newcastle Laboratories, UK), a marker for bile epithelial cells, α-smooth muscle actin (α-SMA; Sigma-Aldrich), TGF-β1 (Santa Cruz, Santa Cruz, CA), or p-Smad2 (Ser465/467; Cell Signaling, Beverly, MA) antibodies, washed, and incubated with EnVision-labeled polymer (Dako, Carpinteria, CA). After diaminobenzidine (Biogenex, San Ramon, CA) development, sections were photographed by using an Axiophot microscope (Nikon, Melville, NY). Control sections were incubated with secondary antibody alone.
Arterial Blood Gas Analysis
Arterial blood was drawn from the femoral artery as previously described,21 and blood gas analysis was performed on an ABL 520 radiometer (Radiometer America, Westlake, OH) in the clinical laboratory of the University of Alabama at Birmingham Hospital. The alveolar–arterial oxygen gradient was calculated as 150 – (PaCO2/0.8) – PaO2.
Microsphere Protocol
The pulmonary microcirculation was evaluated with an established technique. Cross-linked (2.5 × 106) colored polystyrene-divinylbenzene microspheres (size range, 5.5–10 μm; Interactive Medical Technologies, Irvine, CA) were injected through a femoral vein catheter after an aliquot of microspheres was removed to verify the numbers and sizes injected. A blood sample withdrawn from a femoral arterial catheter beginning at the time of femoral vein injection measured microspheres passing through the lung microcirculation. Numbers and sizes of microspheres were assessed by using a Leitz Laborlux microscope (Wetzlar, Germany) with a color video imaging and digital analysis system (Image Pro 5.0; Media Cybernetics, Silver Spring, MD) and counted directly. Intrapulmonary shunting was calculated as a percentage of intrapulmonary shunt fraction, as previously described.21
Cell Culture
NRCs (a kind gift of Dr Nicholas LaRusso, Mayo Clinic Rochester, Rochester, MN) were maintained and passaged in Dulbecco’s modified Eagle medium/F-12 medium (GIBCO, Grand Island, NY) containing 0.0l mL/mL nonessential amino acids, 0.01 mL/mL insulin-transferrin-selenium-S, 0.01 mL/mL lipid concentrate, 0.01 mL/mL vitamin solution, 20 μg/mL gentamicin, 393 ng/mL dexamethasone (Sigma-Aldrich), 30 μg/mL bovine pituitary extract, 25 ng/mL epidermal growth factor, 3.4 μg/mL triiodothyrodine (Sigma-Aldrich), 5% NuSerum IV (Becton Dickinson Labware, Lincoln Park, NJ), and 4.1 μg/mL forskolin (Sigma-Aldrich) on Collagen I Cellware dishes or plates (Becton Dickinson Labware). When cells reached 70% confluence, they were incubated in Dulbecco’s modified Eagle medium without any serum or supplements and stimulated with TGF-β1 (R&D Systems, Minneapolis, MN) at various concentrations (0.5, 2.5, 5.0, and 7.5 ng/mL) for various times (2, 4, 6, 8, and 14 hours). In a set of experiments, NRCs were treated with 10 μg/mL of pan-specific TGF-β polyclonal neutralizing antibody (R&D Systems) or nonspecific rabbit immunoglobulin G (Southern Biotechnology Associates, Birmingham, AL) for 20 minutes and then stimulated by TGF-β1 2.5 ng/mL for 6 hours for Northern blotting or for 24 hours for measurement of ET-1 in cell media and homogenate.
Endothelin 1 Radioimmunoassay
ET-1 concentrations in liver, plasma, and the culture media and cell homogenates from NRCs were measured via radioimmunoassay (Phoenix Pharmaceuticals, Mountain View, CA). Liver tissues and NRCs were homogenized and prepared as previously described.6 Extraction of ET-1 from plasma and the media was accomplished by acidification and elution over Sep-Pak C18 columns (Waters, Milford, MA). Samples were subjected to radioimmunoassay with the use of a rabbit ET-1 antiserum. Recovery from the Sep-Pak C18 columns averaged 90%, and the sensitivity of the assay for ET-1 was 1.5–2.0 pg.
Western Blot Analysis
Tissues were homogenized in radioimmunoprecipitation buffer in the presence of protease inhibitors, as previously described.4,6,7 Equal concentrations of protein from liver were fractionated on Tris-HCl–ready gels (Bio-Rad Laboratories, Hercules, CA) and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Piscataway, NJ). Incubation with primary antibodies for CK19, α-SMA, TGF-β1, p-Smad2, or Smad2/3 (BD Biosciences, Palo Alto, CA) was followed by the addition of horseradish peroxidase–conjugated secondary antibodies and detection with enhanced chemiluminescence.
Laser Capture Microdissection
Liver tissues were immediately frozen in OCT compound (Fisher, Suwanee, GA) and stored at −80°C until use. After rapid staining with H&E, 6-μm sections from frozen sections were microdissected under a PixCell II laser capture microscope with an infrared diode laser (Arcturus Engineering, Mountain View, CA), as previously described.23 Briefly, a specific polymer film mounted on optically transparent caps (CapSure TF-100; Arcturus) was placed on the section. Bile epithelial cells or hepatocytes were captured by focal melting of membranes by the laser beam under visual control. The caps with captured epithelial cells were used as a lid for 500-μL microcentrifuge tubes containing RNA extraction buffer (Arcturus).
RNA Extraction and Amplification
Total RNA from captured epithelial cells was extracted by using a Pico Pure RNA isolation kit (Arcturus) according to the manufacturer’s recommendations. After deoxyribonuclease treatment (Invitrogen, Carlsbad, CA), RNA was eluted and stored at −80°C until use. All total RNA samples were first tested for quality on an Agilent Bioanalyzer 2100B by using an RNA Pico Lab Chip Kit (Agilent Technologies, Palo Alto, CA) and subsequently were amplified with the RiboAmp OA RNA Amplification Kit (Arcturus). The quality of the amplified RNA was again evaluated on an RNA Pico Lab Chip Kit (Agilent Technologies).
Real-Time Quantitative Reverse-Transcription Polymerase Chain Reaction Analysis
Approximately 2 μg of total RNA from bile cholangiocytes or hepatocytes was reverse-transcribed by using the StrataScript first-strand synthesis system (Stratagene, La Jolla, CA). Complementary DNA (cDNA) was amplified with ET-1 and 18S primers and SYBR Green polymerase chain reaction (PCR) master mix (Applied Biosystems, Foster City, CA) by PCR with an iCycler real-time PCR detection system (Bio-Rad) for 40 cycles. Relative RNA levels were calculated by using the iCycler software and a standard equation (Applied Biosystems). ET-1 primers were designed as follows: sense primer, 5′-TCCTGCTCCTCCTTGATG-3′; antisense primer, 5′-TTCCCTTGGTCTGTGGTC-3′ (PCR product, approximately 164 base pairs [bp]). TGF-β1 primers were designed as follows: sense primer, 5′-CTACTGCTTCAGCTCCACAGA-3′; antisense primer, 5′-ACCTTGGGCTTGCGACC-3′ (PCR product, approximately 279 bp). 18S primers were designed as follows: sense primer, 5′-GAAACGGCTACCACATCC-3′; antisense primer, 5′-CACCAGACTTGCCCTCCA-3′ (PCR product, approximately 168 bp). ET-1 and TGF-β1 values were normalized to 18S values in each sample.
