

Abstract
Background:
Late-onset Alzheimer disease (LOAD) is a common disorder with a substantial genetic component. We postulate that many disease susceptibility variants act by altering gene expression levels.
Methods:
We measured messenger RNA (mRNA) expression levels of 12 LOAD candidate genes in the cerebella of 200 subjects with LOAD. Using the genotypes from our LOAD genome-wide association study for the cis-single nucleotide polymorphisms (SNPs) (n = 619) of these 12 LOAD candidate genes, we tested for associations with expression levels as endophenotypes. The strongest expression cis-SNP was tested for AD association in 7 independent case-control series (2,280 AD and 2,396 controls).
Results:
We identified 3 SNPs that associated significantly with IDE (insulin degrading enzyme) expression levels. A single copy of the minor allele for each significant SNP was associated with ∼twofold higher IDE expression levels. The most significant SNP, rs7910977, is 4.2 kb beyond the 3′ end of IDE. The association observed with this SNP was significant even at the genome-wide level (p = 2.7 × 10−8). Furthermore, the minor allele of rs7910977 associated significantly (p = 0.0046) with reduced LOAD risk (OR = 0.81 with a 95% CI of 0.70-0.94), as expected biologically from its association with elevated IDE expression.
Conclusions:
These results provide strong evidence that IDE is a late-onset Alzheimer disease (LOAD) gene with variants that modify risk of LOAD by influencing IDE expression. They also suggest that the use of expression levels as endophenotypes in genome-wide association studies may provide a powerful approach for the identification of disease susceptibility alleles.
GLOSSARY
- AD
= Alzheimer disease;
- CI
= confidence interval;
- GWAS
= genome-wide association study;
- LOAD
= late-onset Alzheimer disease;
- mRNA
= messenger RNA;
- OR
= odds ratio;
- SNP
= single nucleotide polymorphism.
Late-onset Alzheimer disease (LOAD) is the most common cause of dementia in the elderly.1 Despite a substantial estimated genetic component,2 APOE ε4 is the only universally accepted risk factor for LOAD.3–5
Endophenotypes are biologically relevant intermediate quantitative phenotypes.6–8 Gene expression levels may be particularly useful endophenotypes. Polymorphisms in genomic regulatory regions could underlie the genetic basis of complex diseases.9 Analysis of genetic variants that influence gene expression may significantly enhance our understanding of this genetic basis. Recent genome-wide association studies (GWAS) utilizing expression levels in human cells and tissues10–13 suggest a substantial genetic influence on human gene expression levels. We hypothesized that there may be variants that influence AD risk by influencing gene expression levels in the brain. If correct, such variants would associate with both AD risk and brain gene expression levels.
We performed a pilot study using cerebellar gene expression levels of 12 LOAD candidate genes and their cis-single nucleotide polymorphism (SNP) genotypes extracted from our LOAD GWAS.14 Most of these 12 messenger RNAs (mRNAs) encode proteins that are likely to be involved in neurodegeneration.
We identified 3 IDE cis-SNP/transcript associations with study-wide significance. One of these cis-SNPs showed an association with genome-wide significance. This SNP also associated significantly with LOAD risk. Our findings suggest that joint analysis of transcriptome and disease risk with genome data could identify candidate functional variants that influence disease risk by influencing gene expression. The use of brain expression endophenotypes may be a potentially powerful approach in uncovering the genetics of neurologic diseases like LOAD.
METHODS
Subjects and samples for genotyping.
For our GWAS, we genotyped 970 LOAD cases and 1,495 controls from 3 series, collected at Mayo Clinic, Jacksonville, FL (JS_60-80 series), Mayo Clinic, Rochester, MN (RS_60-80 series), and an autopsy-based series (AUT_60-80). The LOAD GWAS subjects had an age at diagnosis/entry of 60-80 years. Four additional series were genotyped for the most significant IDE cis-SNP, rs7910977. Three of these additional series were collected at Mayo Clinic, Jacksonville, FL (JS_80+), Mayo Clinic, Rochester, MN (RS_80+), and an autopsy series (AUT_80+). The fourth additional series was from the National Cell Repository for AD (NCRAD). All subjects are white Americans (appendix e-1: Methods and table e-1 on the Neurology® Web site at www.neurology.org).
