

Abstract
We sought genetic evidence for the involvement of neuronal vascular endothelial growth factor (VEGF) in amyotrophic lateral sclerosis (ALS). Mice expressing human ALS mutant superoxide dismutase-1 (SOD1) were crossed with mice that overexpress VEGF in neurons (VEGF+/+). We report that SOD1G93A/VEGF+/+ double-transgenic mice show delayed motor neuron loss, delayed motor impairment, and prolonged survival compared with SOD1G93A single transgenics. These findings indicate that neuronal VEGF protects against motor neuron degeneration, and may have therapeutic implications for ALS.
Keywords: vascular endothelial growth factor, amyotrophic lateral sclerosis, motor neuron, superoxide dismutase-1, transgenic, neurodegeneration
Introduction
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig's disease, is a devastating degenerative disease of upper and lower motor neurons that affects ∼2 people per 100,000 and produces weakness, muscle atrophy, fasciculations, spasticity, hyporeflexia or hyperreflexia, and extensor plantar responses (Rowland and Shneider, 2001). Existing treatment provides only marginal benefit and death usually occurs within 5 years of onset. In most patients with ALS the cause is unknown, but 5–10% of cases are familial. Patients with familial ALS have been found to have mutations in any of several genes, including Cu/Zn superoxide dismutase-1 (SOD1) (Rosen et al., 1993), alsin (Hadano et al., 2001; Yang et al., 2001), senataxin (Chen et al., 2004), vesicle-associated membrane protein B (Nishimura et al., 2004), and angiogenin (Greenway et al., 2006). Expression of ALS-associated human SOD1 mutations (e.g., SOD1G93A) in mice produces a dominantly inherited syndrome with clinical and histopathological features of ALS, including limb weakness, muscle atrophy, and loss of motor neurons (Gurney et al., 1994).
Recent studies have implicated vascular endothelial growth factor (VEGF) in ALS, and suggest that it might have therapeutic potential. Mice with deletion of the hypoxia-response element in the VEGF promoter region (VEGFδ/δ) develop adult-onset progressive motor neuron degeneration reminiscent of ALS (Oosthuyse et al., 2001). Crossbreeding of VEGFδ/δ with SOD1G93A mice produces VEGFδ/δ/SOD1G93A double mutants with more severe weakness and earlier death than mice carrying the SOD1G93A gene alone (Lambrechts et al., 2003). Intraperitoneal (Zheng et al., 2004) or intracerebroventricular (Storkebaum et al., 2005) administration of VEGF, or intramuscular delivery of a VEGF-expressing lentiviral vector (Azzouz et al., 2004), delays the onset of paralysis, improves motor performance, and prolongs survival in SOD1G93A mice or rats. These findings may have direct clinical relevance, because variations in the human VEGF promoter/leader sequence are associated with reduced levels of circulating VEGF, and confer increased risk for ALS in some populations (Lambrechts et al., 2003).
Materials and Methods
Experiments were performed according to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee. Male mice expressing wild-type human SOD1 [TgN(SOD1)2Gur] and human ALS mutant SOD1 [TgN(SOD1-G93A)1Gur] (Gurney et al., 1994) on a C57BL/6 genetic background were purchased from the Jackson Laboratory (West Grove, PA) and maintained as SOD1G93A hemizygotes. Mice overexpressing human VEGF165 [TgN(NSEVEGF)1651–1653] driven by the rat neuron-specific enolase promoter (VEGF+/+, strain V1) from the Department of Neurology, University of Zurich (Zurich, Switzerland) were maintain on a C57BL/6 background (Wang et al., 2005). SOD1G93A and VEGF+/+ mice were cross-bred and genotyped by PCR of tail-tip DNA, using 5′-AGGAGAGATGAGCTTCCTACAG-3′ and 5′-GATGGCTGGCAA-CTAGAAGGCAC-3′ as primers for VEGF, and 5′-CATCAgCCCTAATCCATCTgA-3′ and 5′-CgCgACTAACAATCAAAgTgA-3′ as primers for SOD1. Age-matched male littermates (n = 6) were used as controls in all experiments.
Western blots were performed to determine the expression levels of VEGF and mutant SOD1 in the spinal cord of wild-type and transgenic mice using rabbit anti-VEGF antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-human SOD1 (#SOD-100; 1:200; Millipore, Temecula, CA), and rabbit anti-mouse/rat SOD1 (#SOD-101; 1:200; Millipore) species-preferring antibodies (Shinder et al., 2001), and a standard protocol as described previously (Jin et al., 2000). Antibodies were removed with stripping buffer, and the membranes were reprobed with anti-actin antibody for control of protein loading.
