

Abstract
Background:
Patients with amnestic mild cognitive impairment (MCI) represent an important clinical group as they are at increased risk of developing Alzheimer disease (AD). 11C-PIB PET is an in vivo marker of brain amyloid load.
Objective:
To assess the rates of conversion of MCI to AD during a 3-year follow-up period and to compare levels of amyloid deposition between MCI converters and nonconverters.
Methods:
Thirty-one subjects with MCI with baseline 11C-PIB PET, MRI, and neuropsychometry have been clinically followed up for 1 to 3 years (2.68 ± 0.6 years). Raised cortical 11C-PIB binding in subjects with MCI was detected with region of interest analysis and statistical parametric mapping.
Results:
Seventeen of 31 (55%) subjects with MCI had increased 11C-PIB retention at baseline and 14 of these 17 (82%) clinically converted to AD during follow-up. Only one of the 14 PIB-negative MCI cases converted to AD. Of the PIB-positive subjects with MCI, half (47%) converted to AD within 1 year of baseline PIB PET, these faster converters having higher tracer-retention values than slower converters in the anterior cingulate (p = 0.027) and frontal cortex (p = 0.031). Seven of 17 (41%) subjects with MCI with known APOE status were ɛ4 allele carriers, this genotype being associated with faster conversion rates in PIB-positive subjects with MCI (p = 0.035).
Conclusions:
PIB-positive subjects with mild cognitive impairment (MCI) are significantly more likely to convert to AD than PIB-negative patients, faster converters having higher PIB retention levels at baseline than slower converters. In vivo detection of amyloid deposition in MCI with PIB PET provides useful prognostic information.
GLOSSARY
- AD
= Alzheimer disease;
- ADAS
= Alzheimer’s Disease Assessment Scale;
- CERAD
= Consortium to Establish a Registry for Alzheimer’s Disease;
- CVLT
= California Verbal Learning Test;
- MCI
= mild cognitive impairment;
- MNI
= Montreal Neurological Institute;
- PIB
= Pittsburgh compound B;
- ROI
= region of interest;
- SPM
= statistical parametric mapping;
- WMS-R
= Wechsler Memory Scale–Revised.
There are various possible causes for memory impairment in patients who present with amnestic mild cognitive impairment (MCI). Frequently, however, there is a neurodegenerative basis to this clinical entity—most commonly Alzheimer disease (AD).1,2 Subjects with MCI are therefore at increased risk of developing dementia, and overall their rate of progression to AD is typically 10%–15% per year.3,4 However, not all subjects with MCI will progress to dementia—some recover, and longitudinal studies have focussed on identifying predictive biomarkers of conversion to AD, including neuropsychological,5 neuroimaging,6–8 and CSF9 markers. In view of the 50% incidence of AD type pathology in subjects with MCI, it follows that any in vivo marker of such pathology could potentially be a candidate for predicting disease progression.
The pathologic hallmarks of AD are extracellular beta-amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles. Neuropathologic studies on postmortem brain tissue have reported an increasing prevalence of AD pathology in elderly nondemented subjects, amnestic MCI, and probable AD cases. However, a difficulty with autopsy studies is that inevitably no longitudinal outcome data are available. PET now provides an opportunity to correlate in vivo the presence of brain amyloid deposition with outcome in health and MCI, as this can be detected using the thioflavin-based radiotracer Pittsburgh compound B (PIB).10 Long-term follow-up studies have the potential to determine the utility of 11C-PIB PET in predicting who will develop AD.11 Furthermore, studies suggest that 11C-PIB may be more sensitive than CSF Aβ and tau measurements for detecting an early AD process in MCI.12
In this study, we sought to determine the rates at which subjects with amnestic MCI with and without an increased amyloid load (as assessed by 11C-PIB PET) converted to clinically probable AD over 1 to 3 years and to compare the levels of amyloid deposition between those PIB-positive subjects with MCI who were faster and slower converters.
METHODS
Subjects.