Standard protocol approvals, registrations, and patient consents.
This study was approved by the appropriate institutional review board and appropriate informed consent was obtained from all participants.
LOAD GWAS.
The LOAD GWAS genotypes were generated using Illumina's HumanHap300-Duo Genotyping BeadChips analyzed with an Illumina BeadLab Station (Illumina, San Diego, CA) at the Mayo Clinic Genotyping Shared Resource according to the manufacturer's protocols. GWAS quality control is discussed in appendix e-1: Methods.
cis-SNP/mRNA expression level associations for 12 LOAD candidate genes.
Autopsy series for mRNA expression analyses.
A total of 200 autopsy-confirmed LOAD subjects from the AUT_60-80 series of our GWAS underwent cerebellar gene expression level measurements. TaqMan® Arrays15,16 were used for the gene expression studies (appendix e-1: Methods).
For follow-up analysis of the most significant IDE cis-SNP rs7910977/transcript association, we measured cerebellar IDE mRNA levels in 3 additional groups composed of autopsy-confirmed LOAD subjects with age at diagnosis ≥80 years, control subjects with age at entry of 60-80 years, and control subjects with age at entry ≥80 years. All control subjects had Braak scores of 2.5 or lower, but many had brain pathology unrelated to AD.
Selection of 12 LOAD candidate genes for expression studies.
We identified cis-SNPs for 12 candidate genes that were genotyped in our LOAD GWAS (table e-2). We define cis-SNPs as variants that either reside within each gene or in its flanking regions (±100 kb). There were 582 cis-SNPs in our LOAD GWAS, of which 564 remained for analysis after the quality control exclusions (appendix e-1).
Statistical analysis.
12 LOAD candidate gene cis-SNP/mRNA expression level association analyses.
Statistical analyses were carried out using PLINK,17 SPlus 8.0, or StatsDirect (v. 2.6.5). The ΔCT values representing brain expression levels of each gene were utilized as endophenotypes. All cis-SNPs were analyzed for association with the expression levels of the corresponding gene using an additive linear regression model, that included minor allele dosage (0, 1, 2) as the primary independent variable, and APOE ε4 dosage (0, 1, 2), age at diagnosis/entry, and gender as covariates. We tested and corrected for population stratification using EIGENSTRAT18 (appendix e-1: Methods).
IDE cis-SNPs/LOAD risk association analyses.
Given that IDE cis-SNPs had the strongest association with expression levels, we tested for their associations with LOAD risk using PLINK.17 We performed logistic regression analyses using a log-additive model for the minor allele (0, 1, or 2 copies), while including covariates for APOE ε4 dosage (0, 1, 2), age at diagnosis/entry, sex, and series. All IDE cis-SNPs/LOAD risk associations were tested in the 3 LOAD GWAS series. The most significant SNP, rs7910977, was also assessed in the 4 additional series described above. To assess the potential that there were differential SNP associations by series, we used logistic regression models to test for a significant series × genotype interaction.
RESULTS
Twelve LOAD candidate gene cis-SNP/transcript associations.
Of the 619 cis-SNP/transcript associations tested in 200 subjects with autopsy-confirmed LOAD with age at diagnosis of 60-80 years, only 3 IDE cis-SNPs achieved p < 0.05 after study-wide Bonferroni correction: rs7910977, rs1999763, and rs10882088. When q-values19–21 are obtained, only the top 3 IDE SNPs had a greater than 95% probability of being true positives. Four (3 IDE, 1 CTNNA3), 9 (7 IDE, 1 CTNNA3, 1 APP), and 15 (10 IDE, 3 CTNNA3, 1 APP, 1 GRN) cis-SNPs had q-values less than 0.1, 0.33, and 0.5. Some of these suggestive results may be genuine associations that could be replicated in a larger study. In this article, we focus on the IDE cis-SNP/transcript associations that were highly significant.
IDE cis-SNPs associate significantly with IDE expression levels.
IDE cis-SNP rs7910977 is 4.2 kb from the 3′ end, rs10882088 is 34.8 kb from the 5′ end, and rs1999763 is within the second intron of IDE. The most significantly associated SNP, rs7910977, had a p value of 2.7 × 10−8, which would be significant even at genome-wide level. IDE expression level in minor allele carriers of each cis-SNP is roughly twice that in major homozygotes (table 1, figure 1). Adjustment for population stratification had essentially no effect on the IDE cis-SNP/transcript associations (appendix e-1: Results, table e-3).