For the rotarod testing, mice were trained for 1 week to perform on an accelerating rotarod (Ugo Basile, Comerio, Italy). Baseline performance was measured at age 60 d, before disease onset, and repeated every 10 d. Each test consisted of three trials, and the best performance (longest duration on the rod without falling, up to a maximum of 180 s) was recorded.
Perfusion-fixed spinal cords were embedded in paraffin, and serial transverse sections (7 μm) through the lumbar spinal cord were cut. Every fifth section was collected, deparaffinized, and stained with cresyl violet. Immunostaining for VEGF and choline acetyltransferase (ChAT) was performed using monoclonal anti-VEGF (1:200; Millipore) and goat polyclonal ant-ChAT (1:500; Millipore), and visualized with diaminobenzidine and hydrogen peroxide.
Quantitative data were expressed as mean ± SEM and ANOVA and Student's t test were used for statistical analysis, with p < 0.05 considered significant.
Results
If functional VEGF deficiency is an important pathogenic factor for ALS, then overexpression of VEGF should attenuate associated deficits. To test this prediction, we crossed SOD1G93A mutant mice with transgenic (VEGF+/+) mice that selectively overexpress hVEGF165 in neurons (Wang et al., 2005). PCR of mouse-tail genomic DNA showed a band corresponding to mutant SOD1 in SOD1G93A mice, and a band representing hVEGF165 in VEGF+/+ mice, which were absent in wild-type littermates (Fig. 1a). VEGF immunohistochemistry and Western blotting demonstrated increased VEGF expression in both ventral horn motor neurons and dorsal horn neurons in the lumbar spinal cord of 60-d-old SOD1G93A/VEGF+/+ compared with 60-d-old (presymptomatic) SOD1G93A mice (Fig. 1b,c). Mutant SOD1 protein was detected in both SOD1G93A and SOD1G93A/VEGF+/+ double-transgenics, and the VEGF+/+ mutation did not appear to affect the abundance of either the endogenous (murine) or transgenic (mutant human) form of SOD1 (Fig. 1d).
Figure 1.
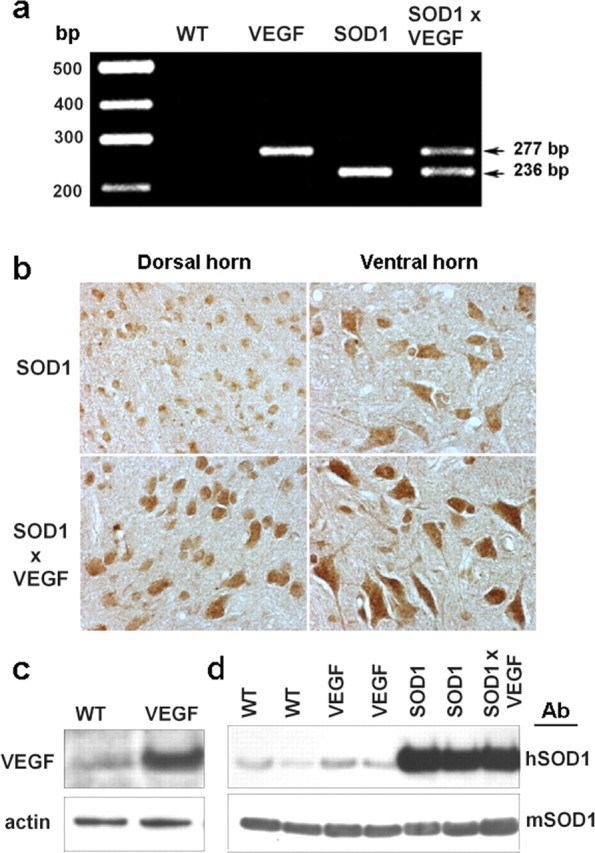
Neuronal overexpression of hVEGF165 in SOD1G93A/VEGF+/+ mice. a, PCR of mouse-tail genomic DNA shows a 236 bp band corresponding to mutant hSOD1 in SOD1G93A mice, a 277 bp band representing hVEGF165 in VEGF+/+ mice, and both bands in SOD1G93A/VEGF+/+ double transgenics. b, VEGF immunohistochemistry in lumbar spinal cord shows increased neuronal expression of VEGF (brown) in both ventral and dorsal horns of VEGF+/+ mice. c, Western blot confirmed that VEGF was overexpressed in the spinal cord in VEGF+/+ mice. d, SOD1 mutant protein was detected in both SOD1G93A and SOD1G93A/VEGF+/+ mice, and the VEGF+/+ mutation did not affect SOD1 abundance, whether the latter was measured using an anti-human SOD1-preferring (hSOD1, top) or anti-mouse/rat SOD1-preferring (mSOD1, bottom) antibody. Note that anti-hSOD1 detects some mSOD1 (Shinder et al., 2001). WT, Wild-type.
