

Abstract
Cardiac hypertrophy is a significant independent risk factor for increased mortality, comprising of maladaptive changes in cellular, molecular and metabolic processes that ultimately lead to heart failure. However, cardiac hypertrophy represents a continuum from physiological to compensatory to pathological hypertrophy, so that treatment modalities aimed to shift hypertrophy towards the physiological phenotype would represent an attractive therapeutic strategy. Many of the physiological changes caused by thyroid hormone (TH) treatment may provide direct benefit to the failing heart. Recent experimental studies have shown that TH rapidly activates pro-survival PKB/Akt-mTOR signaling pathways, thus providing cytoprotection and increasing synthesis of normal contractile proteins and metabolic enzymes. TH induces a normal physiological phenotype by binding to nuclear TH receptors that regulate expression of specific genes which promote cell survival and enhance contractile function. Physiological cardiac growth occurs with a coordinated angiogenic response that normalizes myocardial perfusion during hypertrophy, and recent studies support a significant role for TH and its endothelial cell surface integrin receptors and nuclear receptors in neovascularization during TH-induced hypertrophy. The present review examines these molecular mechanisms and intracellular signaling pathways activated in thyroid hormone-induced cardiac hypertrophy that support its therapeutic potential in the treatment of heart disease.
Keywords: physiological hypertrophy, heart failure, protein translation, apoptosis, neovascularization
1. Introduction
Hypertrophy of the human heart is a clinical diagnosis defined by an increase in myocardial mass that has been described as the single most important independent risk factor for increased mortality (Vakili et al. 2001; Levy et al. 1990). On the other hand, not all cardiac hypertrophy is associated with depressed contractile function, and this has been designated as “compensatory” or adaptive hypertrophy when increased ventricular wall thickness can normalize wall stress and preserve systolic function, as might occur during initial phases of hypertensive disease. Numerous clinical studies have documented the progression of compensated cardiac hypertrophy with normal contractile function to depressed cardiac function (described as “decompensation”) ultimately resulting in heart failure, a highly lethal disease with eight-year mortality (American Heart Association, Heart Disease & Stroke Statistics, 2009). In contrast to this description of “pathological” hypertrophy, increased myocardial mass can also occur without adverse functional consequences in certain conditioned athletes and has been defined as “physiological” hypertrophy. Recent reviews have put into perspective the molecular and cellular basis for the continuum between physiological and pathological cardiac hypertrophy (Dorn 2007; Dorn & Force 2005). Understanding the molecular and biochemical changes that occur in physiological and pathological cardiac hypertrophy may direct the development of therapeutic strategies to treat patients with heart failure. Therefore, the induction of physiological hypertrophic responses including increased synthesis of normal contractile proteins and mitochondria, normalization of metabolic pathways and chamber geometry, reduction of fibrosis, and neovascularization in proportion with myocardial growth, could confer benefit to the pathologically hypertrophied heart. The observation that thyroid hormone (TH) or its analogues activate many of the beneficial aspects of physiological hypertrophy has raised the possibility of their therapeutic utility in the treatment of the post-infarcted heart or in heart failure. This topic has been the subject of several recent reviews (Dillmann 2009; Galli et al. 2009; Pantos et al. 2008; 2009a).
2. TH-induced physiological cardiac hypertrophy
Cardiac growth in response to thyroid hormones (L-thyroxine, T4; and 3,5,3′-tri-iodo-L-thyronine, T3) has been defined as physiological hypertrophy. At the basic cellular level, hypertrophy involves the enlargement of the cardiac myocyte; however, at the organ level, hypertrophic growth requires the proportional proliferative growth of other cell types within the heart including those of the vasculature. Thus, to elicit a physiological growth response, thyroid hormones must stimulate cellular proliferation as well as cardiomyocyte enlargement in a manner that produces normal myofibrillar assembly. Early studies showed that hypertrophy of the heart in response to TH resulted from increased rates of protein synthesis due to increases in translationally active ribosomes, a process that reflects an increase in “efficiency” of the translational machinery, and to a greater “capacity” of protein translation that involves increased cellular content of active ribosomes and mRNAs, requiring transcriptional activation by TH (reviewed by Morgan et al. 1987). Furthermore, thyroid hormones regulate expression of specific genes activated in normal maturational growth, and prevent the expression of the “fetal” gene program characteristic of pathological hypertrophy (reviewed by Dillmann 2002; Klein & Ojamaa 2001). Thus, the effects of TH on cellular processes are diverse and complex, are cell type specific and involve multiple regulatory mechanisms. It is this diversity of actions of THs that has attracted attention in the development of these molecules as potential therapeutic agents.
3. Signaling pathways activated in cardiac hypertrophy
There is substantial evidence that stimuli associated with pathological or maladaptive cardiomyocyte hypertrophy involve activation of heterotrimeric G protein-coupled receptors (GPCR) signaling primarily through Gαq and include agonists such as angiotensin II, endothelin-1 and catecholamines (reviewed by Clerk et al. 2007; Dorn & Force 2005; Molkentin & Dorn 2001). In contrast, physiological or adaptive hypertrophy appears to be mediated by activation of phosphatidylinositol 3′ kinase (PI3K-Class IA p110α-isoform) and signaling through protein kinase B, PKB/Akt, in response to ligands acting principally via membrane receptor tyrosine kinases, such as IGF-1 and insulin (McMullen et al. 2004; reviewed by Oudit et al. 2004 and Naga Prasad et al. 2003); (Figure A shows schematic representation of these signaling events). Shioi et al. (2000) first showed that PI3K determined heart size using transgenic mice with targeted overexpression of a constitutively active p110α isoform of PI3K (caPI3Kα) resulting in increased heart size; conversely, overexpression of a catalytically inactive PI3Kα (dnPI3Kα) molecule resulted in smaller hearts with no observable myocyte necrosis, apoptosis, interstitial fibrosis or contractile dysfunction. In response to pressure overload, the smaller hearts of dnPI3Kα transgenic mice hypertrophied to a similar extent as wildtype hearts; however, in response to exercise training (swimming), the dnPI3Kα mutant protein was able to significantly blunt the hypertrophic response in these mice, thus supporting a role for PI3Kα in mediating physiological but not pathological hypertrophy (McMullen et al. 2003). Further evidence supporting a role for the PI3K/Akt pathway in regulating cardiac hypertrophy came from cardiac-specific inactivation of a phosphatase (PTEN, phosphatase and tensin homolog on chromosome 10) that negatively regulates the PI3K/Akt pathway by dephosphorylating 3′-phosphorylated phosphoinositides. Thus, hearts of transgenic mice expressing an inactive PTEN protein showed significant hypertrophy (Crackower et al. 2002).