Northern Blot Analysis
Total NRC RNA was prepared by lysis in TRIzol reagent (GIBCO), electrophoresed, and transferred to Nytran membranes (Schleicher & Schuell, Keene, NH) followed by hybridization in Quikhyb (Stratagene) with a 0.5-kilobase rat ET-1 cDNA (a gift of Dr Tom Quertermous, Vanderbilt University) or a 0.5-kilobase rat glyceraldehyde-3-phosphate dehydrogenase cDNA (American Type Culture Collection, Manassas, VA) labeled with the DECAprime II kit (Ambion, Austin, TX). Imaging and quantification of signals normalized to glyceraldehyde-3-phosphate dehydrogenase were performed with phosphorimaging.
Reverse-Transcription Polymerase Chain Reaction Analysis
Five micrograms of total RNA from NRCs was reverse-transcribed and amplified by using the ProSTAR first-strand reverse-transcription PCR (RT-PCR) kit (Stratagene). Sequences for the primers were designed on the basis of the published sequences for ET-124 and TGF-β type I and II receptors16 as follows: ET-1 (PCR product, approximately 489 bp)—sense primer, 5′-TTGTGGCTTTCCAAGGAGCTCCAG-3′; antisense primer, 5′-TTGCTGATGGCCTCCAACCTTC-3′; TGF-β I receptor (PCR product, approximately 450 bp)—sense primer, 5′-GCACCATCTTCAAAAACAGG-3′; antisense primer, 5′-TCTTCACAGCAACTTCTTCT-3′; TGF-β II receptor (PCR product, approximately 385 bp)—sense primer, 5′-TCGGAATACACCACCAG-3′; antisense primer, 5′-AAGATCTTGACAGCCACGGT-3′; and α-actin (PCR product, approximately 400 bp)—sense primer, 5′-GACATGACAGACTACCTCAT-3′; antisense primer, 5′-AGACAGCACTGTGTTGGCAT-3′. PCR amplification reactions were performed in a PerkinElmer 2400 PCR machine (Foster City, CA), and RT-PCR products were electrophoresed on an agarose gel to show the amplified bands. Initial PCR amplification products from each set of primers were sequenced using an ABI PRISM Model 377 automated sequencer (Applied Biosystems, Foster City, CA) in a sequencing facility (University of Alabama at Birmingham, Birmingham, AL) to ensure accuracy.
Endothelin 1 Promoter Constructs, Transient Transfection, and Luciferase Reporter Gene Assay
A plasmid (HPPET-LUC) containing −2459 to +165 bp of the human ET-1 promoter was provided by Dr Catherine Aversa (Bristol-Myers Squibb Pharmaceutical, Princeton, NJ).25 Plasmid pET-649 was constructed by insertion of the SacI-BglII restriction fragment from HPPET-LUC (−649 to +165 bp) into the luciferase reporter vector pGL2-Basic (Promega, Madison, WI). The insert for plasmid pET-184 (−184 to +165 bp) was generated from HPPET-LUC by PCR by using the sense primer 5′-GCGGGCGTCTGCTTCTGAAGTT-3′ and the antisense primer 5′-ATCTCAAAGCGATCCTTCAGCC-3′. The insert was cloned into pCR2.1 (Invitrogen) and sequenced to verify promoter orientation and sequence accuracy. Subsequently, the SacI-XhoI fragment was subcloned into pGL2-Basic. Plasmid pET-949 contains the full putative Smad-binding element, whereas the pET-184 does not.18 The plasmid pRL-tk (Promega) was cotransfected in each experiment as a control for transfection efficiency. Transient cotransfection of either pET-649 and pRL-tk or pET-184 and pRL-tk into cultured NRCs was performed by using the TransFast Transfection Reagent (Promega) according to the manufacturer’s protocol. Briefly, the transfection mixture containing DNA (target plasmid/control plasmid, 40:1) and the transfection reagent was added into cells. After incubation, transfected cells were then subjected to various treatments. Luciferase activity was measured in the cellular extracts by using a Dual-Luciferase Reporter Assay Kit (Promega). After treatment, the cell lysates were collected after the addition of cell culture lysis reagent (Promega). The relative light units were then determined in a luminometer (MEM Instrument Inc., Hamden, CT) for a total of 10 seconds after a 5-second delay.
Densitometry and Statistics
The density of autoradiographic signals was assessed with an Astra 1200s scanner (UMAX, Fremont, CA) and quantitated with Scion ImagePC software (Scion, Frederick, MD). Data were analyzed with the Student t test or analysis of variance with Bonferroni correction for multiple comparisons between groups. Simple regression analysis was used to evaluate correlations between liver and/or plasma ET-1 content and liver CK19, TGF-β1, and p-Smad2 levels. Measurements are expressed as means ± SE. Statistical significance was designated as P < .05.
Results
Physiological Assessment for Hepatopulmonary Syndrome After Common Bile Duct Ligation and Thioacetamide Administration
To evaluate the development of HPS in relation to hepatic and pulmonary alterations in models of biliary and nonbiliary cirrhosis, we measured liver histology, portal venous pressure, spleen weight, arterial blood gases, and microsphere shunting through the pulmonary microcirculation at serial time points after CBDL and TAA administration. Trichrome staining of the liver (Figure 1) showed no morphological damage in the control group throughout the experiment period. After CBDL, bile duct proliferation and periductal fibrosis were seen within 1 week that progressed to overt biliary cirrhosis by 3 weeks. TAA-treated animals also developed progressive hepatic fibrosis with bridging by 2 weeks and micronodular cirrhosis by 8 weeks, although bile duct proliferation was not a prominent feature. A progressive increase in portal venous pressure and spleen weight was observed in both CBDL- and TAA-treated animals (Table 1). HPS developed only after CBDL, as reflected by the development of a widened alveolar arterial oxygen gradient and increased shunting of microspheres through the dilated pulmonary microcirculation beginning 2 weeks after ligation, as previously observed (Table 14,21). No significant changes in arterial blood gas or the pulmonary microcirculation occurred after TAA administration.
Figure 1.