Thirty-one subjects fulfilling the Petersen et al. criteria3 for amnestic MCI were studied. Fourteen of these subjects with MCI (mean age 66.6 years: SD 9.6) were recruited from UK Hospitals (Imperial College Healthcare NHS Trust [London], The National Hospital for Neurology and Neurosurgery [London], St. Margaret’s Hospital [Epping, and Victoria Hospital [Swindon]). The remaining 17 subjects (mean age 71.7 years: SD 5.3) were recruited from the University Hospital of Turku, Finland. The demographic data of subjects with MCI and controls according to center are presented in table e-1 on the Neurology® Web site at www.neurology.org. The majority of subjects with MCI enrolled to the study were newly diagnosed cases. All subjects with MCI had a neurologic examination, neuropsychological assessment, and routine blood analysis. The clinical criteria for MCI were operationalized as 1) subjective memory complaint by the patient, preferably corroborated by an informant, 2) objective memory impairment as assessed by performance below age-matched normals on at least one neuropsychological measure of memory, 3) relatively normal performance in other cognitive domains, 4) intact activities of daily living, and 5) no dementia. A strict cutoff score was not applied for the definition of objective memory impairment. However, the appropriate age-matched normative values were used when determining evidence for objective memory impairment on performance on tests of memory including word list savings (%) of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD)13 test battery, Wechsler Memory Scale–Revised (WMS-R)14 (Turku), Alzheimer’s Disease Assessment Scale (ADAS) word list learning with delayed recall,15 California Verbal Learning Test (CVLT),16 and immediate and delayed recall of a modified complex figure17 (London).
Twenty-seven of the 31 subjects had their 11C-PIB PET and MRI at baseline as part of previous studies comparing 11C-PIB uptake in MCI and controls.18,19
11C-PIB PET data from 26 controls (14 London20 [mean age 64.6 years: SD 6.3], 12 Turku21 [mean age 66.2 years: SD 6.8]) were compared with our MCI group data. The controls were healthy volunteers without any history or evidence of neurologic or psychiatric disease. All underwent clinical examination and had no evidence of memory impairment on neuropsychological testing. Informed written consent was obtained from all participating subjects and ethical approval was obtained from the Ethics Committees of Hammersmith and Queen Charlotte’s Hospitals and the Southwest Finland Health Care District.
Clinical follow-up.
The subjects with MCI were clinically followed for periods of 1 to 3 years after their baseline 11C-PIB PET. All subjects with MCI who had a PIB PET scan at baseline were reviewed at follow-up. Over this follow-up period, there was either no change in their diagnosis and they still fulfilled criteria for MCI (nonconverters) or their symptoms and clinical performance declined to the extent that they fulfilled National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association criteria22 for a diagnosis of probable AD (converters).
Brain imaging.
MRI.
All subjects had T1-weighted volumetric MRI to allow structure-function coregistration with 11C-PIB PET images. Additionally, T2-weighted MRI was performed to exclude structural lesions.
11C-PIB PET.
UK.
All subjects with MCI and healthy volunteers were scanned at the Cyclotron Building, Hammersmith Hospital, London, using a Siemens ECAT EXACT HR+ camera in 3-dimensional acquisition mode.23 Prior to the injection of 11C-PIB, a 10-minute transmission scan was performed to measure tissue attenuation of 511 keV γ-radiation. The 11C-PIB PET protocol has been previously described.20 Subjects with MCI received 367 ± 25 MBq 11C-PIB injected IV, followed by a 90-minute dynamic emission scan. 11C-PIB was manufactured and supplied by GE Healthcare, Hammersmith Imanet, UK.
Turku.
Subjects with MCI and healthy volunteers were scanned with a GE Advance camera, using a previously described protocol.24 The dynamic PET emission scans were acquired in 3-dimensional mode during a 90-minute period and transmission scans performed to measure tissue attenuation of 511 keV γ-radiation. 11C-PIB was manufactured by Turku PET Centre, Finland. The mean injected dose of 11C-PIB for subjects with MCI was 487 ± 44 MBq.
Image analysis.
Creation of 11C-PIB target region: Cerebellar cortex ratio images.
All UK and Turku PIB PET images were analyzed at a central site (Cyclotron Building, Hammersmith Hospital, London), by a standardized method of analysis previously described.20 In summary, using the cerebellar cortex as a reference region, we produced parametric ratio images of PIB retention by normalizing the 60-minute to 90-minute (late) summation images to the mean cerebellar cortical uptake value over this same period. Image analysis was performed using Analyze (Mayo Clinic, MN) and Statistical Parametric Mapping software (SPM99; Wellcome Department of Imaging Neuroscience, University College London). Using individual T1-weighted MRIs as a template, each parametric image was normalized into Montreal Neurological Institute (MNI) space and PIB retention assessed by both region of interest (ROI) analysis and statistical parametric mapping (SPM).