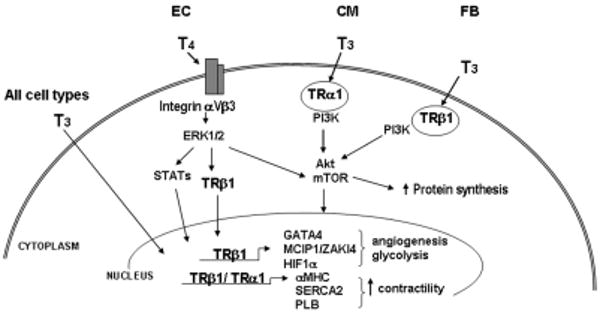
Figure. Schematic representation of thyroid hormone signaling in cardiac hypertrophy.
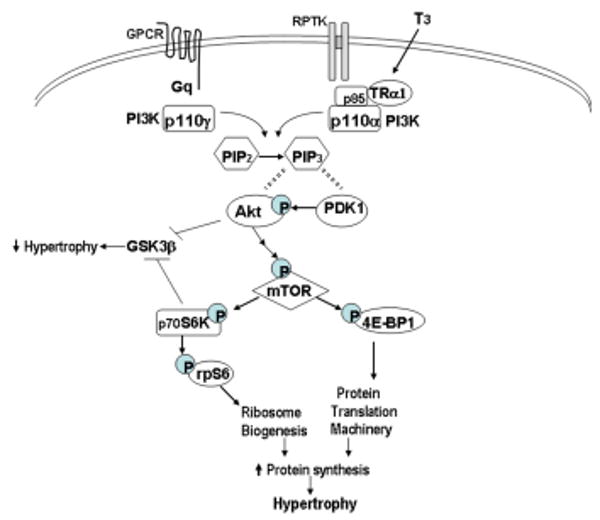
(A) In physiological hypertrophy, T3 binding to cytoplasmic TRα1 or growth factor binding to receptor protein tyrosine kinases (RPTK) activates PI3K isoform p110α through direct interactions with the p85 subunit of PI3K. p110α phosphorylates phosphatidylinositol PIP2 at the 3′ position to produce PIP3, which serves to bind phosphatidylinositol-dependent kinase (PDK1) and Akt, allowing PDK1 to phosphorylate Akt. Activation of Akt then leads to activation of mTOR (mammalian target of rapamycin) which regulates protein synthesis through downstream targets including S6 kinase (p70S6K) and initiation factor eIF4E binding protein (4E-BP1), resulting in increased rates of protein translation and ribosome biosynthesis, leading to cardiomyocyte hypertrophy. Akt and S6K also phosphorylate and inhibit glycogen synthase kinase (GSK3β), an inhibitor of protein synthesis, thereby promoting cardiac hypertrophy. In contrast, pathological hypertrophic agonists (i.e., angiotensin, catecholamines) acting through G-protein coupled receptors (GPCR) and Gq, activate the Akt/mTOR pathway by associating with the p110γ isoform of PI3K.
(B) Reported effects of thyroid hormones (T3, L-tri-iodothyronine; T4, L-thyroxine) on various cell types including endothelial cells (EC), cardiomyocytes (CM) and fibroblasts (FB). T3 activation of the PI3K/Akt/mTOR signal transduction pathway as been shown to be initiated by binding to cytoplasmic thyroid hormone receptors (TRα1 and TRβ1), resulting in increased protein synthesis and activation of a hypertrophic gene program. In endothelial cells, T4 has been reported to bind to a plasma membrane integrin receptor (αVβ3) and to activate the mitogen-activated protein kinase (ERK1/2) signaling pathway, leading to TRβ1 phosphorylation and/or translocation to the nucleus where its transcriptional activity is enhanced. Furthermore, T3 can enter the nucleus directly in virtually all cells and bind to nuclear TRs that regulate expression of target genes that increase cardiac contractility, stimulate angiogenesis, regulate cell metabolism and provide cytoprotection. (MCIP, inhibitor of calcineurin; HIF, hypoxia-inducible factor; MHC, myosin heavy chain; PLB, phospholamban; SERCA2, sarcoplasmic reticulum calcium ATPase; GATA4, transcription factor).
The changes in heart size elicited by PI3K activation are mediated by the immediate downstream effector kinase, PKB/Akt, that was shown to be activated in cardiomyocytes of the caPI3Kα transgenic mice and inhibited in the dnPI3Kα mice (Shioi et al. 2000). Furthermore, studies in transgenic mice with cardiac-restricted overexpression of constitutively active PKB/Akt lead to a significant increase in heart size, thus supporting a role for PKB/Akt in the regulation of cardiac growth (Condorelli et al. 2002; Shioi et al. 2002). PKB/Akt mediates the hypertrophic response by regulating several downstream targets including glycogen synthase kinase-3β (GSK-3β) (Antos et al. 2002; Michael et al. 2004) which is a negative regulator of cell growth (reviewed in Murphy & Steenbergen, 2005), and mammalian target of rapamycin (mTOR), a master modulator of protein synthesis (reviewed by Wang & Proud, 2006). Rapamycin, an inhibitor of mTOR activity, attenuates pressure overload hypertrophy induced by aortic constriction, and is partially effective in regressing established hypertrophy (McMullen et al. 2004). Downstream targets of mTOR are the S6 kinases (p70/85 and p54/56), the translation initiation factor binding protein (4E-BP1) and elongation factor (eEF2) kinase. 4E-BP1 plays a key regulatory role in the initiation of protein translation by binding to and inhibiting eukaryotic initiation factor 4E (eIF4E) initiation complex formation. Activation of mTOR results in phosphorylation of 4E-BP1 and eEF2 kinase that relieves their inhibitory effect on eIF4E and eEF2, respectively, thus enabling translation to proceed (reviewed by Tee & Blenis 2005). The S6 kinases (i.e., p70S6K) are also important regulators of protein translation by virtue of their ability to phosphorylate the 40S ribosomal subunit protein S6, which enables the translation of mRNAs containing a 5′ terminal oligopyrimidine (TOP) track that are important in ribosome biogenesis (Jefferies et al. 1997).