Table 1 Significant IDE expression level associations with top 3 IDE cis-SNPs
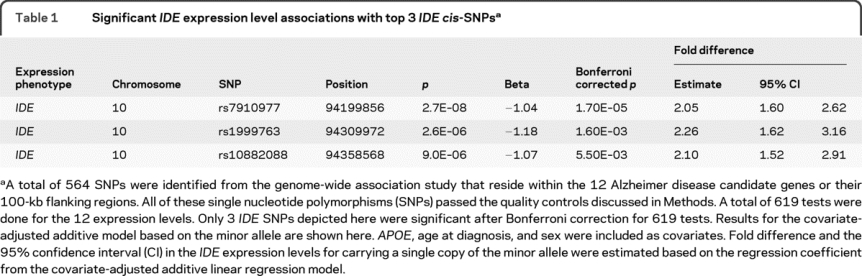
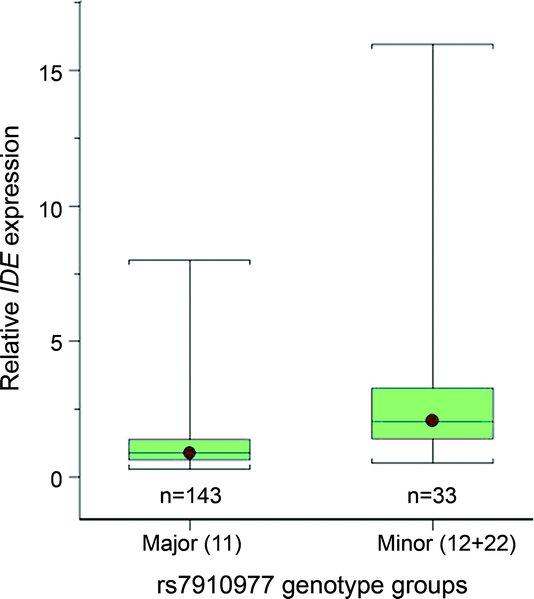
Figure 1 Relative IDE expression levels by rs7910977 genotypes
IDE expression levels relative to the mean of the major homozygotes are shown separately for the major homozygotes (11) and the minor allele carriers (12 + 22) of the IDE SNP rs7910977. Relative IDE expression levels for each subject were calculated using the 2-ΔΔCT method. The whiskers depict the smallest and largest data points for each box plot.
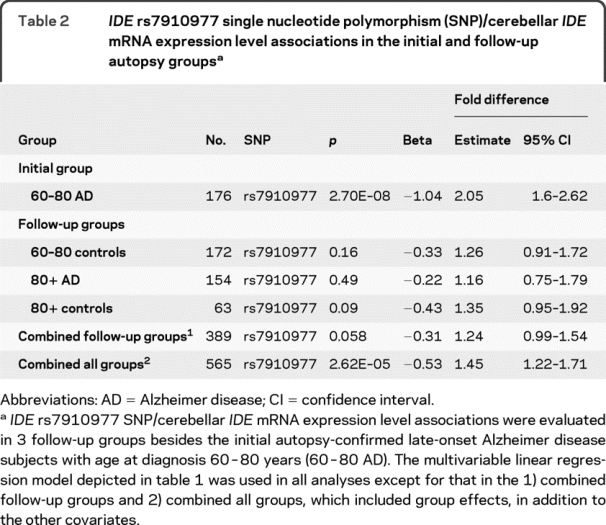
To follow up the most significant IDE cis-SNP rs7910977/IDE transcript association observed in the subjects with LOAD from our GWAS, we measured cerebellar IDE mRNA expression and genotyped rs7910977 in 3 additional groups from the autopsy series, collectively composed of 389 subjects (table 2). In each of the additional groups tested, rs7910977 minor allele was associated with elevated IDE transcript levels (table 2), but the fold expression level difference in each group was smaller than that in the initial 60-80 year AD group (2.05-fold, 95% confidence interval [CI] 1.6-2.62). When all 3 follow-up groups were combined, the fold difference was 1.24 (95% CI 0.99-1.54, p = 0.058). When the data from all 4 groups were combined, the fold difference associated with each copy of the minor allele of rs7910977 was 1.45 (95% CI 1.22-1.71) and the association continued to be highly significant (p = 2.6 × 10−5).