Statistical analysis.
ROI analysis.
Using an in-house probabilistic brain atlas25 that creates an individualized anatomic template for each subject, we quantified average PIB retention (left and right) in the anterior cingulate, posterior cingulate, frontal, temporal, parietal, and occipital cortex.
All PIB PET dynamic data (London and Turku) were acquired over 90 minutes and expressed as a 60-minute to 90-minute target region: cerebellar ratio of PIB retention, minimizing between-site variability by using an internal reference for each subject. Individually, PIB retention ratio values for each subject with MCI were compared to that of the control mean of their scanning site. We considered PIB binding to be increased in each subject with MCI if ≥2 SD greater than the control mean in all 6 of our predefined ROIs.
Increases in PIB retention in the subjects with MCI relative to their control mean were described as a percentage change. Additionally, we used univariate analysis of variance to detect significant differences in regional PIB binding between MCI and control groups, with the 2 different PET centers being set as fixed factors, and using the p plot method and Hochberg correction to control for multiple comparisons.26 Using these methods, we also compared PIB retention between MCI converters and nonconverters. Finally, we considered PIB-positive subjects with MCI alone and compared differences in regional PIB binding in the PIB-positive faster converters (conversion to AD within 1 year follow-up) to that in the combined group of PIB-positive slower converters (conversion to AD after more than 1 year) and PIB-positive nonconverters (no conversion to date).
A secondary statistical analysis was performed using age as a covariate, the rest of the analysis being otherwise identical.
APOE ɛ4 status.
APOE status was available in 17 of our 31 subjects with MCI (Turku cohort). For these subjects, we applied binomial chi-square tests to assess the relationship between APOE ɛ4 status and rates of conversion.
Statistical tests were performed using SPSS for Windows 14.0 statistical software.
SPM.
In addition to ROI analysis, we also interrogated PIB ratio images with SPM in order to localize at a voxel level clusters of significant differences in mean PIB retention between MCI converters and nonconverters, and between PIB-positive faster and PIB-positive slower and nonconverters. We smoothed our normalized PIB ratio images using a Gaussian kernel of 6 mm. Between-group comparisons were made using a voxel threshold of p < 0.0001 (converters vs nonconverters) and p < 0.01 (faster converters vs slower and nonconverters), with an extent threshold of 200 voxels. We did not apply grand mean or proportional scaling to the data, and the 2 scanning sites were considered as covariates when performing the analysis. Any resulting clusters with a corrected p value of <0.05 are reported.
RESULTS
ROI analysis.
Baseline.
Subjects with MCI and controls.
The mean PIB retention ratio values in the subjects with MCI are shown in table 1. When compared to controls, the subjects with MCI showed a 41% increase in PIB retention in the anterior cingulate (p < 0.01), 40% increase in the posterior cingulate (p < 0.01), 37% increase in the frontal cortex (p < 0.01), 36% increase in the parietal cortex (p < 0.01), 28% increase in the temporal cortex (p < 0.01), and 20% increase in the occipital cortex (p < 0.01). Significance levels remained unchanged after applying age as a covariate.
Table 1 Demographic data and 11C-PIB mean retention ratio values in MCI subgroups
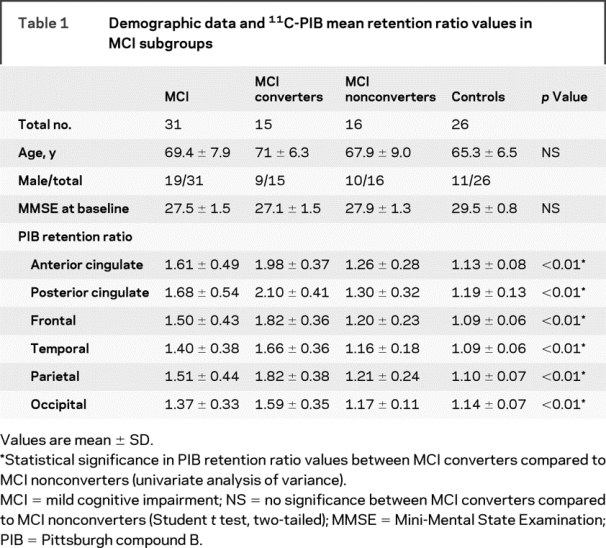
Individually, 17 of 31 (55%) subjects with MCI had significantly increased PIB retention values compared to controls at baseline in all ROIs. Two other subjects with MCI demonstrated isolated increases in PIB retention in the anterior cingulate and frontal cortex (see Discussion).