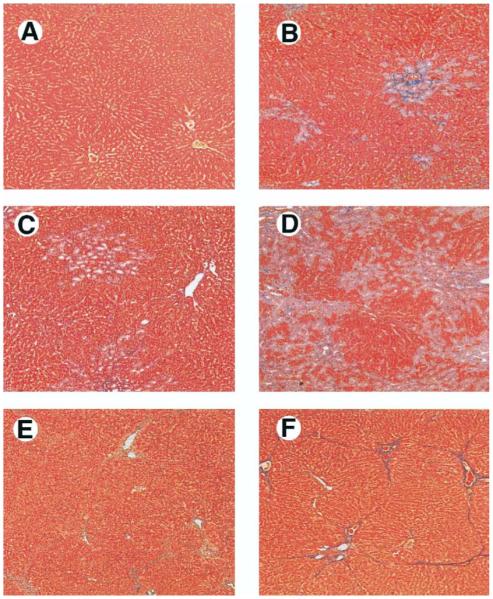
Liver histology in common bile duct ligation (CBDL)-treated and thioacetamide (TAA)-treated animals. Compared with control animals (A), 1-week CBDL animals developed bile duct proliferation and periductal fibrosis (B). Two-week CBDL animals had progressive duct proliferation with expansion of portal tracts and bridging fibrosis (C). Three-week CBDL animals developed biliary cirrhosis (D). TAA-treated animals had progression from focal hepatocyte necrosis and bridging fibrosis with minimal inflammation at 2 weeks (E) to micronodular cirrhosis at 8 weeks (F) (Masson’s trichrome stain; original magnification, 40×).
Table 1.
Physiological Analysis and ET-1 Levels in CBDL- and TAA-Treated Animals
| CBDL |
TAA |
|||||
|---|---|---|---|---|---|---|
| Variable | Control | 1 Wk | 2 Wk | 3 Wk | 2 Wk | 8 Wk |
| PVP (mm Hg) | 7.4 ± 0.3 | 11.1 ± 0.7a | 14.0 ± 0.6a | 15.2 ± 0.9a | 9.6 ± 0.8a | 12.6 ± 1.8a |
| Spleen weight (g) | 0.7 ± 0.1 | 0.8 ± 0.1 | 1.3 ± 0.1a | 1.6 ± 0.1a | 0.6 ± 0.1 | 0.9 ± 0.1a |
| AaPo2 (mm Hg) | 7.5 ± 1.5 | 7.8 ± 2.7 | 17.9 ± 1.7a | 19.4 ± 2.9a | 10.4 ± 3.7 | 12.9 ± 2.7 |
| Intrapulmonary shunt fraction (%) | 5.8 ± 0.1 | 6.1 ± 1.3 | 12.0 ± 2.4a | 14.0 ± 3.0a | 6.1 ± 0.1 | 6.6 ± 0.1 |
| Liver ET-1 (ng/g tissue) | 2.4 ± 0.3 | 5.2 ± 0.8a | 5.7 ± 0.7a | 8.6 ± 1.6a | 2.5 ± 0.5 | 3.0 ± 0.4 |
| Plasma ET-1 (pg/mL) | 6.9 ± 0.2 | 20.0 ± 1.0a | 17.4 ± 0.7a | 22.6 ± 2.5a | 8.9 ± 0.4 | 8.6 ± 1.0 |
NOTE. Values are mean ± SE (n = 5–8 animals per group).
ET, endothelin; CBDL, common bile duct ligation; TAA, thioacetamide; PVP, portal venous pressure; AaPo2, alveolar–arterial oxygen gradient.
P < .05 compared with normal control.
Quantification and Hepatic Immunohistochemical Localization of Endothelin 1 After Common Bile Duct Ligation and Thioacetamide Administration
To evaluate ET-1 production in relation to the development of HPS, we measured hepatic and plasma ET-1 levels in CBDL- and TAA-treated animals (Table 1). Hepatic ET-1 levels began to increase within 1 week after CBDL and progressively increased to a maximum of 3.5-fold control levels after 3 weeks, as previously described. This increase was followed by a progressive increase in plasma ET-1 levels of similar magnitude to that previously observed.6,26 In contrast, hepatic and plasma ET-1 levels were unchanged after TAA administration.
To assess the distribution of ET-1 in the liver, we performed immunohistochemical staining for ET-1, CK19, and α-SMA (Figure 2). In control animals, the predominant ET-1 staining was in the sinusoidal regions, with minimal bile duct and hepatocyte staining. Within 1 week after CBDL, there was a substantial increase in biliary ET-1 staining that persisted over the 3-week time course. The distribution of ET-1 staining was similar to that observed for the cholangiocyte marker CK19. There was also a smaller focal increase in ET-1 staining in the periductal regions after CBDL in a distribution consistent with stellate cell or portal myofibroblast staining based on α-SMA staining. In 2- and 8-week TAA-treated animals (Figure 3), ET-1 staining in lobular regions was observed in a sinusoidal distribution similar to that of controls and was also seen in cells adjacent to fibrous bands in areas where myofibroblasts accumulate, according to α-SMA staining.
Figure 2.

Immunohistochemical localization of endothelin-1 (ET)-1, cytokeratin 19 (CK19), and α-smooth muscle actin (α-SMA) in liver sections from CBDL animals. Representative images are shown of ET-1 (top), CK19 (middle), and α-SMA (bottom) in normal (A, E, and I), 1-week (B, F, and J), 2-week (C, G, and K), and 3-week (D, H, and L) CBDL liver. In normal liver (A), ET-1 staining was most prominent in a pattern consistent with sinusoidal endothelial staining. After CBDL (B–D), staining increased markedly in the cytoplasm of biliary epithelial cells and focally in periductal regions. CK19 stained the biliary epithelium in normal (E) and CBDL (F–H) animals in a pattern similar to CBDL ET-1 staining. In normal liver (I), α-SMA staining was most prominent in vascular smooth muscle. After CBDL (J–L), α-SMA staining increased in the periductal regions (but not in biliary epithelium) in a pattern consistent with stellate cells or portal myofibroblast staining (original magnification, 40×).
Figure 3.
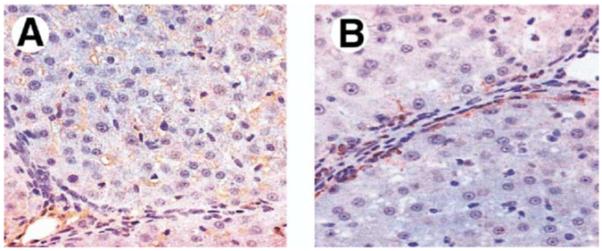
Immunohistochemical localization of ET-1 and α-SMA in liver sections from 8-week TAA-treated animals. ET-1 staining (A) was found in lobular regions in a sinusoidal distribution similar to that of normal controls and additionally in cells adjacent to fibrous bands. α-SMA staining (B) was seen in stellate cells adjacent to fibrous bands in a pattern similar to perifibrous ET-1 staining (original magnification, 40×).