Table 2 IDE rs7910977 single nucleotide polymorphism (SNP)/cerebellar IDE mRNA expression level associations in the initial and follow-up autopsy groups
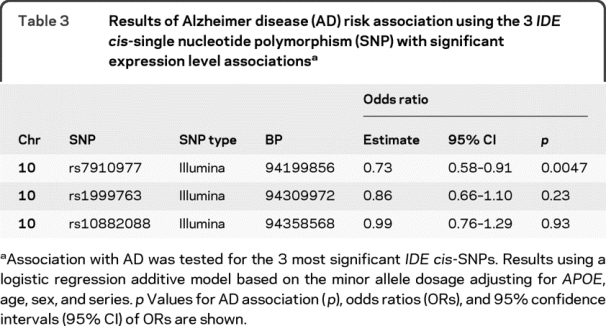
IDE cis-SNP/LOAD risk associations are less robust than IDE expression level associations.
We tested for association with LOAD risk using the 24 IDE cis-SNPs from our LOAD GWAS (table e-4). The logistic regression p values for the top 3 IDE expression cis-SNPs are 0.0047, 0.23, and 0.93 for the rs7910977, rs1999763, and rs10882088 SNPs (table 3). While only rs7910977 demonstrated significant association with AD, the odds ratio (OR) estimates for all 3 IDE SNPs indicate a protective effect for the minor allele. These results are biologically consistent with the higher IDE expression levels associating with the minor alleles of these SNPs.
Table 3 Results of Alzheimer disease (AD) risk association using the 3 IDE cis-single nucleotide polymorphism (SNP) with significant expression level associations
IDE cis-SNP rs7910977 shows consistent association with LOAD risk in 7 independent series.
In addition to the 3 LOAD GWAS series, we genotyped the rs7910977 SNP in 4 more case-control series (figure 2 and table e-5). The OR estimates for each of the series are protective, though only JS_60-80 series achieves significance. The OR estimate for the rs7910977 SNP minor allele in the combined series is 0.81 (95% CI 0.70-0.94, p = 0.0046). This LOAD association was obtained in 2,280 patients with AD and 2,396 controls, whereas the genome-wide significant rs7910977/IDE expression association was obtained in 176 subjects. The strength of association for LOAD risk is much less robust than that for the IDE expression phenotype.

Figure 2 Odds ratio plots of Alzheimer disease association with rs7910977 SNP in 7 late-onset Alzheimer disease case-control series
The odds ratio (OR) and 95% confidence interval (CI) of Alzheimer disease (AD) association for the rs7910977 SNP were obtained using a logistic regression approach using age at diagnosis/entry, APOE 4 genotype, and sex as covariates. The series names are shown to the left and the OR estimates (95% CI) to the right of each series. In the combined analysis, series effects were included as covariates. We also formally tested for a series effect by including “series × genotype” variables in the model and determined no significant evidence for a series × genotype effect.
Series with younger ages at diagnosis/entry (JS_60-80, RS_60-80, AUT_60-80) appear to harbor a stronger effect for the rs7910977/AD association compared to the older series (JS_80+, RS_80+, AUT_80+). However, the comparison of heterogeneity by series on the rs7910977/AD association did not reach significance (data not shown).