Follow-up.
Subjects with MCI.
During a 12–36-month follow-up period after their baseline PIB PET scan, 15 (4 London, 11 Turku) of the 31 subjects with MCI (48%) clinically converted to AD. Fourteen of these 15 converters were PIB-positive at baseline, making the percentage conversion rate in the PIB-positive subgroup 82% (14 out of 17) over this same follow-up period. One converter had small increases in PIB retention in the anterior cingulate and frontal cortex alone. When assessed by center, the percentage conversion rate in the PIB-positive subgroup was 57% for London (4 out of 7) and 100% for Turku (all 10 PIB-positive subjects with MCI converted to AD).
MCI converters vs nonconverters.
Compared to MCI nonconverters, the converters showed higher PIB retention in all cortical brain regions (p < 0.01 both with and without correction for age) (table 1).
MCI PIB-positive faster vs combined PIB-positive slower and nonconverters.
As a group, the PIB-positive faster converters had higher PIB retention values compared to the combined subgroup of slower and nonconverters in the anterior cingulate (p = 0.027) and frontal cortex (p = 0.031), although this did not survive correction for multiple comparisons (table 2). Figure 1 illustrates individual ratio values in PIB-positive faster, slower, and nonconverting subjects with MCI compared to controls. In total, 8 (47%) of the 17 PIB-positive subjects with MCI were faster converters, having converted to a diagnosis of clinically probable AD within 1 year of follow-up after their baseline 11C-PIB PET scan.
Table 2 Demographic data and 11C-PIB mean retention ratio values in PIB-positive MCI subgroups
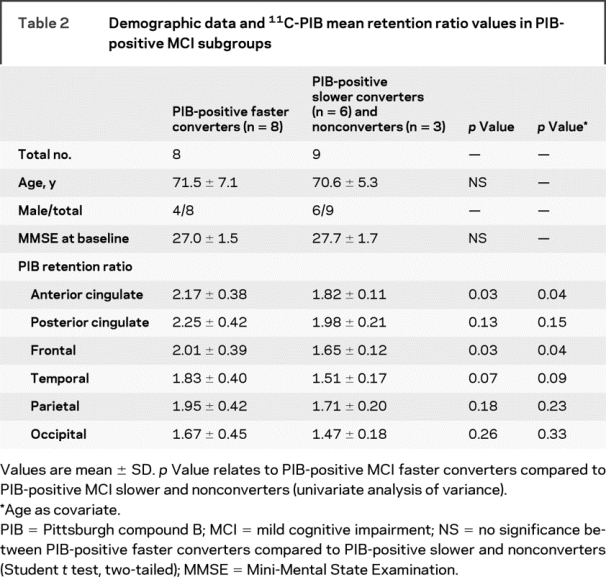
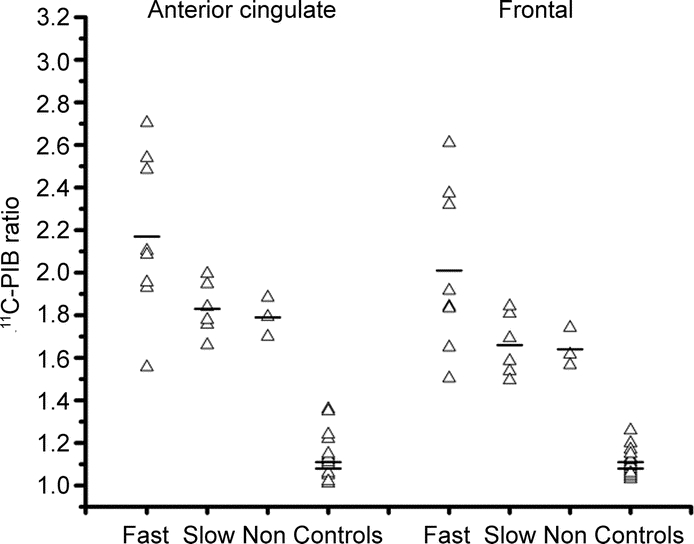
Figure 1 Amyloid deposition in mild cognitive impairment (MCI) subgroups
Scatter plot showing the distribution of individual Pittsburgh compound B (PIB) retention values in PIB-positive MCI subgroups: fast, slow, and nonconverters compared to controls (top line = Turku control mean; bottom line = London control mean).