In principle, transgenic overexpression of VEGF could mistakenly appear to affect the course of disease in SOD1G93A mice if it acted during development to increase the baseline number of spinal cord motor neurons. To address this possibility, we stained lumbar spinal cord sections from 4-week-old (presymptomatic) adult mice with an antibody against the motor neuron marker ChAT, and counted cells in wild-type, VEGF+/+, SOD1G93A, and SOD1G93A/VEGF+/+ mice. However, no differences were observed among these four groups (Fig. 2a,b).
Figure 2.

Equivalence of spinal cord motor neuron numbers in 4-week-old wild-type (WT), VEGF+/+, SOD1G93A, and SOD1G93A/VEGF+/+ mice. a, Sections through the ventral horn of the lumbar spinal cord show similar numbers of ChAT-immunopositive cells. b, Cell counts show no differences in ChAT-immunopositive cell counts among the four groups of mice.
The most striking histopathological finding in patients with ALS and in SOD1G93A mice is loss of motor neurons in the anterior horns of the spinal cord. Compared with wild-type littermates (or VEGF+/+ mice), SOD1G93A mice show prominent depletion of anterior horn motor neurons (Fig. 3a,b). However, motor-neuron loss was markedly reduced in SOD1G93A/VEGF+/+ double transgenics. Motor impairment was delayed in onset in SOD1G93A/VEGF+/+ mice, although its rate of progression, once manifested, was not appreciably altered. Thus, rotarod performance began to decline ∼20 d later in SOD1G93A/VEGF+/+ than in SOD1G93A mice, but deteriorated at the same rate thereafter (Fig. 3c). Finally, compared with SOD1G93A mice, SOD1G93A/VEGF+/+ mice showed a delay in the onset of mortality (149 vs 129 d), and prolonged average (159 ± 3 vs 136 ± 3 d) and maximal (167 vs 143 d) survival (Fig. 3d).
Figure 3.
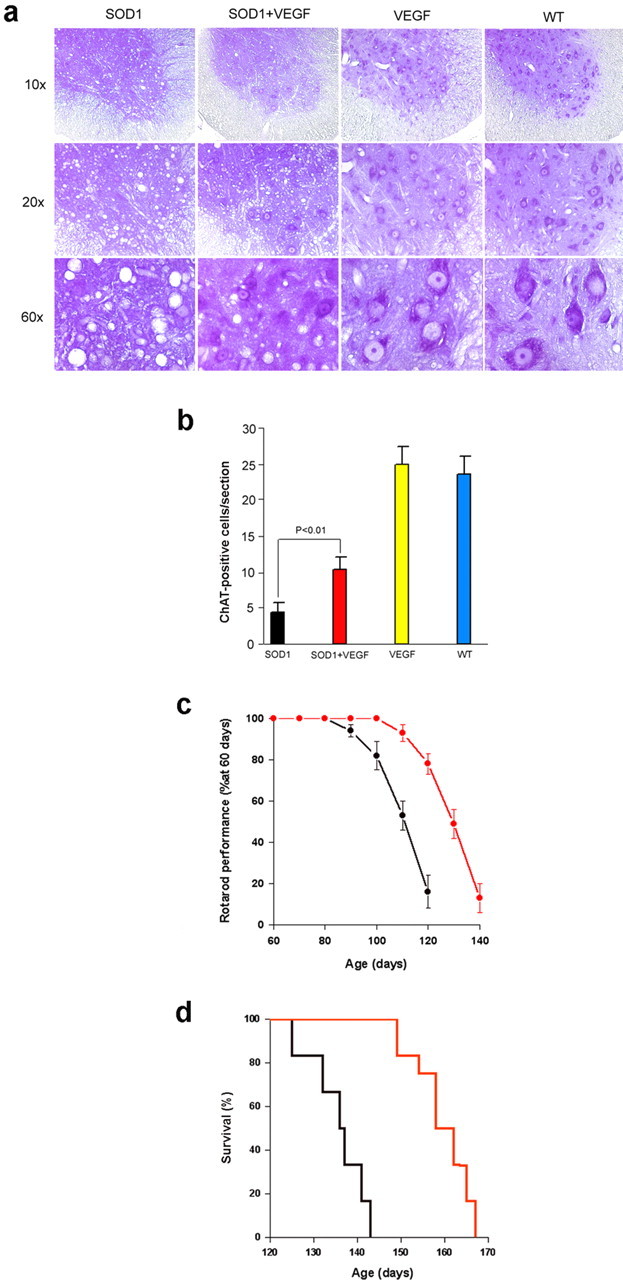
Effect of transgenic overexpression of hVEGF165 on the course of disease in ALS transgenic (SOD1G93A) mice. a, Cresyl violet-stained sections through the ventral horn of lumbar spinal cord show partial preservation of neurons in SOD1G93A/VEGF+/+ compared with SOD1G93A mice. b, Survival of cholinergic (ChAT-immunopositive) cells was also increased in SOD1G93A/VEGF+/+ compared with SOD1G93A mice. c, Impairment of rotarod peformance, measured as latency to fall on an accelerating rotarod and expressed as a percentage of performance at age 60 d ± SEM, was delayed in SOD1G93A/VEGF+/+ (red) compared with SOD1G93A (black) mice. d, Onset of mortality was delayed, and mean and maximal survival was prolonged in SOD1G93A/VEGF+/+ (red) compared with SOD1G93A (black) mice. n = 6 per group. WT, Wild-type.
Discussion