To quantify the relative increase in cholangiocytes, stellate cells, and myofibroblasts after CBDL and TAA administration in relation to hepatic ET-1 levels, we performed Western blot analysis for CK19 and α-SMA in liver tissue (Figure 4A). After CBDL, CK19 levels significantly increased and were approximately 20-fold control values at 3 weeks. α-SMA levels also increased after CBDL to approximately 5-fold control values at 3 weeks, but the increase was significantly less than that seen for CK19. After TAA administration, CK19 levels did not change, and α-SMA levels increased approximately 2-fold at 8 weeks. In addition, we found a strong positive linear correlation between liver CK19 levels and liver and plasma ET-1 levels in CBDL animals (Figure 4B).
Figure 4.
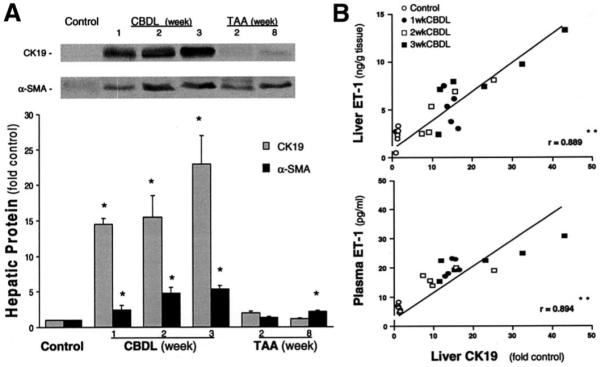
Liver CK19 and α-SMA protein levels in CBDL- and TAA-treated animals and correlation of liver CK19 levels with liver and plasma ET-1 levels in CBDL animals. (A) The top panel shows representative liver CK19 and α-SMA immunoblots, and the bottom graph summarizes Western blot results. Single bands of approximately 40 kilodaltons for CK19 or 42 kilodaltons for α-SMA were seen. Values are expressed as means ± SE (n = 5–7 animals for each group). *P < .05 compared with normal controls. (B) The relation between liver CK19 content and liver or plasma ET-1 levels in control and CBDL animals. The linear correlation coefficient (r) was significant for each comparison. **P < .0001.
Quantitation of Endothelin 1 Messenger RNA Expression in Cholangiocytes and Hepatocytes After Common Bile Duct Ligation
To confirm that biliary epithelial cells express ET-1 in vivo after CBDL, we used laser capture microdissection (LCM) to obtain cholangiocytes and hepatocytes, followed by isolation and amplification of RNA and quantitative real-time RT-PCR (Figure 5). LCM allows selective capture of cells of interest on the basis of morphology, and the subsequent purity of cholangiocyte RNA preparations was confirmed by documenting that these cells express CK19 and connexin 43 and not α-SMA (data not shown). Cholangiocyte ET-1 messenger RNA (mRNA) levels increased significantly within 1 week after CBDL to 3.5-fold control values and remained increased through 3 weeks. In contrast, there was no significant change in hepatocyte ET-1 levels after CBDL.
Figure 5.
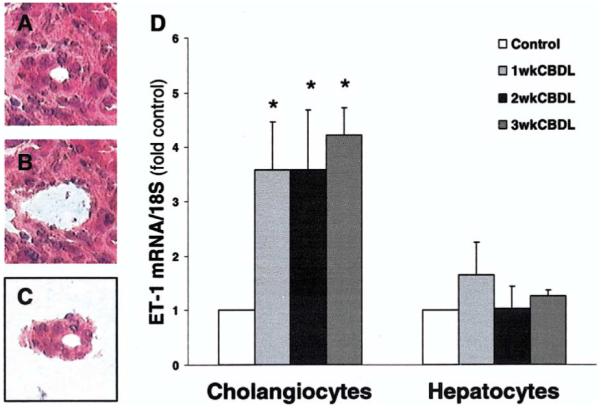
Laser capture microdissection (LCM) of cholangiocytes and hepatocytes and ET-1 mRNA analysis from CBDL liver. Representative H&E-stained images from CBDL liver show a bile ductule cross section before (A) and after (B) LCM and after capture to a cap (C). The graph summarizes ET-1 mRNA levels in total RNA amplified after isolation from cholangiocytes and hepatocytes from 1-, 2-, and 3-week CBDL liver (D). Data are expressed relative to 18S levels as means ± SE and are from 4 to 5 real-time quantitative RT-PCR assays. *P < .05 compared with normal controls (n = 4–5 animals for each group).
Quantification and Localization of Hepatic and Cholangiocyte Transforming Growth Factor β1 and Hepatic Phospho-Smad2 After Common Bile Duct Ligation
To explore potential mediators of increased cholangiocyte ET-1 expression after CBDL, we performed Western blot analysis and immunohistochemistry for TGF-β1, p-Smad2, or Smad2/3 in liver from CBDL animals and assessed TGF-β1 mRNA levels in isolated cholangiocytes by using real-time quantitative RT-PCR analysis. There was a progressive increase in liver TGF-β1 levels relative to control beginning 1 week after CBDL and reaching a maximum of 9-fold control at 3 weeks (Figure 6A). The increase in TGF-β1 levels was accompanied by a progressive increase in hepatic p-Smad2 levels (Figure 6B), but there was no change in total hepatic Smad2/3 levels. To assess whether changes in TGF-β1 or p-Smad2 levels might be important in the increases in liver ET-1 levels observed in experimental HPS, we sought a correlation between liver ET-1 content and liver TGF-β1 or p-Smad2 levels in CBDL animals (Figure 6C). There was a strong positive linear correlation between liver ET-1 levels and liver TGF-β1 or p-Smad2 levels after CBDL.
Figure 6.

Liver transforming growth factor (TGF)-β1 and phospho-Smad2 (p-Smad2) protein levels and correlation of liver ET-1 content with liver TGF-β1 or p-Smad2 levels in CBDL animals. Single bands of approximately 50 kilodaltons consistent with TGF-β1 (A) and 58 kilodaltons consistent with p-Smad2 or Smad2/3 (B) are seen. Graphs summarize TGF-β1 and p-Smad2 protein levels in liver. Values are expressed as means ± SE (n = 5–7 animals for each group). *P < .05 compared with normal controls. The relation is shown between ET-1 levels and TGF-β1or p-Smad2 levels in liver from control and CBDL animals (C). The linear correlation coefficient (r) was significant for each comparison. **P < .0001. A graphical summary is shown of TGF-β1 mRNA levels in cholangiocytes from control and 1-, 2-, and 3-week CBDL liver (D). Data are expressed relative to 18S levels as means ± SE and are from 4 to 5 real-time quantitative RT-PCR assays. P < .05 compared with normal control (n = 4–5 animals for each group).
By immunohistochemistry (Figure 7), TGF-β1 staining was minimal in normal animals. After CBDL, TGF-β1 staining increased most notably in the cytoplasm of cholangiocytes beginning within 1 week. There was also a focal increase in staining in periductal cells and a distribution consistent with sinusoidal staining. Smad2 phosphorylation and nuclear localization were also minimal in normal animals. After CBDL, p-Smad2 staining increased and was found most prominently in cholangiocyte nuclei, as well as in the nuclei of some periductal cells and periportal hepatocytes.
Figure 7.