APOE ɛ4 status.
An APOE ɛ4 allele was present in 41% (7 out of 17) of the subjects with MCI in whom the APOE ɛ4 status was known. Six of these 7 APOE ɛ4 carriers were PIB-positive. There was an association between APOE ɛ4 status in the PIB-positive subjects with MCI and rate of conversion, with all 4 of the faster converters with known genotype (100%) being APOE ɛ4 carriers (p = 0.035; Pearson chi-square). Two out of 6 (one-third) of the PIB-positive slow converters were APOE ɛ4 carriers.
SPM.
MCI converters vs nonconverters.
SPM detected significantly higher 11C-PIB uptake in the MCI converters compared to nonconverters in the anterior and posterior cingulate, frontal, temporal, and parietal cortices, with local maxima in the frontal cortex bilaterally (figure 2).

Figure 2 Statistical parametric mapping analysis: Localization of increased 11C-PIB retention in mild cognitive impairment (MCI) converters
Surface rendering is used to illustrate the cortical areas (red-yellow) where 11C-PIB retention is significantly increased in MCI converters compared to MCI nonconverters. (Corrected p value at cluster level <0.001.)
MCI PIB-positive faster converters vs combined PIB-positive slower and nonconverters.
SPM confirmed the findings from ROI analysis; significantly higher PIB binding was detected in the PIB-positive faster converters compared to the combined group of PIB-positive slower and nonconverters and was most marked in the superior frontal cortex. SPM additionally localized significantly increased 11C-PIB binding in the temporal cortex of faster compared to slower and nonconverters (figure 3).

Figure 3 Statistical parametric mapping: Comparison of 11C-PIB retention in PIB-positive mild cognitive impairment (MCI) subgroups
Sagittal (A) and coronal sections (B and C) illustrating increased 11C-PIB retention in anterior cingulate, frontal, and temporal cortex in the PIB-positive MCI faster converters compared to PIB-positive slower converters and nonconverters. Corrected p value at cluster level <0.001.
DISCUSSION
In this 11C-PIB PET study, we found that 17 of 31 (55%) subjects with amnestic MCI had significantly increased PIB retention compared to controls. This finding is consistent with the prevalence of amyloid deposition previously reported in 11C-PIB PET studies in subjects with amnestic MCI.27,28 When compared to nonconverters, MCI converters were found to have significantly increased PIB retention in all designated ROIs. Individually, 15 of 31 subjects with MCI (48% of total) converted to a diagnosis of AD during a follow-up period which ranged from 1 to 3 years (2.9 years ± 0.5 in converters; 2.5 years ± 0.7 in nonconverters). Fourteen of these 15 subjects were PIB-positive at baseline.
A recent PET study11 reported that one third of their 21 subjects with MCI, all PIB positive, converted to AD over 8 months. Only 12 of 21 of their subjects with MCI were categorized as amnestic, but all 7 converters were of the amnestic subtype. Eighty-two percent of our PIB-positive amnestic subjects with MCI converted to a diagnosis of AD over a 3-year follow-up period. The conversion rates to AD in PIB-positive compared to PIB-negative subjects with MCI in these 2 studies suggest that in vivo detection of amyloid deposition may provide useful prognostic information with respect to stratifying those amnestic patients with MCI at increased risk of dementia.
In contrast to population-based studies in MCI, where some subjects recover,29 all our subjects with MCI continue to have evidence of memory impairment.
A recent neuropathologic study in subjects with MCI reported that the majority had “prodromal or incipient” AD with diffuse neocortical amyloid deposits and frequent medial temporal lobe neurofibrillary tangles, albeit at insufficient levels to fulfill AD criteria.1 One way to investigate the pathologic changes present in the earliest phase of AD is to study familial AD mutation carriers. Recently, 11C-PIB PET studies have been performed in such patients and interestingly point mutations30 and deletions31 in both symptomatic and asymptomatic carriers of the presenilin-1 gene and duplication of the amyloid precursor protein locus32 have all been shown to lead to increases in 11C-PIB uptake, which are predominantly striatal—a pattern distinct from that seen in sporadic AD. Follow-up studies will reveal how the pattern of 11C-PIB uptake will evolve with the development and progression of clinical symptoms.