The ability of transgenically expressed neuronal VEGF to postpone the onset of motor neuron disease in ALS mice is consistent with previous evidence for a protective effect of VEGF in this disorder, and may shed new light on the mechanisms involved. Because VEGF overexpression in our VEGF+/+ and SOD1G93A/VEGF+/+ mice was confined to neurons, neuronal overexpression of VEGF seems sufficient for protection, consistent with the observation that selective neuronal infection with a VEGF-expressing lentiviral vector moderates the course of disease in SOD1G93A mice (Azzouz et al., 2004). Because VEGF is a secreted protein, this does not necessarily imply that motor neurons are the cellular targets of neuronal VEGF in SOD1G93A/VEGF+/+ mice. However, autocrine (Ogunshola et al., 2002) and direct neuroprotective (Jin et al., 2000) effects of VEGF are well documented, and transgenic neuronal overexpression of the VEGFR2 receptor also prolongs survival in SOD1G93A mice (Storkebaum et al., 2005), so the mechanism of action of VEGF in this murine model of ALS could be exclusively neuronal.
It is also notable that early onset of VEGF therapy, as achieved in our SOD1G93A/VEGF+/+ transgenic mice or in SOD1G93A mice given a VEGF-expressing lentivirus vector beginning at 21 rather than 90 d of age (Azzouz et al., 2004) produces more marked (17–30%) extension of survival than does treatment started at later times (7–8%) (Zheng et al., 2004; Storkebaum et al., 2005). This could relate to differences in the levels of VEGF achieved in these different models, but it could also imply that VEGF induces early adaptations that reduce the sensitivity of SOD1G93A motor neurons to degeneration in advance of its onset.
Footnotes
This work was supported by United States Public Health Service Grants AG21980 to K.J. and NS44921 to D.A.G.
References
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Hardiman O. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown RH, Jr, Scherer SW, Rouleau GA, Hayden MR, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29:166–173. doi: 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–394. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunshola OO, Antic A, Donoghue MJ, Fan SY, Kim H, Stewart WB, Madri JA, Ment LR. Paracrine and autocrine functions of neuronal VEGF in the CNS. J Biol Chem. 2002;277:11410–11415. doi: 10.1074/jbc.M111085200. [DOI] [PubMed] [Google Scholar]
- Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kilic E, Kilic U, Weber B, Bassetti CL, Marti HH, Hermann DM. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain. 2005;128:52–63. doi: 10.1093/brain/awh325. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- Zheng C, Nennesmo I, Fadeel B, Henter JI. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2004;56:564–567. doi: 10.1002/ana.20223. [DOI] [PubMed] [Google Scholar]