

Mitochondrial toxicity (MT) can occur as a consequence of HIV infection and from treatment with highly active antiretroviral therapy (HAART).1 Toxicity is additive and manifested by signs including lipodystrophy and lactic acidosis. HIV and HAART are also reported in association with skeletal myopathy and mitochondrial abnormalities on muscle biopsy.2 This is in contrast to mitochondrial disorders associated with nuclear or mitochondrial DNA (mtDNA) mutations, in which patients may present with syndromic constellations of dysfunction in metabolically active tissues such as the brain and skeletal muscles. Chronic progressive external ophthalmoplegia (CPEO) is a mitochondrial syndrome with gradual onset of ptosis and ophthalmoparesis.3 Thus far, phenotypes resembling genetic mitochondrial disorders have not been reported in association with acquired mitochondrial toxicity from disease or drugs. We report 3 cases of HIV-infected patients with long exposure to HAART who developed clinical syndromes resembling CPEO.
Case series.
See the table for details. The patients were men, between the ages of 52 and 58 years, with long duration of HIV infection (16-24 years) and exposure to HAART (10–15 years). They developed MT manifested by lipodystrophy and also developed ptosis. Two patients had ophthalmoparesis and elevated serum lactate. One patient developed fatigue, migraines, and exertional myalgia, and another had dysphagia and diplopia on lateral gaze without clear ophthalmoparesis on examination. None of the patients had a family history of mitochondrial disease.
Table Patient characteristics
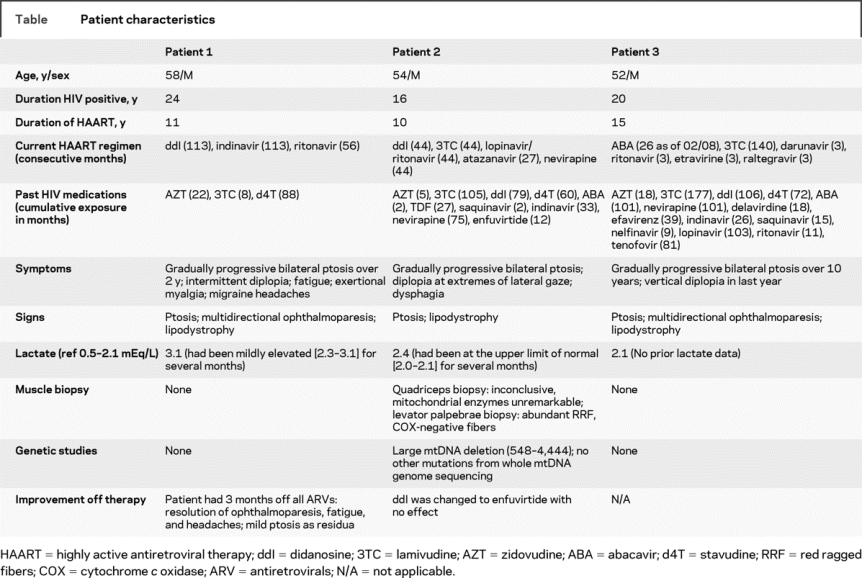
During a 3-month period off all antiretroviral agents, patient 1 had marked amelioration of fatigue, headaches, and ptosis. Significant improvement in ptosis and resolution of ophthalmoparesis was demonstrated on examination. This patient started a new antiretroviral regimen without didanosine (ddI) and remained stable with only mild ptosis.
Patient 2 had a period on a ddI-sparing HAART regimen and did not experience improvement. Quadriceps muscle biopsy demonstrated mild nonspecific changes including increased subsarcolemmal staining on oxidative stains and cytochrome c oxidase (COX)-negative fibers. Mitochondrial respiratory chain enzyme analysis was unremarkable. Six months later, a levator palpebrae biopsy collected during an oculoplastics procedure for ptosis demonstrated the majority of fibers were ragged-red on modified Gomori trichrome stain, and occasional COX-negative fibers. Genetic analysis revealed a 3.9-kb mtDNA deletion (547-4,443) affecting approximately 40%-60% of mtDNA in both biopsies.
Patient 3 did not return for reassessment; therefore, further investigations or clinical data are not available.
Discussion.