In a longitudinal 11C-PIB PET follow-up of patients with early AD over 2 years,33 researchers found that the amyloid load in their AD group remained relatively stable. This finding, in addition to PIB PET findings in familial AD cases, suggests that amyloid deposition occurs early in the clinical evolution of AD. It is less clear, however, at what stage amyloid deposition, as detectable by PIB PET, plateaus in any given patient during the clinical transition from MCI to AD.
We found that our PIB-positive more rapid converters had a significantly increased amyloid burden in the anterior cingulate and frontal cortices compared to our PIB-positive slower and nonconverters that fell within the reported range for patients with AD.20 This suggests that our MCI faster converters may have already reached their amyloid load plateau. While the slower converters had lower levels of amyloid load, these were also within the reported Alzheimer range, and so may represent a “prodromal or incipient” AD phase at the time of scanning.
Alternatively, some separate factor, for example, APOE status or levels of education,34 may have influenced the difference in rates of conversion and amyloid burden between the 2 groups. Postmortem studies have shown correlations between the presence of an APOE ɛ4 allele and higher Aβ burden in the brains of patients with sporadic AD.35,36 A separate study found that levels of Aβ protein deposition varied according to APOE genotype in elderly subjects both with and without dementia, the highest mean values being associated with the presence of at least one ɛ4 allele.37 A limitation of this current study is that we did not possess APOE ɛ4 status in all of our subjects with MCI, given that a previous 11C-PIB PET study in AD, examining the association between clinical severity and amyloid plaque load, reported APOE ɛ4 status to be a possible confounding factor.38 Here, the authors found an association between dementia severity and 11C-PIB uptake in AD, ROI analysis showing that individuals with more severe dementia had higher 11C-PIB uptake bilaterally in the frontal and anterior cingulate cortices and putamina. In our cohort, we found an association between APOE ɛ4 status in our PIB-positive subjects with MCI and rate of clinical conversion to AD, with all faster converters being APOE ɛ4 carriers.
Two subjects showed small selective increases in PIB retention in the anterior cingulate and frontal cortex alone. Patient 1 is an 80-year-old woman whose clinical diagnosis during a 3-year follow-up period has remained MCI. Patient 2 is a 64-year old man who at his 3-year follow-up had clinically converted to AD. A number of 11C-PIB PET studies have demonstrated increased amyloid deposition with or without evidence of objective memory impairment in asymptomatic elderly controls.39,40 However, given their documented memory impairment, it is more likely that the focally increased amyloid burden in patient 1 represents preclinical AD. Further clinical and PIB PET follow-up in this patient will determine the significance of this increased amyloid burden, while further PIB PET follow-up in all our subjects with MCI will allow us to follow, in vivo, the progression over time in amyloid deposition and its relevance to the clinical conversion to AD.
AUTHOR CONTRIBUTIONS
Statistical Analysis conducted by A. Okello, Imperial College London, UK.
ACKNOWLEDGMENT
The authors thank Hope McDevitt, Andreanna Williams, James Anscombe, and Andrew Blyth for help with scanning.
DISCLOSURE
Dr. Okello has received honoraria from Elan for image analysis services and receives salary support from the Alzheimer’s Research Trust. Dr. Koivunen reports no disclosures. Dr. Edison has received speaker honoraria from Novartis and has received salary support from the Medical Research Council as a clinical research fellow. Dr. Archer has received salary support from the Alzheimer’s Research Trust. Dr. Turkheimer has received research support from The Royal Society [JP0871550 (Principal Investigator)]; CRUK-EPSRC (Engineering and Physical Sciences Research Council) [C2536/A10337 (Co-Principal Investigator)]; EPSRC [EP/E049451/1 (Co-Principal Investigator)]; and from the Medical Research Council [U1200.04.004.000001.01 (Principal Investigator)]. Dr. Någren reports no disclosures. Dr. Bullock serves as Co-editor of CNS Neuroscience and Therapeutics. Dr. Walker has received funding for travel from GE Healthcare to attend the European Congress of Psychiatry, Lisbon; has received consultancy and speaker fees and research