4. Signaling mechanisms in TH-induced physiological cardiac hypertrophy
The well characterized actions of thyroid hormones on gene expression fail to adequately explain the observed effects of TH on cardiomyocyte hypertrophy. Evidence has accumulated to unequivocally show that receptors for thyroid hormone (TRs) not only function as transcriptionally active proteins, but they also participate in cytoplasmic-initiated signaling processes resulting in unique biological responses as illustrated in Figure A & B (reviewed by Davis et al. 2008; Furuya et al. 2009; Moeller et al. 2006). TRs are derived from two genes that are alternatively spliced to give rise to four T3-binding receptor isoforms, of which TRα1 and TRβ1 are predominant in the cardiomyocyte with TRα1 expressed in both nuclear and cytosolic compartments (Kinugawa et al. 2005; Kenessey et al. 2006). Simoncini et al. (2000) were first to report T3-dependent TR-mediated activation of phosphatidylinositol 3′-kinase (PI3K) activity in human endothelial cells, and to subsequently publish that direct physical interaction of TRα1 with the p85α regulatory subunit of PI3K resulted in Akt phosphorylation and activation of endothelial nitric oxide synthase (Hiroi et al. 2006). Similar TH-dependent TR-mediated PI3K activation has been described in other cell types including human fibroblasts (Cao et al. 2005), pituitary cells (Storey et al. 2006) and cardiomyocytes (Kenessey & Ojamaa 2006). In experimental animal studies, Kuzman et al (2005) showed increased phosphorylation of Akt, S6 kinase (p70S6K) and mTOR in hypertrophied hearts of thyroxine treated rats. In light of these novel observations, extranuclear signaling activities of TH and its receptors offers a potential mechanism by which this hormone induces cardiomyocyte hypertrophy independent of its regulatory effects on gene expression.
In studies of cultured primary rat cardiomyocytes, we showed that wortmannin, a specific PI3K inhibitor, blocked T3-stimulated protein synthesis as measured by reduced [3H]-leucine incorporation into cellular protein, and prevented T3-induced myocyte hypertrophy (Kenessey & Ojamaa 2006). This response was initiated by T3-dependent activation of PI3K through a direct protein-protein interaction of cytosolic TRα1 and PI3K (Figure A). Furthermore, we have shown that TRα1 and PI3K are present in plasma membrane lipid raft fractions that are enriched in caveolin-3, a scaffold protein of caveolae, and that a complex of TRα1-PI3K-caveolin-3 co-immunoprecipitated from purified cardiomyocyte plasma membranes, suggesting their localization to caveolae (Kenessey & Ojamaa, unpublished data). A downstream target of PI3K and dependent on its enzymatic product, L-α-phophatidylinositol-3,4,5-triphopsphate (PIP3), is the phosphoinositide-dependent kinase, PDK1, and its target, PKB/Akt (Shioi et al. 2002; reviewed by Tee & Blenis 2005). T3 treatment resulted in Akt phosphorylation, and subsequent phosphorylation of mTOR in a time frame consistent with its location downstream of Akt in the signaling pathway. Two downstream targets of mTOR, 4E-BP1 and S6 kinases, are important regulators of protein translation, and their regulation by phosphorylation enables protein synthesis to proceed. We showed that T3 treatment of cardiomyocytes resulted in phosphorylation of S6K (p70S6K), followed by sustained phosphorylation of S6 ribosomal protein, and increased phosphorylation of 4E-BP1. These effects were blocked by inhibitors of PI3K and mTOR, further supporting a role for the PI3K-Akt-mTOR signaling pathway in T3-induced cardiomyocyte hypertrophy.
5. Other growth factors in TH-induced cardiac hypertrophy
Is there a role for other growth factors in mediating thyroid hormone-induced physiological cardiac hypertrophy, including those known to produce pathological hypertrophy such as angiotensin II, endothelin and catecholamines? As discussed above, myocyte hypertrophy initiated by these diverse stimuli results from the activation of partially overlapping signaling pathways that lead to an increase in protein synthesis. Several reports have suggested that the rennin-angiotensin-system (RAS) contributes to cardiac hypertrophy in hyperthyroidism, with increased expression and activity of renin and angiotensin II in cardiac tissue of TH-treated animals (Kobori et al. 1997; 1999). Treatment with angiotensin receptor (AT1R) antagonists or angiotensin converting enzyme (ACE) inhibitors has been shown to attenuate TH-induced cardiac hypertrophy (Hu et al. 2003; Kobori et al. 1997; Pantos et al. 2005). Whether these effects are attributable to direct actions of TH and RAS in the myocardium or to hemodynamic changes known to occur in hyperthyroidism remain unclear. A recent study has provided evidence that TH-induced cardiomyocyte hypertrophy through Akt-mTOR mechanism can be inhibited by AT1R blockade (Diniz et al. 2009). However, it remains to be reconciled how TH signaling through AT1R –Gq linked pathways (known to induce pathological hypertrophy) can induce a physiological hypertrophic phenotype.