Immunohistochemical localization of TGF-β1 and p-Smad2 in liver sections from 1-, 2-, and 3-week CBDL animals. Representative images of TGF-β1 (top) and p-Smad2 (bottom) are shown in normal (A and E), 1-week (B and F), 2-week (C and G), and 3-week (D and H) CBDL liver. In normal animals (A), TGF-β1 staining was minimal and was not observed in ductular epithelium. After CBDL (B–D), TGF-β1 staining increased most notably in the cytoplasm of cholangiocytes beginning within 1 week. There was also a focal increase in staining in periductal cells and in a distribution consistent with sinusoidal staining. Smad2 phosphorylation and nuclear localization were also minimal in normal animals (E). After CBDL (F–H), p-Smad2 staining increased and was found most prominently in cholangiocyte nuclei (arrows) and in the nuclei of some periductal cells and periportal hepatocytes.
To directly assess whether cholangiocytes express TGF-β1 mRNA, we measured levels in cholangiocytes obtained by LCM (Figure 6D). There was a significant progressive increase in cholangiocyte TGF-β1 mRNA levels beginning at 1 week and reaching a maximum of 4-fold control at 3 weeks.
TGF-β Receptor Expression and Effects of Exogenous TGF-β1 on ET-1 mRNA and Protein Levels, and Promoter Activity in NRCs
To determine whether ET-1 expression can be regulated by TGF-β1, we evaluated TGF-β receptor mRNA expression and the effects of TGF-β1 on ET-1 mRNA, protein levels, and ET-1 promoter activity in NRCs. NRCs expressed signals for both TGF-β I and II receptors by RT-PCR (Figure 8A). TGF-β1 treatment resulted in a dose- and time-dependent increase in ET-1 mRNA levels with a maximal 2-fold increase relative to control seen at a dose of 2 to 5 ng/mL after 4 to 6 hours (Figure 8B). The increase in ET-1 mRNA was accompanied by a significant increase of similar magnitude in ET-1 protein levels in cell culture media (Figure 8C and D). The increases in ET-1 mRNA and protein were inhibitable with a specific anti–TGF-β antibody. To assess the effects of TGF-β1 on ET-1 promoter activity, we transiently transfected 2 promoter constructs (pET-184, lacking the complete putative Smad-binding element, and pET-649, which contains the complete Smad-binding element) into NRCs (Figure 8E). In initial studies, basal promoter activity was compared in pGL2-Basic constructs with or without the addition of the ET promoter segment. Luciferase production was minimal (0.15 relative light units) in constructs lacking the ET-1 promoter segment and was not altered by the addition of TGF-β1 (data not shown). In cells transfected with the pET-649 construct, there was a significant 2.5-fold increase in ET-1 promoter activity, normalized for transfection efficiency, after TGF-β1 stimulation. This effect was inhibited by a specific anti–TGF-β antibody. In contrast, cells transfected with the shorter pET-184 construct showed no increase in promoter activity after TGF-β1 stimulation.
Figure 8.
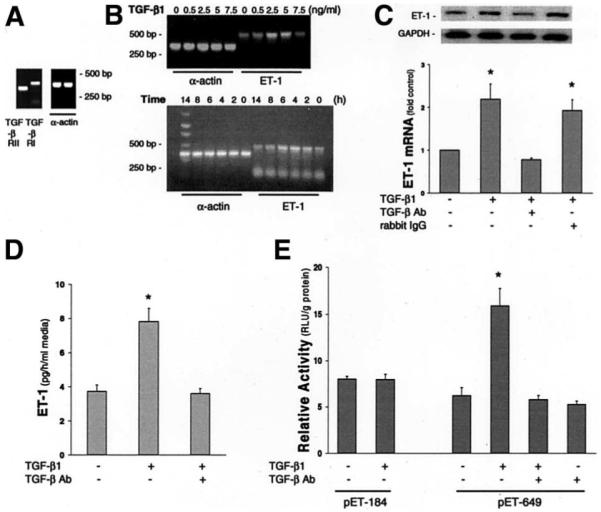
Evaluation of TGF-β1 modulation of ET-1 expression in NRCs. (A) RT-PCR analysis shows the expression of the TGF-β type I and II receptors in NRCs. (B) Dose and time course RT-PCR analysis of ET-1 mRNA levels relative to α-actin mRNA levels after TGF-β1 stimulation of NRCs. (C) NRC ET-1 Northern blot analysis. Top panel insert shows representative ET-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Northern blots in control NRCs and in NRCs incubated with TGF-β1 alone or TGF-β1 after pretreatment with a specific anti–TGF-β antibody (TGF-β Ab) or a nonspecific rabbit immunoglobulin G (10 μg/mL). Bottom graph summarizes effects of TGF-β1 stimulation on ET-1 mRNA levels in NRCs normalized to GAPDH mRNA levels. ET-1 mRNA levels are expressed relative to control as means ± SE (n = 4–9 for each group). *P < .05 compared with control. (D) NRC ET-1 radioimmunoassay. NRCs were treated with TGF-β1 in the presence or absence of a specific TGF-β Ab. The graph summarizes effects of TGF-β1 on ET-1 production in NRCs. ET-1 levels are expressed as means ± SE (n = 4–9 for each group). *P < .05 compared with control. (E) Analysis of TGF-β1 effects on ET-1 promoter activity in NRCs by using 2 promoter constructs (pET-184 and pET-649) determined by a luciferase reporter gene assay normalized for transfection efficiency. Data are expressed in relative light units (RLU) per gram of protein as mean ± SE (n = 6 for each group). *P < .05 compared with the control that was transfected without any treatment.
Discussion
In this study, we assessed hepatic ET-1 production and regulation in experimental HPS induced by CBDL in comparison to nonbiliary cirrhosis induced by TAA administration where HPS does not develop. We found a progressive increase in hepatic production and plasma levels of ET-1 after CBDL that did not occur after TAA administration. The increase in hepatic ET-1 production was found predominately in biliary epithelium according to immunohistochemistry and on direct assessment of cholangiocyte ET-1 expression by LCM and real-time qualitative RT-PCR. An increase in TGF-β1 production also occurred in cholangiocytes and adjacent portal areas after CBDL. This paralleled the increase in cholangiocyte ET-1 and was accompanied by evidence of TGF-β–mediated signaling in cholangiocytes based on p-Smad2 localization and levels. Finally, exogenous TGF-β1 stimulates ET-1 gene expression and peptide production in NRCs. These findings define cholangiocyte production as a major source of ET-1 after CBDL and support a role for TGF-β1 in modulating ET-1 expression. These results provide insight into the mechanisms of the unique susceptibility of CBDL animals to experimental HPS.