support from GE Healthcare, consultancy fees from Bayer Healthcare, and research support from Lundbeck. Dr. Kennedy reports no disclosures. Dr. Fox receives research support from the Medical Research Council [#G0601846 and # G0801306 (Principal Investigator)], the NIH [#AG024904 (Co-investigator)], the Alzheimer’s Research Trust [#ART/RF/2007/1 (Principal Investigator)], and the EPSRC (Engineering and Physical Sciences Research Council) [#GR/S48837/01 (Principal Investigator)]; has received research support (through the Dementia Research Centre) from the Alzheimer’s Research Trust; has received honoraria for image analysis services and/or consultancy fees from Elan, Wyeth, Lundbeck, Abbott, Eisai, IXICO, Neurochem, and GE Healthcare; and serves on a scientific advisory board for Alzforum. Dr. Rossor serves on a Safety Monitoring Committee for Wyeth (for which he receives payment), serves as an editorial board member for the Journal of Neurology, Neurosurgery and Psychiatry (Editor in Chief), and receives research support from the Department of Health [UK Coordinating Center for DeNDRoN (Director)], the Medical Research Council, and the Alzheimer’s Research Trust. Dr. Rinne serves as an Associate Editor of the Journal of Alzheimer’s Disease; serves as a consultant to GE Healthcare (and its subsidiary Turku Imanet Ltd.), Bristol-Meyers-Squibb, Wyeth, Elan, and AC Immune; and has received research support from the Academy of Finland [# 205954 (Principal Investigator)]. Dr. Brooks serves or has served on scientific advisory boards for CeNes, Synosia, Genentech, GlaxoSmithKline, and Orion; receives funding for travel from TEVA; serves or has served as an editorial board member for the Journal of Neural Transmission, Brain, Movement Disorders, the Journal of Neurology, Neurosurgery and Psychiatry, and Synapse; has received speaker honoraria from GlaxoSmithKline and Orion Pharma; serves as Head of Neurology for GE Healthcare; and receives research support from the Alzheimer’s Research Trust and the Medical Research Council Clinical Sciences Centre. This work was conducted in collaboration with Imanet, GE Healthcare.
Supplementary Material
Address correspondence and reprint requests to Professor David J. Brooks, Cyclotron Building, Hammersmith Hospital, Du Cane Road, London W12 0NN, UK david.brooks@csc.mrc.ac.uk
Editorial, page 744
Supplemental data at www.neurology.org
e-Pub ahead of print on July 8, 2009, at www.neurology.org.
Supported by the Medical Research Council, the Sigrid Juselius Foundation, Academy of Finland, and clinical grant of Turku University Hospital (EVO). UCLH/UCL receives funding from the NIHR Biomedical Research Centres funding scheme.
Disclosure: Author disclosures are provided at the end of the article.
Received January 25, 2009. Accepted in final form May 13, 2009.
REFERENCES
- 1.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 2006;63:665–672. [DOI] [PubMed] [Google Scholar]
- 2.Mufson EJ, Chen EY, Cochran EJ, et al. Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol 1999;158:469–490. [DOI] [PubMed] [Google Scholar]
- 3.Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol 2001;58:1985–1992. [DOI] [PubMed] [Google Scholar]
- 4.Boyle PA, Wilson RS, Aggarwal NT, et al. Mild cognitive impairment: risk of Alzheimer disease and rate of cognitive decline. Neurology 2006;67:441–445. [DOI] [PubMed] [Google Scholar]
- 5.Tierney MC, Szalai JP, Snow WG, et al. Prediction of probable Alzheimer’s disease in memory-impaired patients: a prospective longitudinal study. Neurology 1996;46:661–665. [DOI] [PubMed] [Google Scholar]
- 6.Jack CR, Jr., Petersen RC, Xu YC, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999;52:1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chetelat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC. Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology 2003;60:1374–1377. [DOI] [PubMed] [Google Scholar]
- 8.Yuan Y, Gu ZX, Wei WS. Fluorodeoxyglucose-positron-emission tomography, single-photon emission tomography, and structural MR imaging for prediction of rapid conversion to Alzheimer disease in patients with mild cognitive impairment: a meta-analysis. AJNR Am J Neuroradiol Epub 2008. [DOI] [PMC free article] [PubMed]
- 9.Bouwman FH, Schoonenboom SN, van der Flier WM, et al. CSF biomarkers and medial temporal lobe atrophy predict dementia in mild cognitive impairment. Neurobiol Aging 2007;28:1070–1074. [DOI] [PubMed] [Google Scholar]