DISCUSSION
Functional variants that influence disease risk by influencing gene expression may have a substantial contribution to the genetics of common diseases.9 Identification of variants that associate both with AD and gene expression levels will provide important supportive evidence for the role of expression-SNPs in conferring disease risk. To test this hypothesis, we performed a pilot study, where we tested 564 cis-SNPs from our LOAD GWAS14 for association with the transcripts of 12 LOAD candidate genes measured in the cerebellum of subjects with LOAD. We identified 3 IDE cis-SNPs that reached study-wide significance. The IDE cis-SNP rs7910977/transcript associations achieved genome-wide significance even in the small number of samples that were analyzed (n = 176). In these samples, IDE mRNA levels in minor allele carriers of rs7910977 were approximately twice those in noncarriers. This SNP also associated (p = 0.0046) with reduced risk of LOAD when 2,280 LOAD cases and 2,396 controls from 7 independent series were collectively analyzed. In a separate study (M. Allen, personal communication), the association of rs7910977 with plasma amyloid ß 40 (Aß40) and Aß42 was analyzed in 949 individuals from Vis and 930 individuals from Korcula, 2 isolated populations recently analyzed for multiple disease-related traits using genome-wide scans.22–24 In the combined data from these 1,879 subjects, rs7910977 showed association with reduced Aß40 (β ± SE = −0.124 ± 0.05, p = 0.011) and suggestive association with reduced plasma Aß42 levels (meta-analysis β ± SE = −0.07 ± 0.05, p = 0.18). Thus, rs7910977 showed strong association with elevated IDE expression and reduced risk of LOAD in the current study and, in a separate study, it showed significant association with reduced levels of Aβ, a substrate of IDE that is implicated in LOAD pathogenesis.
The IDE cis-SNP rs7910977 is in complete LD with rs6583817, which resides in an evolutionarily conserved region of IDE in intron 12. This region is expected to harbor putative transcription factor binding sites. Furthermore, the minor allele of rs6583817 (tagged by the minor allele of rs7910977) is predicted to alter the binding of transcription factors in a way that would increase expression of IDE mRNA (appendix e-1: Results).
In this study, we analyzed mRNAs in LOAD brain tissue obtained at autopsy to improve detection of brain-specific cis-SNP/transcript associations that would go undetected in peripheral tissues or lymphoblastoid cell lines. Because LOAD causes profound neuronal cell loss and astrogliosis that may alter mRNA levels in affected regions, mRNAs were measured in the cerebellum, which is largely unaffected by LOAD pathology. Since our goal was to identify expression differences due to genetic effects rather than those that occur as a result of neurodegeneration or neuronal loss, cerebellar tissue was chosen for RNA extractions. Our search for cis-SNP/transcript associations relevant to LOAD in cerebellum presupposes that at least some LOAD SNPs will influence gene expression similarly in brain regions both affected and unaffected by AD pathology. Some genetic variants may alter expression similarly in affected and unaffected brain regions and even in peripheral tissues and lymphoblastoid cell lines. It is possible, however, that some SNPs will only affect expression in brain regions affected by AD pathology. To fully exploit mRNA levels as LOAD endophenotypes it will, therefore, be important to search for strong cis-SNP/transcript associations in a variety of brain regions, tissues, and cell lines from both LOAD and unaffected control subjects.
Our results suggest that the rs7910977/IDE transcript association may be context dependent. Rs7910977 was consistently associated with elevated IDE transcript in all groups tested (table 2). In our combined data, each copy of the minor allele of rs7910977 was associated with a 1.45-fold elevation in IDE transcript level (p = 2.6 × 10−5), but the elevation of 2.05-fold observed initially in the 60-80 year AD group was considerably greater than that observed in any of the 3 follow-up groups (80+ AD, 60-80 controls, 80+ controls).
Our strong association with gene expression levels obtained using <200 subjects that is biologically congruent with the AD risk association in >4,500 subjects provides evidence that 1) joint analysis of disease risk and brain gene expression levels may lead to the identification of candidate functional AD variants and 2) the use of gene expression endophenotype may be a more powerful approach than the use of the disease phenotype, likely due to increased underlying genetic homogeneity of the endophenotype and the increased statistical power of the quantitative phenotype. In fact, our expression analyses would have had greater than 99% power to detect an association at genome-wide significance, even if the SNP association led to a 1.5-fold change rather than the twofold differences that were observed. On the other hand, even with the thousands of cases and controls in the LOAD/SNP association analyses, we only would have had 47% power to detect the observed association at genome-wide significance. Indeed, the LOAD GWAS reported to date,14,25–31 including our study, utilize thousands of subjects to achieve genome-wide significance, whereas the expression endophenotype approach has led to results at this level of significance utilizing hundreds of subjects.11–13,32,33 Thus, the expression GWAS approach may be a more powerful strategy to identify the genetic underpinnings of common, complex diseases.
We suggest the use of the expression GWAS as a complementary approach to the more traditional disease GWAS. This is because not all functional variants are expected to influence gene expression and the disease GWAS approach screens the whole genome for risk variants regardless of the underlying biology. Furthermore, the disease GWAS, in conjunction with the expression GWAS, will allow the cross-validation of the association results from both approaches, thereby leading to the identification of a set of plausible candidate functional disease variants influencing gene expression.