6. TH-regulated transcription maintains physiological cardiac phenotype
As illustrated in Figure A, GPCR agonists that induce pathological hypertrophy also activate the PI3K-Akt signaling pathway; however, the activated PI3K isoform (p110γ) differs from the isoform (p100α) activated by growth factors mediating physiological hypertrophy. PI3K-p110γ is activated by recruitment to the plasma membrane by βγ subunits of activated Gq protein, whereas p110α is principally activated by binding to ligand-activated growth factor receptors. Intracellular signaling initiated by these distinct types of stimuli that ultimately result in either physiological or pathological hypertrophy converge at the Akt-mTOR junction to increase protein synthesis. Therefore, the mechanisms by which increased protein synthesis result in distinct forms of hypertrophy (adaptive/physiological or maladaptive/pathological) must necessarily involve stimulus-dependent regulation of gene expression, producing unique cardiac phenotypes. In this regard, thyroid hormones acting through nuclear receptors are perhaps the only known stimuli positioned to simultaneously regulate cardiac gene expression that translates into a normal phenotype and to induce cardiomyocyte hypertrophy by increasing protein synthesis.
TH regulation of cell phenotype can be classified into several modes of TH action: (1) effects mediated through TH receptors (TRα1, TRβ1) localized in the nucleus that interact with specific DNA elements on T3-responsive genes (historically defined as genomic action), or (2) effects initiated by T3-binding to cytosolic TRs with activation of intracellular signaling pathways including PI3K-Akt-mTOR, or (3) effects that are independent of TRs and initiated by TH binding to plasma membrane receptors (i.e., integrin αVβ3), resulting in an intracellular signaling response. These genomic and non-genomic activities of TH have been the topic of recent reviews (Davis et al. 2008; Moeller et al. 2006). All modes of TH action, whether initiated in the nucleus, cytosol or plasma membrane have the potential to influence gene expression. Recent studies focusing on cytosolic TR-initiated actions of TH showed that activation of the PI3K-Akt-mTOR pathway was necessary for TH induction of genes encoding hypoxia inducible factor (HIF-1α) and calcineurin inhibitor protein, ZAKI-4α (also known as modulatory calcineurin-interacting protein, MCIP2) (Cao et al. 2005; Moeller et al. 2005). HIF is a key mediator of angiogenesis and metabolic adaptation to hypoxia by increasing the expression of genes involved in glycolysis, glucose transport, cell survival, antioxidant enzymes and mitochondrial function (reviewed by Loor & Schumacker 2008). Whether other TH-induced cardiac genes are regulated in a non-genomic PI3K-dependent manner remains to be determined.
Bergh et al. (2005) have recently identified integrin αVβ3 as a cell surface receptor for TH. Binding of L-thyroxine (T4) or T3 (with lower affinity) resulted in rapid phosphorylation of mitogen-activated protein kinase (MAPK/ERK1/2), and its subsequent nuclear translocation and phosphorylation of TRβ1, thereby increasing TRβ1 transcriptional activity (Davis et al. 2000). Whether TH signaling initiates at integrins on cardiomyocytes or on other cardiac cell types such as endothelial cells has not been investigated. Integrins are a class of receptors that physically connect the intracellular sarcomeric contractile machinery to extracellular matrix proteins at specific plasma membrane sites comprising of both structural and signaling proteins, that play a significant role in mediating mechanical stress-induced cardiac hypertrophy (reviewed by Brancaccio et al. 2006; Samarel, 2005). It may, therefore, be interesting to consider a role for TH acting via integrin signaling to downstream targets such as Akt/GSK3β and ERK1/2, in modulating the hypertrophic and cell survival responses to hemodynamic overload.
The physiological cardiac phenotype induced by TH is mediated principally through its actions on specific genes by binding to nuclear TRs (reviewed by Klein & Ojamaa 2001; Dillmann 2002 & 2009). These T3-responsive genes encode both structural and regulatory proteins including myofibrillar proteins, such as the myosin heavy chains (MHC α and β), and the sarcoplasmic reticulum proteins, calcium-activated ATPase (SERCA2) and phospholamban. The increase in diastolic and systolic cardiac function observed with hyperthyroidism can be largely explained by changes in expression of these proteins (Kiss et al. 1994; Ladenson et al. 1992; Mintz et al. 1991; Rohrer et al. 1991). TH regulation of collagen gene expression in cardiac fibroblasts is likely responsible for the absence of fibrosis in TH-induced myocardial hypertrophy (Yao & Eghbali 1992). Other TH-responsive genes such as those of the adrenergic receptor complex including β-adrenergic receptors, guanine-nucleotide regulatory proteins, and adenylyl cyclases, contribute to increased adrenergic activity of the heart with hyperthyroidism resulting in increased heart rate, widened pulse pressure and increased cardiac output (Hoit et al. 1997; Ojamaa et al. 2000). Genes encoding several plasma-membrane ion transporters such as Na+/K+-ATPase, Na+/Ca2+ exchanger, and voltage-gated K+ channels are regulated by TH, leading to increases in inotropy and chronotropy (Gick et al. 1990; Gloss et al. 2001; Ojamaa et al. 1999; Reed et al. 2000). De et al. (2004) used a limited cDNA microarray analysis (588 genes) to study differentially expressed genes in TH-induced hypertrophied rat hearts. The results confirmed previously documented TH-regulated genes, but also identified TH regulation of genes involved in glucose and lipid metabolism. Recently, studies of microRNAs have shown that a cardiac-specific microRNA (miR-208) encoded by an intron of the αMHC gene is required for cardiomyocyte hypertrophy and its expression is regulated by TH (van Rooij et al. 2007). Hearts of miR-208 knockout mice resemble hyperthyroid hearts with physiological hypertrophy, absence of fibrosis and reduced expression of βMHC. These studies implicate miR-208 in the TH-signaling pathway and provide a novel mechanism by which TH regulates cardiac function and hypertrophy (reviewed by van Rooij & Olson 2007).