- 10.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 11.Forsberg A, Engler H, Almkvist O, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging 2008;29:1456–1465. [DOI] [PubMed] [Google Scholar]
- 12.Koivunen J, Pirttila T, Kemppainen N, et al. PET amyloid ligand [11C]PIB uptake and cerebrospinal fluid beta-amyloid in mild cognitive impairment. Dement Geriatr Cogn Disord 2008;26:378–383. [DOI] [PubMed] [Google Scholar]
- 13.Morris JC, Mohs RC, Rogers H, Fillenbaum G, Heyman A. Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) clinical and neuropsychological assessment of Alzheimer’s disease. Psychopharmacol Bull 1988;24:641–652. [PubMed] [Google Scholar]
- 14.Wechsler D. Wechsler Memory Scale–Revised manual. San Antonio: The Psychological Corporation; 1987. [Google Scholar]
- 15.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984;141:1356–1364. [DOI] [PubMed] [Google Scholar]
- 16.Delis DC, Kramer JH, Kaplan E, et al. California Verbal Learning Test Adult version. San Antonio: The Psychological Corporation; Harcourt Brace & Company; 1987. [Google Scholar]
- 17.Becker JT, Boller F, Saxton J, McGonigle-Gibson KL. Normal rates of forgetting of verbal and non-verbal material in Alzheimer’s disease. Cortex 1987;23:59–72. [DOI] [PubMed] [Google Scholar]
- 18.Okello A, Edison P, Archer HA, et al. Microglial activation and amyloid deposition in mild cognitive impairment. Neurology 2009;72:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kemppainen NM, Aalto S, Wilson IA, et al. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology 2007;68:1603–1606. [DOI] [PubMed] [Google Scholar]
- 20.Edison P, Archer HA, Hinz R, et al. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology 2007;68:501–508. [DOI] [PubMed] [Google Scholar]
- 21.Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338. [DOI] [PubMed] [Google Scholar]
- 22.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 23.Brix G, Zaers J, Adam LE, et al. Performance evaluation of a whole-body PET scanner using the NEMA protocol: National Electrical Manufacturers Association. J Nucl Med 1997;38:1614–1623. [PubMed] [Google Scholar]
- 24.Kemppainen NM, Aalto S, Wilson IA, et al. Voxel-based analysis of PET amyloid ligand [11C]PIB uptake in Alzheimer disease. Neurology 2006;67:1575–1580. [DOI] [PubMed] [Google Scholar]
- 25.Hammers A, Allom R, Koepp MJ, et al. Three-dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp 2003;19:224–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turkheimer FE, Smith CB, Schmidt K. Estimation of the number of “true” null hypotheses in multivariate analysis of neuroimaging data. Neuroimage 2001;13:920–930. [DOI] [PubMed] [Google Scholar]
- 27.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68:1718–1725. [DOI] [PubMed] [Google Scholar]
- 28.Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab 2005;25:1528–1547. [DOI] [PubMed] [Google Scholar]
- 29.Larrieu S, Letenneur L, Orgogozo J, et al. Incidence and outcome of mild cognitive impairment in a population-based prospective cohort. Neurology 2002;59:1594–1599. [DOI] [PubMed] [Google Scholar]
- 30.Klunk WE, Price JC, Mathis CA, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 2007;27:6174–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koivunen J, Verkkoniemi A, Aalto S, et al. PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain 2008;131:1845–1853. [DOI] [PubMed] [Google Scholar]
- 32.Remes AM, Laru L, Tuominen H, et al. Carbon 11-labeled Pittsburgh compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol 2008;65:540–544. [DOI] [PubMed] [Google Scholar]
- 33.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 2006;129:2856–2866. [DOI] [PubMed] [Google Scholar]
- 34.Kemppainen NM, Aalto S, Karrasch M, et al. Cognitive reserve hypothesis: Pittsburgh Compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer’s disease. Ann Neurol 2008;63:112–118. [DOI] [PubMed] [Google Scholar]
- 35.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993;90:9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 1993;11:575–580. [DOI] [PubMed] [Google Scholar]
- 37.Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med 1995;333:1242–1247. [DOI] [PubMed] [Google Scholar]
- 38.Grimmer T, Henriksen G, Wester HJ, et al. Clinical severity of Alzheimer’s disease is associated with PIB uptake in PET. Neurobiol Aging Epub 2008. [DOI] [PubMed]
- 39.Pike K, Savage G, Villemagne V, et al. β-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain 2007;130:2837–2844. [DOI] [PubMed] [Google Scholar]
- 40.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006;67:446–452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.