ET-1 is established as an important modulator of hepatic fibrosis and intrahepatic vascular resistance in liver disease.27 Several studies have documented that the hepatic ET-1 system is up-regulated in human and experimental cirrhosis, including CBDL.6,12 The major hepatic cell types recognized to produce ET-1 in the liver include endothelial cells28 and stellate cells.29,30 In the setting of cirrhosis, ET-1 production has also been reported in hepatocytes28 and biliary epithelium,6,11,12 but stellate cell production has been considered the major source. Increased circulating levels of ET-1 have also been found in decompensated liver disease in the setting of marked hyperdynamic changes and ascites,31,32 and hepatic production is thought to contribute to increased circulating levels in liver disease.28 Our prior studies and the current work show that CBDL seems to be unique relative to other cirrhotic models—specifically, TAA-induced nonbiliary cirrhosis—in that hepatic production and plasma ET-1 levels significantly increase within 1 week after CBDL before the development of cirrhosis or a hyperdynamic state.10 Our prior work implicates hepatic production as the source of circulating ET-1 on the basis of the increase in hepatic venous ET-1 levels after CBDL,9 the coordinate increase in hepatic and plasma ET-1 levels,6,10 and the lack of significant increases in ET-1 in other organs after CBDL.6 In the current study, we localized the major increase in hepatic ET-1 staining to the biliary epithelium after CBDL, with a less prominent increase in periductal regions, consistent with myofibroblast or stellate cell staining. This finding correlates with the significantly greater increase in bile ducts relative to stellate cells after CBDL and also provides an explantation for the lack of increase in hepatic and circulating ET-1 levels in TAA cirrhosis, where bile duct proliferation is minimal at 8 weeks. The concept that ductal epithelium may express ET-1 and also other cytokines is in line with the findings that gallbladder33 and renal ductular epithelium16 are established to express ET-1 and with the recognition of biliary epithelium as an important effector cell that produces chemokines and cytokines that influence liver damage in the setting of bile duct injury.34 In addition, our results support that a relatively modest increase in bile duct mass, as seen at 1 week after CBDL, may be sufficient to increase hepatic and circulating ET-1 levels.
To directly confirm that cholangiocytes express ET-1 in vivo after CBDL, we used LCM to obtain isolated populations of cholangiocytes and hepatocytes. This technique has been increasingly used to isolate and amplify RNA from specific cell types in tissues while avoiding the potential confounding effects related to cell isolation.35 We then used real-time RT-PCR to quantify ET-1 mRNA levels. Cell type–specific mRNAs were measured to ensure the purity of isolated cells, and hepatocytes served as a control cell type on the basis of the lack of hepatocyte ET-1 staining after CBDL by immunohistochemistry. We found a significant increase in ET-1 mRNA levels in cholangiocytes but not hepatocytes after CBDL, in line with our immunohistochemical findings and with prior findings in isolated hepatocytes from CBDL animals.36 This analysis directly confirms an increase in ET-1 mRNA in cholangiocytes after CBDL.
The regulation of cholangiocyte ET-1 production has not been well characterized. One potential modulator of ET-1 expression during liver injury is TGF-β1. TGF-β1 is recognized to increase ET-1 mRNA and protein production in several cell types, including renal ductular epithelium, where endogenous TGF-β1 production modulates ET-1 expression in an autocrine fashion through its specific receptors.16 Enhanced TGF-β1 expression is also well established in experimental and human liver damage,13,37 particularly in and around bile ducts in experimental biliary cirrhosis38,39 and in human bile duct disorders.40,41 In this study, we have shown that hepatic TGF-β1 levels increase over a similar time course relative to ET-1 after CBDL and that the increase in TGF-β1 occurs in cholangiocytes and in periductal cells. We also found evidence of TGF-β1 activation and signaling in cholangiocytes in vivo; there was an increase in hepatic p-Smad2 levels and localization in cholangiocyte nuclei after CBDL. The increase in liver TGF-β1 and p-Smad2 levels shows a strong direct correlation with hepatic ET-1 levels.
To test whether TGF-β1 directly influences rat cholangiocyte ET-1 expression, we used a well-characterized nontransformed cholangiocyte cell line that maintains features of cholangiocytes in vivo. In these cells, we found that TGF-β1 administration increased ET-1 promoter activity and ET-1 expression. Our finding that the pET-649, but not the pET-184, ET-1 promoter construct is responsive to TGF-β1 is consistent with the recent finding that a Smad-binding element located at −193/−171 of the ET-1 promoter is critical for TGF-β1–mediated induction of ET-1 expression.18 Although we have not confirmed our findings in freshly isolated cholangiocytes from CBDL animals, because these cells cannot be maintained in culture for sufficient periods of time to assess ET-1 peptide production or promoter activity, our results document that TGF-β1 modulates ET-1 in rat cholangiocytes. Together, these data support a potential role for TGF-β1 in mediating cholangiocyte ET-1 expression after CBDL. Other mediators that may influence cholangiocyte ET-1 production include factors that influence cholangiocyte proliferation,42 most notably intracellular adenosine 3′,5′-cyclic monophosphate levels,43 as well as tumor necrosis factor α,44 both of which have been reported to increase ET-1 expression. Finally, posttranslational modifications, particularly changes in ET-converting enzyme production, may also influence ET-1 levels in cholangiocytes, as occurs in hepatic stellate cells.15 These are important areas for future study.
A central issue in studies of experimental HPS is how the findings may provide insight into human disease. Our prior and current findings indicate that a combination of pulmonary endothelial ETB receptor overexpression and increased cholangiocyte-derived ET-1 production and release contribute to eNOS-mediated NO production and vasodilatation in the pulmonary microvasculature. Our findings here support that TGF-β1may drive ET-1 production in cholangiocytes and that the relatively modest degree of cholangiocyte proliferation in 1-week CBDL animals is sufficient to increase hepatic ET-1 levels. These results suggest that hepatic ET-1 production might increase in any form of chronic liver disease in which a degree of cholangiocyte proliferation develops in the setting of increased TGF-β1 activity. Because both of these events are found across a wide spectrum of differing etiologies of chronic liver disease in humans, ET-1–mediated HPS would not be expected to be confined to cholestatic disorders. Indirect support for this concept comes from the finding of significant ET-1 staining in proliferating bile ducts in noncholestatic human cirrhosis.12 In addition, chronic bile duct disorders in adults are often characterized by bile duct injury and loss rather than marked proliferation, a situation clearly distinct from CBDL, and would not be expected to uniformly be associated with increased hepatic ET-1 production. Important areas for future translational studies include defining whether hepatic ET-1 production is increased in human HPS and fully characterizing the mechanisms that lead to ET-1 and ETB receptor alterations in experimental models.
Acknowledgments
Supported by Grant DK02030 and a VA Merit Review grant (to M.B.F.).