7. TH-induced neovascularization in cardiac hypertrophy
Physiological cardiac hypertrophy requires proportional increases in myocardial capillary density, whereas pathological hypertrophy arising from heart diseases such as aortic stenosis, dilated or ischemic cardiomyopathy has been shown to be associated with decreased capillarity (Karch et al. 2005; Rakusan et al. 1992). Therefore, in physiological or compensatory cardiac hypertrophy, growth promoting signals must simultaneously induce hypertrophy and angiogenic growth factor expression to maintain the balance between myocyte growth and coronary angiogenesis. This has been clearly illustrated in a transgenic mouse model of Akt-induced myocardial hypertrophy in which enhanced angiogenesis was associated with mTOR-dependent induction of myocardial vascular endothelial growth factor (VEGF) and angiopoietin-2 expression during the compensatory phase of hypertrophy; whereas inhibition of angiogenesis resulted in an accelerated conversion from compensatory hypertrophy to heart failure (Shiojima et al. 2005; reviewed by Shiojima & Walsh 2006). Thus, induction of angiogenic growth factors such as VEGF is an important component in compensatory or physiological hypertrophy, and several angiogenic transcription factors including GATA4 and hypoxia-inducible factor (HIF-1α) have been reported to be activated by hypertrophic stimuli including thyroid hormone (Heineke et al. 2007; Ma et al. 2004; Moeller et al. 2005; Molkentin et al. 1994; Sano et al. 2007). Early anatomical studies documented a coordinated angiogenic response to TH-induced myocardial growth in experimental animal models, suggesting that formation of new capillaries and arterioles served to normalize myocardial perfusion during hypertrophy (Chilian et al. 1985; Tomanek & Busch 1998). Recent studies have elucidated potential molecular mechanisms underlying TH-induced angiogenesis. Using a chick chorioallantoic membrane model of angiogenesis, Davis et al. (2004) showed that the proangiogenic effect of T4 was initiated at the endothelial cell membrane and was dependent on increased fibroblast growth factor (FGF2) expression and secretion. These authors subsequently reported the identification of integrin αVβ3 as the endothelial cell surface receptor for T4 and the initiation site for T4-induced activation of ERK1/2 signaling and induction of angiogenesis (Bergh et al. 2005). A potential role for nuclear TRs in regulating coronary angiogenesis has also been recently proposed. Studying transgenic mouse models, Makino et al. (2009) reported that cardiac capillary density was significantly reduced in TRβ deficient mice but not in TRα KO mice or cardiomyocyte-specific TRβ KO mice, suggesting that TRβ expression in endothelial cells plays a role in angiogenesis. These authors showed that coronary endothelial cells (EC) isolated from TRβ KO mice had reduced ability to form capillary networks than EC from either wildtype or TRα KO mice. Furthermore, in a pressure-overload model, T3 administration restored capillary density in the ventricular myocardium, normalized TRβ levels, induced coronary endothelial cell KDR/Flk1 (VEGF receptor) expression, and significantly increased coronary vascular conductance. Taken together, these data support a significant role for TH and its endothelial cell surface integrin receptors and nuclear TRs in neovascularization during TH-induced physiological hypertrophy.
8. TH protects cardiomyocytes from cell death during hypertrophy
Cardiac myocyte death is an irreversible cellular event occurring during stress-induced hypertrophy that can lead to decompensation and heart failure (reviewed by Diwan & Dorn 2006). In contrast, stimuli that promote physiological or adaptive hypertrophy signal principally through the PI3K and PKB/Akt pathway that is consistently associated with increased rates of protein synthesis and with cytoprotection (Matsui & Rosenzweig 2005). Cytoprotection occurs in part as a result of inhibition of cell death mediators, thus preventing apoptosis which is a highly regulated process involving either an extrinsic death receptor pathway or an intrinsic mitochondrial pathway that culminates in the degradation of proteins and cleavage of DNA. Recent studies have shown that activation of prosurvival kinases of the PI3K/Akt and ERK1/2 signaling pathways can protect the heart against ischemia reperfusion injury (reviewed by Hausenloy & Yellon 2004). Kuzman et al. (2005) showed that thyroid hormone treatment of cardiomyocytes cultured in serum-depleted conditions prevented cell apoptosis through an Akt-mediated mechanism. These authors subsequently showed that T3 treatment following myocardial infarction in experimental animals reduced cardiomyocyte apoptosis in the infarct border area which was associated with activation of the Akt signaling pathway (Chen et al. 2008). Using a Langendorff-rat heart model of ischemia reperfusion injury, Pantos et al. (2009) showed that T3 treatment during the reperfusion period significantly enhanced the recovery of function and reduced myocyte apoptosis. These data support an anti-apoptotic function of TH that may be important in preventing adverse cellular responses during TH-induced growth of the heart.
9. Therapeutic potential of TH treatment of patients with pathological hypertrophy or heart failure
If the beneficial characteristics of physiological hypertrophy were imposed on the maladaptive hypertrophied heart, then progression to heart failure may be averted. Konhilas et al. (2006) tested this hypothesis using exercise as the physiological growth stimulus, and found that the severity of the hypertrophic cardiomyopathy was significantly reduced. Since exercise may not be appropriate for heart failure patients, other more suitable molecular targets are necessary. TH treatment stimulates physiological cardiac hypertrophy with many molecular features that could be beneficial to the decompensated hypertrophied heart. In experimental animal models of myocardial ischemia, treatment strategies with TH or thyroid hormone analogues have shown improved contractile function, increased capillarity, and reduced cardiomyocyte apoptosis (reviewed by Pantos et al. 2008). The human experience with TH treatment strategies has also shown promise. Recently, a randomized, placebo-controlled trial using short-term treatment (3 days) with T3 replacement therapy in patients with chronic heart failure and low-T3 syndrome showed significant increases in stroke volume and left-ventricular end-diastolic volume with significant improvements in the neurohumoral profile including plasma levels of noradrenaline, B-type natriuretic peptide and aldosterone (Pingitore et al. 2008). Studies using longer-term treatment with L-T4 treatment of patients with idiopathic dilated cardiomyopathy showed improvement in cardiac contractility, exercise performance and circulatory function (Moruzzi et al. 1996). Emerging experimental and clinical evidence support a critical role of TH in maintaining normal cardiovascular function, and thus the potential therapeutic utility of TH (L-T3, L-T4 or thyroid analogues) in the treatment of decompensated “pathological” cardiac hypertrophy or heart failure has taken on renewed significance (reviewed by Galli et al. 2009).