CPEO-like syndromes have not been previously reported in association with mitochondrial toxicity in HIV/HAART. Patient 1 did not have a muscle biopsy although his symptoms clearly improved with cessation, followed by alteration of therapy, suggesting that HAART-related MT may have contributed to his syndrome. Patient 2 did not improve upon HAART cessation but had a muscle biopsy diagnostic for mitochondrial disease showing evidence of a 3.9-kb mtDNA deletion in both quadriceps and levator biopsies, which was previously reported with CPEO,4 suggestive of underlying genetic mitochondrial disease. We therefore suggest that these patients may have had subclinical mitochondrial disease that was unmasked by the additive MT of HIV infection and HAART. Nucleoside analogues such as ddI and stavudine are likely to be implicated, given their well-described time-dependent and dose-dependent MT5 and partial reversibility.6 All patients in this series received these agents and in one patient symptoms improved off therapy and did not recur on a regimen that excluded ddI and stavudine.
While other mitochondrial syndromes such as Kearns-Sayre syndrome are characterized by early age at onset,7 CPEO usually has a later age at onset and is typically benign and slowly progressive.3 Late-onset CPEO likely occurs when mtDNA abnormalities with low expressivity reach a critical level of heteroplasmy producing phenotypic expression, and this may be accelerated by MT from aging or environmental exposures. HIV infection and HAART likely present more intense MT that may be capable of phenotypically exposing potentially pathogenic mtDNA abnormalities. HIV-negative individuals with late-onset CPEO may have equally important sources of MT that have not yet been identified.
CPEO-like syndromes may occasionally occur in patients with long duration of HIV infection and exposure to HAART. These syndromes may be from accumulated mitochondrial toxicity or toxicity superimposed upon a preexisting subclinical genetic mitochondrial disorder. Improvement may occur by withdrawing offending agents or by use of alternative drug regimens. Patients with HIV with long disease duration and exposure to HAART may provide a model for future study of mitochondrial toxicity and mtDNA heteroplasmy in CPEO. Attempts should be made to identify other sources of mitochondrial toxicity in HIV-negative patients with late-onset CPEO and suspicion should be heightened for a CPEO-like syndrome in long-term HAART-treated HIV survivors.
H.C.F.C. was supported by salary awards from the Michael Smith Foundation for Health Research and the Canadian Institutes for Health Research.
Disclosure: The authors report no disclosures.
Received October 8, 2008. Accepted in final form February 13, 2009.
Address correspondence and reprint requests to Dr. Michelle M. Mezei, Adult Metabolic Diseases Clinic, Vancouver General Hospital, 4th Floor, 2775 Laurel St., Vancouver, BC, V5Z 1M9, Canada; mezei@interchange.ubc.ca
REFERENCES
- 1.Moyle G. Mechanisms of HIV and nucleoside reverse transcriptase inhibitor injury to mitochondria. Antivir Ther 2005;10 suppl 2:M47–M52. [PubMed] [Google Scholar]
- 2.Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med 1990;322:1098–1105. [DOI] [PubMed] [Google Scholar]
- 3.Danta G, Hilton RC, Lynch PG. Chronic progressive external ophthalmoplegia. Brain 1975;98:473–492. [DOI] [PubMed] [Google Scholar]
- 4.Moraes CT, Andreetta F, Bonilla E, Shanske S, DiMauro S, Schon EA. Replication-competent human mitochondrial DNA lacking the heavy-strand promoter region. Mol Cell Biol 1991;11:1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venhoff N, Setzer B, Melkaoui K, Walker UA. Mitochondrial toxicity of tenofovir, emtricitabine and abacavir alone and in combination with additional nucleoside reverse transcriptase inhibitors. Antivir Ther 2007;12:1075–1085. [PubMed] [Google Scholar]
- 6.Ananworanich J, Nuesch R, Cote HC, et al. Changes in metabolic toxicity after switching from stavudine/didanosine to tenofovir/lamivudine-a staccato trial substudy. J Antimicrob Chemother 2008;61:1340–1343. [DOI] [PubMed] [Google Scholar]
- 7.Schmiedel J, Jackson S, Schafer J, Reichmann H. Mitochondrial cytopathies. J Neurol 2003;250:267–277. [DOI] [PubMed] [Google Scholar]