Abbreviations used in this paper
- bp
base pair
- CBDL
common bile duct ligation
- CK19
cytokeratin 19
- eNOS
endothelial nitric oxide synthase
- ET
endothelin
- HPS
hepatopulmonary syndrome
- LCM
laser capture microdissection
- NRC
normal rat cholangiocyte
- PCR
polymerase chain reaction
- p-Smad2
phospho-Smad2
- RT-PCR
reverse-transcription polymerase chain reaction
- α-SMA
α-smooth muscle actin
- TAA
thioacetamide
- TGF
transforming growth factor
References
- 1.Fallon M, Abrams G. Pulmonary dysfunction in chronic liver disease. Hepatology. 2000;32:859–865. doi: 10.1053/jhep.2000.7519. [DOI] [PubMed] [Google Scholar]
- 2.Lange PA, Stoller JK. The hepatopulmonary syndrome. Ann Intern Med. 1995;122:521–529. doi: 10.7326/0003-4819-122-7-199504010-00008. [DOI] [PubMed] [Google Scholar]
- 3.Schenk P, Schoniger-Hekele M, Fuhrmann V, Madl C, Silberhumer G, Muller C. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology. 2003;125:1042–1052. doi: 10.1016/s0016-5085(03)01207-1. [DOI] [PubMed] [Google Scholar]
- 4.Fallon MB, Abrams GA, Luo B, Hou Z, Dai J, Ku DD. The role of endothelial nitric oxide synthase in the pathogenesis of a rat model of hepatopulmonary syndrome. Gastroenterology. 1997;113:606–614. doi: 10.1053/gast.1997.v113.pm9247483. [DOI] [PubMed] [Google Scholar]
- 5.Carter EP, Hartsfield CL, Miyazono M, Jakkula M, Morris KG, Jr, McMurtry IF. Regulation of heme oxygenase-1 by nitric oxide during hepatopulmonary syndrome. Am J Physiol Lung Cell Mol Physiol. 2002;283:L346–L353. doi: 10.1152/ajplung.00385.2001. [DOI] [PubMed] [Google Scholar]
- 6.Luo B, Abrams GA, Fallon MB. Endothelin-1 in the rat bile duct ligation model of hepatopulmonary syndrome: correlation with pulmonary dysfunction. J Hepatol. 1998;29:571–578. doi: 10.1016/s0168-8278(98)80152-9. [DOI] [PubMed] [Google Scholar]
- 7.Luo B, Liu L, Tang L, Zhang J, Stockard C, Grizzle W, Fallon M. Increased pulmonary vascular endothelin B receptor expression and responsiveness to endothelin-1 in cirrhotic and portal hypertensive rats: a potential mechanism in experimental hepatopulmonary syndrome. J Hepatol. 2003;38:556–563. doi: 10.1016/s0168-8278(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Luo B, Chen SJ, Abrams GA, Fallon MB. Endothelin-1 stimulation of endothelial nitric oxide synthase in the pathogenesis of hepatopulmonary syndrome. Am J Physiol. 1999;277:G944–G952. doi: 10.1152/ajpgi.1999.277.5.G944. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Zhang M, Luo B, Abrams GA, Fallon MB. Biliary cyst fluid from common bile duct ligated rats stimulates eNOS in pulmonary artery endothelial cells: a potential role in hepatopulmonary syndrome. Hepatology. 2001;33:722–727. doi: 10.1053/jhep.2001.22701. [DOI] [PubMed] [Google Scholar]
- 10.Luo B, Liu L, Tang L, Zhang J, Ling Y, Fallon MB. ET-1 and TNF-α in HPS: analysis in prehepatic portal hypertension and biliary and nonbiliary cirrhosis in rats. Am J Physiol Gastrointest Liver Physiol. 2004;286:G294–G303. doi: 10.1152/ajpgi.00298.2003. [DOI] [PubMed] [Google Scholar]
- 11.Koda W, Harada K, Tsuneyama K, Kono N, Sasaki M, Matsui O, Nakanuma Y. Evidence of the participation of peribiliary mast cells in regulation of the peribiliary vascular plexus along the intrahepatic biliary tree. Lab Invest. 2000;7:1007–1017. doi: 10.1038/labinvest.3780106. [DOI] [PubMed] [Google Scholar]
- 12.Pinzani M, Milani S, DeFranco R, Grappone C, Caligiuri A, Gentilini A, Tosti-Guerra C, Maggi M, Failli P, Ruocco C, Gentilini P. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology. 1996;110:534–548. doi: 10.1053/gast.1996.v110.pm8566602. [DOI] [PubMed] [Google Scholar]
- 13.Saperstein L, Jirtle R, Farouk M, Thompson HJ, Chung KS, Meyers WC. Transforming growth factor-beta 1 and mannose 6-phosphate/insulin-like growth factor-II receptor expression during intrahepatic bile duct hyperplasia and biliary fibrosis in the rat. Hepatology. 1994;19:412–417. [PubMed] [Google Scholar]
- 14.Eakes AT, Olson MS. Regulation of endothelin synthesis in hepatic endothelial cells. Am J Physiol. 1998;274:G1068–G1076. doi: 10.1152/ajpgi.1998.274.6.G1068. [DOI] [PubMed] [Google Scholar]
- 15.Shao R, Shi Z, Gotwals PJ, Koteliansky VE, George J, Rockey DC. Cell and molecular regulation of endothelin-1 production during hepatic wound healing. Mol Biol Cell. 2003;14:2327–2341. doi: 10.1091/mbc.02-06-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schnermann J, Zhu X, Shu X, Yang T, Huang Y, Kretzler M, Briggs J. Regulation of endothelin production and secretion in cultured collecting duct cells by endogenous transforming growth factor-β. Endocrinology. 1996;137:5000–5008. doi: 10.1210/endo.137.11.8895374. [DOI] [PubMed] [Google Scholar]
- 17.Dijke Pt, Hill CS. New insights into TGF-β-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Pascual F, Redondo-Horcajo M, Lamas S. Functional cooperation between Smad proteins and activator protein-1 regulates transforming growth factor-β-mediated induction of endothelin-1 expression. Circ Res. 2003;92:1288–1295. doi: 10.1161/01.RES.0000078491.79697.7F. [DOI] [PubMed] [Google Scholar]
- 19.Easter DW, Wade JB, Boyer JL. Structural integrity of hepatocyte tight junctions. J Cell Biol. 1983;96:745–749. doi: 10.1083/jcb.96.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chojkier M, Groszmann RJ. Measurement of portal-systemic shunting in the rat by using gamma-labeled microspheres. Am J Physiol. 1981;240:G371–G375. doi: 10.1152/ajpgi.1981.240.5.G371. [DOI] [PubMed] [Google Scholar]
- 21.Fallon MB, Abrams GA, McGrath JW, Hou Z, Luo B. Common bile duct ligation in the rat: a model of intrapulmonary vasodilatation and hepatopulmonary syndrome. Am J Physiol. 1997;272:G779–G784. doi: 10.1152/ajpgi.1997.272.4.G779. [DOI] [PubMed] [Google Scholar]
- 22.Goldner J. A modification of Masson trichrome technique for routine laboratory purposes. Am J Pathol. 1938;14:237–243. [PMC free article] [PubMed] [Google Scholar]