10. Conclusions
Thyroid hormone treatment induces physiological cardiomyocyte hypertrophy by activation of intracellular signaling pathways which preserve cell survival, maintain normal myocyte protein expression, reduce fibrosis and promote neovascularization proportional to organ growth. These diverse pleiotropic actions of TH may provide the basis for its therapeutic utility in the setting of congestive heart failure or following myocardial ischemic injury.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antos CL, McKinsey TA, Trey N, Kutschke S, McAnally J, Shelton JM, Richardson JA, Hill JA, Olson EN. Activated glycogen synthase-3β suppresses cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2002;99:907–912. doi: 10.1073/pnas.231619298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ. Integrin αVβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinol. 2005;146:2864–2871. doi: 10.1210/en.2005-0102. [DOI] [PubMed] [Google Scholar]
- Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54:146–51. doi: 10.2337/diabetes.54.1.146. [DOI] [PubMed] [Google Scholar]
- Brar BK, Stephanou A, Pennica D, Latchman DS. CT-1 mediated cardioprotection against ischaemic re-oxygenation injury is mediated by PI3 kinase, Akt and MEK1/2 pathways. Cytokine. 2001;16:93–6. doi: 10.1006/cyto.2001.0951. [DOI] [PubMed] [Google Scholar]
- Brancaccio M, Hirsch E, Notte A, Selvetella G, Limbo G, Tarone G. Integrin signalling, The tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Buerke M, Murohara T, Skurk C, Nuss C, Tomaselli K, Lefer AM. Cardioprotective effect of insulin-like growth factor 1 in myocardial ischemia followed by reperfusion. Proc Natl Acad Sci USA. 1995;92:8031–5. doi: 10.1073/pnas.92.17.8031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YF, Kobayashi S, Chen J, Redetzke RA, Said S, Liang Q, Gerdes AM. Short term triiodo-L-thyronine treatment inhibits cardiac myocyte apoptosis in border area after myocardial infarction in rats. J Mol Cell Cardiol. 2008;44:180–187. doi: 10.1016/j.yjmcc.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarainen S, Sugden PH. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol. 2007;212:311–322. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Laccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MVG, Napoli C, Sadoshima J, Croce CM, Ross J. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci USA. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsh E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Davis PJ, Leonard JL, Davis FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2009;29:211–218. doi: 10.1016/j.yfrne.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Davis FB, Mousa SA, O'Connor L, Mohamed S, Lin HY, Cao HJ, Davis PJ. Proangiogenic action of thyroid hormone is fibroblast growth factor-dependent and is initiated at the cell surface. Circ Res. 2004;94:1500–1506. doi: 10.1161/01.RES.0000130784.90237.4a. [DOI] [PubMed] [Google Scholar]
- Davis PJ, Shih A, Lin HY, Martino LJ, Davis FB. Thyroxine promotes association of mitogen-activated protein kinase and nuclear thyroid hormone receptor (TR) and causes serine phosphorylation of TR. J Biol Chem. 2000;275:38032–38039. doi: 10.1074/jbc.M002560200. [DOI] [PubMed] [Google Scholar]
- De K, Ghosh G, Datta M, Konar A, Bandyopadhyay J, Bandyopadhyay D, Bhattacharya S, Bandyopadhyay A. Analysis of differentially expressed genes in hyperthyroid-induced hypertrophied heart by cDNA microarray. J Endocrinol. 2004;182:303–314. doi: 10.1677/joe.0.1820303. [DOI] [PubMed] [Google Scholar]
- Dillmann WH. Cellular action of thyroid hormone on the heart. Thyroid. 2002;12:447–452. doi: 10.1089/105072502760143809. [DOI] [PubMed] [Google Scholar]
- Dillmann W. Cardiac hypertrophy and thyroid hormone signaling. Heart Fail Rev. 2009 doi: 10.1007/s10741-008-9125-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz GP, Carneiro-Ramos MS, Barreto-Chaves MLM. Angiotensin type I receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3b/mROR signaling pathway. Basic Res Cardiol. 2009 doi: 10.1007/s00395-009-0043-1. 9 July ahead of print. [DOI] [PubMed] [Google Scholar]
- Diwan A, Dorn GW., II Decompensation of cardiac hypertrophy, cellular mechanism and novel therapeutic targets. Physiol. 2006;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- Dorn GW., II The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- Dorn GW, II, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya F, Lu C, Guigon CJ, Cheng SY. Nongenomic activation of phosphatidylinositol 3-kinase signaling by thyroid hormone receptors. Steroids. 2009;74:628–634. doi: 10.1016/j.steroids.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli E, Pingitore A, Iervasi G. The role of thyroid hormone in the pathophysiology of heart failure, clinical evidence. Heart Fail Rev. 2009 doi: 10.1007/s10741-008-9126-6. ahead of print. [DOI] [PubMed] [Google Scholar]
- Gloss B, Trost S, Bluhm W, Sanson E, Clark R, Winfein R, Janzen K, Giles W, Chassande O, Samarut J, Dillmann WD. Cardiac ion channel expression and contractile function in mice with deletion of thyroid receptor alpha or beta. Endocrinol. 2001;142:544–550. doi: 10.1210/endo.142.2.7935. [DOI] [PubMed] [Google Scholar]
- Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004;94:960–6. doi: 10.1161/01.RES.0000122392.33172.09. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury, targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Heineke J, Auger-Messier M, Xu J, Oka T, Sargent MA, York A, Klevitsky R, Vaikunth S, Suncan SA, Aronow BJ, Robbins J, Cromblehol TM, Molkentin JD. Cardiomyocyte GATA4 functions as a stress-responsive regulator of angiogenesis in the murine heart. J Clin Invest. 2007;117:3198–3210. doi: 10.1172/JCI32573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi Y Y, Kim HH, Ying H, Furuya F, Hyang Z Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci USA. 2006;103:14104–14109. doi: 10.1073/pnas.0601600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LW, Benvenuti LA, Liberti EA, Carneiro-Ramos MS, Barreto-Chaves MLM. Thyroxine-induced cardiac hypertrophy, influence of adrenergic nervous system versus renin-angiotensin system on myocyte remodeling. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1473–1480. doi: 10.1152/ajpregu.00269.2003. [DOI] [PubMed] [Google Scholar]
- Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5-TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997;16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch R, Neumann F, Ullrich R, Neumuller J, Podesser BK, Neumann M, Schreiner W. The spatial pattern of coronary capillaries in patients with dilated, ischemic or inflammatory cardiomyopathy. Cardiovasc Pathol. 2005;14:135–144. doi: 10.1016/j.carpath.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Kenessey A, Ojamaa K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J Biol Chem. 2006;281:20666–20672. doi: 10.1074/jbc.M512671200. [DOI] [PubMed] [Google Scholar]
- Kenessey A, Sullivan EA, Ojamaa K. Nuclear localization of protein kinase C-α induces thyroid hormone receptor-α1 expression in the cardiomyocyte. Am J Physiol Heart Circ Physiol. 2006;290:H381–H389. doi: 10.1152/ajpheart.00576.2005. [DOI] [PubMed] [Google Scholar]
- Kinugawa K, Jeong MY, Bristow MR, Long CS. Thyroid hormone induces cardiac myocyte hypertrophy in a thyroid hormone receptor α1-specific manner that requires TAK1 and p38 mitogen-activated protein kinase. Mol Endocrinol. 2005;19:1618–1628. doi: 10.1210/me.2004-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss E, Jakab G, Kranias EG, Edes I. Thyroid hormone-induced alterations in phospholamban protein expression, regulatory effects on sarcoplasmic reticulum Ca2+ transport and myocardial relaxation. Circ Res. 1994;75:245–251. doi: 10.1161/01.res.75.2.245. [DOI] [PubMed] [Google Scholar]
- Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509. doi: 10.1056/NEJM200102153440707. [DOI] [PubMed] [Google Scholar]
- Kobori H, Ichihara A, Miyashita Y, Hayashi M, Saruta T. Local renin-angiotensin system contributes to hyperthyroidism-induced cardiac hypertrophy. J Endocrinol. 1999;160:43–47. doi: 10.1677/joe.0.1600043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobori H, Ichihara A, Suzuki H, Takenaka T, Miyashita Y, Hayashi M, Saruta T. Role of the renin-angiotensin system in cardiac hypertrophy induced in rats by hyperthyroidism. Am J Physiol. 1997;273:H593–H599. doi: 10.1152/ajpheart.1997.273.2.H593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konhilas JP, Watson PA, Maass A, Boucek DM, Horn T, Stauffer BL, Luckey SW, Rosenberg P, Leinwand LA. Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res. 2006;98:540–548. doi: 10.1161/01.RES.0000205766.97556.00. [DOI] [PubMed] [Google Scholar]
- Kuzman JA, Gerdes AM, Kobayashi S, Liang Q. Thyroid hormone activates Akt and prevents serum starvation-induced cell death in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2005;39:841–844. doi: 10.1016/j.yjmcc.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Kuzman JA, Vogelsang KA, Thomas TA, Gerdes AM. L-Thyroxine activates Akt signaling in the heart. J Mol Cell Cardiol. 2005;39:251–258. doi: 10.1016/j.yjmcc.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Ladenson PW, Sherman IS, Baughman RL, Ray PE, Feldman AM. Reversible alterations in myocardial gene expression in a young man with dilated cardiomyopathy and hypothyroidism. Proc Natl Acad Sci USA. 1992;89:5251–5255. doi: 10.1073/pnas.89.12.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implication of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Different. 2008;15:686–690. doi: 10.1038/cdd.2008.13. [DOI] [PubMed] [Google Scholar]
- Ma Y, Freitag P, Zhou J, Brune B, Frede S, Fandrey J. Thyroid hormone induces erythropoietin gene expression through augmented accumulation of hypoxia-inducible factor-1. Am J Physiol Integr Comp Physiol. 2004;287:R600–R607. doi: 10.1152/ajpregu.00115.2004. [DOI] [PubMed] [Google Scholar]
- Makino A, Suarez J, Wang H, Belke DD, Scott BT, Dillmann WH. Thyroid hormone receptor-b is associated with coronary angiogenesis during pathological cardiac hypertrophy. Endocrinol. 2009;150:2008–2015. doi: 10.1210/en.2008-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function, the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38:63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- McMullen JR, Sherwood MC, Tarnasvski L, Zhang L, Dorfman AL, Shioi T, Izumo S. inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circ. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase (p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J Biol Chem. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- Michael A, Haq S, Chen X, Hsich, Cui L, Walters B, Shao Z, Bhattacharya K, Kilter H, Huggins G, Andreucci M, Periasamy M, Solomon RN, Liao R, Patten R, Molkentin JD, Force T. Glycogen synthase kinase-3β regulates growth, calcium homeostasis, and diastolic function in the heart. J Biol Chem. 2004;279:21383–21393. doi: 10.1074/jbc.M401413200. [DOI] [PubMed] [Google Scholar]
- Mintz G, Pizzarello R, Klein I. Enhanced left ventricular diastolic function in hyperthyroidism, noninvasive assessment and response to treatment. J Clin Endocrinol Metab. 1991;73:146–150. doi: 10.1210/jcem-73-1-146. [DOI] [PubMed] [Google Scholar]