Until recently, APOE was the only locus robustly associated with LOAD.3 Additional promising genes that have weaker association with LOAD are now being identified as GWAS are performed on case-control series of sufficient size. Among these are SORL1,34 GAB2,26 and PCDH11X.14 With the recent publication of 2 powerful genome-wide association studies, 3 more susceptibility genes have been identified: CLU, PICALM, and CR1.30,31 Our results suggest that analysis of cis-SNPs in these genes for association with their transcript levels in brain may lead to a better understanding of their mechanism of action.
IDE also degrades Aβ and influences its level in the brain.35–37 IDE is also located near regions of LOAD linkage on chromosome 10q,38,39 making it an excellent LOAD candidate gene. This has prompted at least 19 studies investigating the association of IDE variants with LOAD and its endophenotypes.40 Some of these studies showed highly significant association, and others did not. In addition, no published GWAS on LOAD14,25–31 has identified variants within IDE as risk variants for LOAD at a level of significance that remained noteworthy after correction for multiple testing. All of these findings reinforce our conclusion that IDE is a LOAD gene with variants that have strong effects on IDE expression and weaker effects on LOAD risk. By chance, the weak association of IDE variants with LOAD risk occasionally reaches statistical significance in some case-control studies; however, the size of the previously published case-control series lacks the power needed to show significant association consistently.
The strong IDE cis-SNP/transcript association that we observed was not evident in a study32 which investigated associations between 366,140 SNPs and 14,078 transcripts from the cerebral cortex of 193 neuropathologically normal subjects. It is important to emphasize that IDE expression level data were available for only 70 of the 193 subjects in this study, making it considerably less powerful than our dataset (178 samples) to detect IDE cis-SNP/transcript associations. Other reasons for this discrepancy may be related to differences in the SNPs tested in the 2 studies, the brain region studied, the method of mRNA quantification, or the fact that we analyzed LOAD brains. This group recently reported their analysis of brain transcriptome levels from 176 cases with pathologic diagnosis of AD and 188 neuropathologically normal subjects for 8,650 transcripts.33 Given that IDE was not among the 8,650 transcripts reported, we are unable to compare our results on this gene with this study.
Recent GWAS of transcripts from lymphoblastoid cell lines11–13 and human cerebral cortex32,33 indicate that many cis-SNPs show strong association that achieves genome-wide significance when hundreds of samples are analyzed. These studies identifying many genes with strong cis-SNP/transcript associations fit well with our finding that 1 of 564 cis-SNPs tested for association with 12 transcripts showed genome-wide significance. Thus our results support the concept that genes with strong cis-SNP/transcript associations can be analyzed effectively for association with disease using the most significant cis-SNP from each gene. Because the number of SNPs with such strong, functionally meaningful associations will be small relative to the number of SNPs tested in a typical GWAS, analysis for association with disease will require less correction for multiple testing and have greater power to detect weak disease associations than a typical GWAS. It should be possible to identify these SNPs relatively efficiently, for testing in follow-up studies of large case control series, by performing GWAS of the whole transcriptome in affected and unaffected brain regions from several hundred LOAD cases and controls. Our results provide strong evidence that IDE is a LOAD gene with variants that reduce risk of LOAD by increasing IDE expression that would thereby reduce Aβ. More generally, they suggest that the use of brain mRNA levels as endophenotypes in LOAD GWAS may be a powerful way to identify LOAD susceptibility alleles.
ACKNOWLEDGMENT
The authors thank the patients and their families.
STUDY FUNDING
Supported by NIH grants: NIA [R01 AG18023 to N.R.G.-R. and S.G.Y.]; Mayo Alzheimer's Disease Research Center: [P50 AG16574 to R.C.P., D.W.D., N.R.G.-R., and S.G.Y.]; Mayo Alzheimer's Disease Patient Registry: [U01 AG06576 to R.C.P.]; NIA [AG25711, AG17216, AG03949 to D.W.D.]. This project was also generously supported by the Robert and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program [to R.C.P., D.W.D., N.R.G.-R., S.G.Y.] and by the Palumbo Professorship in Alzheimer's Disease Research [to S.G.Y.]. N.E.-T. is the recipient of NIA [F32 AG20903], NIH [KL2 RR024151], Johnnie B. Byrd and Siragusa Foundation grants. K.M. and O.B. thank the Alzheimer's Research Trust (ART) for funding.