- 23.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 24.Tchekneva E, Lawrence M, Meyrick B. Cell-specific differences in ET-1 system in adjacent layers of main pulmonary artery. A new source of ET-1. Am J Physiol Lung Cell Mol Physiol. 2000;278:L813–L821. doi: 10.1152/ajplung.2000.278.4.L813. [DOI] [PubMed] [Google Scholar]
- 25.Inoue A, Yanagisawa M, Takuwa Y, Mitsui Y, Kobayashi M, Masaki T. The human preproendothelin-1 gene. J Biol Chem. 1989;264:14954–14959. [PubMed] [Google Scholar]
- 26.Ling Y, Zhang J, Luo B, Song D, Liu L, Tang L, Stockard C, Grizzle W, Ku D, Fallon MB. The role of endothelin-1 and the endothelin B receptor in the pathogenesis of experimental hepatopulmonary syndrome. Hepatology. 2004;39:1593–1602. doi: 10.1002/hep.20244. [DOI] [PubMed] [Google Scholar]
- 27.Thirunavukkarasu C, Yang Y, Subbotin VM, Harvey SAK, Fung J, Gandhi CR. Endothelin receptor antagonist TAK-044 arrests and reverses the development of carbon tetrachloride induced cirrhosis in rats. Gut. 2004;53:1010–1019. doi: 10.1136/gut.2003.026534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kamath P, Carpenter H, Lloyd R, McKusick M, Steers J, Nagorney D, Miller V. Hepatic localization of endothelin-1 in patients with idiopathic portal hypertension and cirrhosis of the liver. Liver Transplant. 2000;6:596–602. doi: 10.1053/jlts.2000.9735. [DOI] [PubMed] [Google Scholar]
- 29.Tieche S, De Gottardi A, Kappeler A, Shaw S, Sagesser H, Zimmermann A, Reichen J. Overexpression of endothelin-1 in bile duct ligated rats: correlation with activation of hepatic stellate cells and portal pressure. J Hepatol. 2001;34:38–45. doi: 10.1016/s0168-8278(00)00031-3. [DOI] [PubMed] [Google Scholar]
- 30.Shao R, Yan W, Rockey D. Regulation of endothelin-1 synthesis by endothelin-converting enzyme during wound healing. J Biol Chem. 1999;274:3228–3234. doi: 10.1074/jbc.274.5.3228. [DOI] [PubMed] [Google Scholar]
- 31.Asbert M, Gines A, Gines P, Jimenez W, Claria J, Salo J, Arroyo V, Rivera F, Rodes J. Circulating levels of endothelin in cirrhosis. Gastroenterology. 1993;104:1485–1491. doi: 10.1016/0016-5085(93)90360-o. [DOI] [PubMed] [Google Scholar]
- 32.Moore K, Wendon J, Frazer M, Krani J, Williams R, Badr K. Plasma endothelin immunoreactivity in liver disease and the hepatorenal syndrome. N Engl J Med. 1992;327:1774–1777. doi: 10.1056/NEJM199212173272502. [DOI] [PubMed] [Google Scholar]
- 33.Fouassier L, Chinet T, Robert B, Carayon A, Balladur P, Mergey M, Paul A, Poupon R, Capeau J, Barbu V, Housset C. Endothelin-1 is synthesized and inhibits cyclic adenosine monophosphate-dependent anion secretion by an autocrine/paracrine mechanism in gallbladder epithelial cells. J Clin Invest. 1998;101:2881–2888. doi: 10.1172/JCI2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramm G, Hoskins A, Greco S, Pereira T, Lewindon P. Signals for hepatic figrogenesis in pediatric cholestatic liver disease: review and hypothesis. Comp Hepatol. 2004;3:S5. doi: 10.1186/1476-5926-2-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taniai M, Higuchi H, Burgart L, Gores G. p16INK4a promoter mutations are frequent in primary sclerosing cholangitis (PSC) and PSC-associated cholangiocarcinoma. Gastroenterology. 2002;123:1090–1098. doi: 10.1053/gast.2002.36021. [DOI] [PubMed] [Google Scholar]
- 36.Rockey D, Fouassier L, Chung J, Carayon A, Vallee P, Rey C, Housset C. Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells. Hepatology. 1998;27:472–480. doi: 10.1002/hep.510270222. [DOI] [PubMed] [Google Scholar]
- 37.El-Youssef M, Mu Y, Huang L, Stellmach V, Crawford S. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J Pediatr Gastroenterol Nutr. 1999;28:386–392. doi: 10.1097/00005176-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Napoli J, Prentice D, Niinami C, Bishop G, Desmond P, McCaughan G. Sequential increases in the intrahepatic expression of epidermal growth factor, basic fibroblast growth factor and transforming growth factor β in a bile duct ligated rat model of cirrhosis. Hepatology. 1997;26:624–633. doi: 10.1002/hep.510260314. [DOI] [PubMed] [Google Scholar]
- 39.Tao L, Enzan H, Hayashi Y, Miyazaki E, Saibara T, Hiroi M, Toi M, Kuroda N, Naruse K, Jin Y, Guo L. Appearance of denuded hepatic stellate cells and their subsequent myofibroblast-like transformation during the early stage of biliary fibrosis in the rat. Med Electron Microsc. 2000;33:217–230. doi: 10.1007/s007950000022. [DOI] [PubMed] [Google Scholar]
- 40.Baer H, Friess H, Abou-Shady M, Berberat P, Zimmermann A, Gold L, Korc M, Buchler M. Transforming growth factor betas and their receptors in human liver cirrhosis. Eur J Gastroenterol Hepatol. 1998;10:1031–1039. doi: 10.1097/00042737-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Lamireau T, Le Bail B, Boussaire L, Fabre M, Vergenes P, Bernard O, Gautier F, Bioulac-Sage P, Rosenbaum J. Expression of collagens type I and IV, osteonectin and transforming growth factor beta-1 (TGFbeta1) in biliary atresia and paucity of intrahepatic bile ducts in infancy. J Hepatol. 1999;31:248–255. doi: 10.1016/s0168-8278(99)80221-9. [DOI] [PubMed] [Google Scholar]
- 42.LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte proliferation. Liver Int. 2001;21:73–80. doi: 10.1034/j.1600-0676.2001.021002073.x. [DOI] [PubMed] [Google Scholar]
- 43.Woods M, Wood E, Mitchell J, Warner T. Cyclic AMP regulates cytokine stimulation of endothelin-1 release in human vascular smooth muscle cells. J Cardiovasc Pharmacol. 2000;36:S404–S406. doi: 10.1097/00005344-200036051-00118. [DOI] [PubMed] [Google Scholar]
- 44.Woods M, Wood E, Mitchell J, Warner T. Signal transduction pathways involved in cytokine stimulation of endothelin-1 release from human vascular smooth muscle cells. J Cardiovasc Pharmacol. 2000;36:S407–S409. doi: 10.1097/00005344-200036051-00119. [DOI] [PubMed] [Google Scholar]