- Moeller LC, Cao X, Dumitrescu AM, Refetoff S. Cytosolic action of thyroid hormone leads to induction of hypoxia-inducible factor-1α and glycolytic genes. Mol Endocrinol. 2005;19:2955–2963. doi: 10.1210/me.2004-0542. [DOI] [PubMed] [Google Scholar]
- Moeller LC, Cao X, Dumitrescu AM, Seo H, Refetoff S. Thyroid hormone mediated changes in gene expression can be initiated by cytosolic action of the thyroid hormone receptor β through the phosphatidylinositol 3-kinase pathway. Nuc Receptor Signal. 2006;4:1–4. doi: 10.1621/nrs.04020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Kalvakolanu DV, Markham BE. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the a-myosin heavy-chain gene. Mol Cell Biol. 1994;14:4947–4957. doi: 10.1128/mcb.14.7.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Dorn GW., II Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- Morgan HE, Gordon EE, Kira Y, Chua BHL, Russo LA, Peterson CJ, McDermott PJ, Watson PA. Biochemical mechanisms of cardiac hypertrophy. Ann Rev Physiol. 1987;49:533–543. doi: 10.1146/annurev.ph.49.030187.002533. [DOI] [PubMed] [Google Scholar]
- Moruzzi P, Doria E, Agostoni PG. Medium-term effectiveness of L-thyroxine treatment in idiopathic dilated cardiomyopathy. Am J Med. 1996;101:461–467. doi: 10.1016/s0002-9343(96)00281-1. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Inhibition of GSK-3β as a target for cardioprotection, the importance of timing, location, duration and degree of inhibition. Expert Opin Ther Targets. 2005;9:447–456. doi: 10.1517/14728222.9.3.447. [DOI] [PubMed] [Google Scholar]
- Naga Prasad SV, Perrino C, Rockman HA. Role of phosphoinositide 3-kinase in cardiac function and heart failure. Trends Cardiovasc Med. 2003;13:206–212. doi: 10.1016/s1050-1738(03)00080-x. [DOI] [PubMed] [Google Scholar]
- Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449–471. doi: 10.1016/j.yjmcc.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Pantos C, Mourouzis I, Cokkinos DV. Rebuilding the post-infarcted myocardium by activating “physiologic” hypertrophic signaling pathways: the thyroid hormone paradigm. Heart Fail Rev. 2009a doi: 10.1007/s10741-008-9111-0. [DOI] [PubMed] [Google Scholar]
- Pantos C, Mourouzis I, Saranteas T, Clave G, Ligeret H, Noack-Fraissignes P, Renard PY, Massonneay M, Perimenis P, Spanou D, Kostopanagiotou G, Cokkinos D. Thyroid hormone improves postischaemic recovery of function while limiting apoptosis: a new therapeutic approach to support hemodynamics in the setting of ischaemia-reperfusion? Basic Res Cardiol. 2009b;104:69–77. doi: 10.1007/s00395-008-0758-4. [DOI] [PubMed] [Google Scholar]
- Pantos C, Mourouzis I, Xinaris C, Papadopoulou-Daifoti Z, Cokkinos D. Thyroid hormone and “cardiac metamorphosis”: potential therapeutic implications. Pharmacol Ther. 2008;118:277–294. doi: 10.1016/j.pharmthera.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Pantos C, Paizis I, Mourouzis I, Moraitis P, Tzeis S, Karamanoli E, Mourouzis C, Karageorgiou H, Cokkinos DV. Blockade of angiotensin II type 1 receptor diminishes cardiac hypertrophy, but does not abolish thyroxin-induced preconditioning. Horm Metab Res. 2005;37:500–504. doi: 10.1055/s-2005-870317. [DOI] [PubMed] [Google Scholar]
- Rakusan JC, Flanagan MF, Geva T, Southern J, Van Praagh R. Morphometry of human coronary capillaries during normal growth and the effect of age in left ventricular pressure-overload hypertrophy. Circ. 1992;86:2223–2232. doi: 10.1161/01.cir.86.1.38. [DOI] [PubMed] [Google Scholar]
- Reed TD, Babu GJ, Ji Y, Zilberman A, Ver Heyen M, Wuytack F, Periasamy M. The expression of calcium SR transport ATPase and the Na(+)/Ca(2+) exchanger are antithetically regulated during mouse cardiac development in hypo/hyperthyroidism. J Mol Cell Cardiol. 2000;32:453–464. doi: 10.1006/jmcc.1999.1095. [DOI] [PubMed] [Google Scholar]
- Rohrer DK, Hartong R, Dillmann WH. Influence of thyroid hormone and retinoic acid on slow sarcoplasmic reticulum Ca2+ATPase and myosin heavy chain alpha gene expression in cardiac myocytes. J Biol Chem. 1991;266:8638–8646. [PubMed] [Google Scholar]
- Samarel AM. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;289:H2291–H2301. doi: 10.1152/ajpheart.00749.2005. [DOI] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komura I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000;19:2537–2548. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioi T, McMullen JR, Kang PM, douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/Protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt-PKB signaling pathway. Genes Devel. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey NM, Gentile S, Ullah H, Russo A, Muessel M, Erxleben C, Armstrong DL. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone. Proc Natl Acad Sci USA. 2006;103:5197–5201. doi: 10.1073/pnas.0600089103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol. 2005;16:29–37. doi: 10.1016/j.semcdb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Vakili BA, Okin PM, Devereux RB. Prognostic implication of left ventricular hypertrophy. Am Heart J. 2001;141:334–341. doi: 10.1067/mhj.2001.113218. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiol. 2006;21:362–369. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- Yao J, Eghbali M. Decreased collagen gene expression and absence of fibrosis in thyroid hormone-induced myocardial hypertrophy. Circ Res. 1992;71:831–839. doi: 10.1161/01.res.71.4.831. [DOI] [PubMed] [Google Scholar]