DISCLOSURE
Dr. Zou and Dr. Carrasquillo report no disclosures. Dr. Pankratz receives research support from the NIH (NIA P50 AG 16574 [Core Director], NIA U01 AG 06786 [Co-I], NCI R01 CA 128931 [Co-I], NCI P30 CA 15083 [Biostatistician], NCI P50 CA 116201 [Statistician], NCI R01 CA 122340 [Co-I], NCI R01 CA 128978 [Co-I], NCI R01 CA 132879 [Co-PI], NIAID N01 AI 40065 [Co-I], and NIAID R01 AI 48793 [Co-I]). Dr. Belbin, Dr. Morgan, Ms. Allen, Ms. Wilcox, Ms. Ma, Ms. Walker, Ms. Kouri, Mr. Burgess, and Dr. L.H. Younkin report no disclosures. Mr. S.G. Younkin receives support from the NIH (T32-HL007567 [Student] and R01-GM028356 [Student]). Mr. C.S. Younkin, Ms. Bisceglio, and Dr. Crook report no disclosures. Dr. Dickson serves on the editorial boards of the American Journal of Pathology, Journal of Neuropathology and Experimental Neurology, Brain Pathology, Neurobiology of Aging, Journal of Neurology Neurosurgery and Psychiatry, Annals of Neurology, and Neuropathology; and receives research support from the NIH [P50-AG25711 (CL), P50-AG16574 (CL), P50-NS40256 (PI), P01-AG17216 (PI), P01-AG03949 (Co-I), and R01-AG15866 (Co-I)]. Dr. Petersen serves on scientific advisory boards for Elan Corporation, Wyeth, and GE Healthcare; receives royalties from the publication of Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH/NIA (U01 AG 06786 [PI], P50 AG 16574 [PI], U01 AG 024904 [Subcontract PI], and R01 AG11378 [Co-I]). Dr. Graff-Radford serves on a scientific advisory board for Codman; serves on the editorial boards of The Neurologist; and Alzheimer Disease and Therapy; has a patent pending on the Ab40Ab42 ratio as a predictor of Alzheimer disease; received royalties for an article in UpToDate on Normal Pressure Hydrocephalus (ADD DATE); has received honoraria from GE Healthcare on imaging for Movement Disorders; and receives research support from Pfizer, Elan Corporation, Forest Laboratories, Inc., Medivation, Inc. and the NIH (NIA P50AG16574 [Co-I], NIA U24AG26395 [Site PI], NIA U01AG24904 [Site PI], and NIA R01AG06656 [Co-I]). Dr. S.G. Younkin has received honoraria and funding for travel from Eisai Inc.; receives research support from the NIH (AG06656 [PI], AG018023 [PI], and AG16574 [Co-I]); and receives royalty payments from Mayo for licensing of Tg2576 mouse model of Alzheimer disease (US 5877399, issued 03/02/1999; US 6262335, issued 07/17/2001; US 6509515, issued 01/21/2003; and Europe [UK, France and Germany] 0742831, issued 9/29/2004). Dr. Ertekin-Taner has received/receives research support from the NIH (NIA F32 AG20903 [PI] and KL2 RR024151 [trainee]), the Johnnie B. Byrd Foundation, and the Siragusa Foundation.
Supplementary Material
Address correspondence and reprint requests to Dr. N. Ertekin-Taner, Departments of Neurology and Neuroscience, Mayo Clinic College of Medicine, Jacksonville, FL 32224 taner.nilufer@mayo.edu
Supplemental data at www.neurology.org
*These authors contributed equally.
Disclosure: Author disclosures are provided at the end of the article.
Received July 22, 2009. Accepted in final form November 5, 2009.
REFERENCES
- 1.Rice DP, Fillit HM, Max W, Knopman DS, Lloyd JR, Duttagupta S. Prevalence, costs, and treatment of Alzheimer's disease and related dementia: a managed care perspective. Am J Manag Care 2001;7:809–818. [PubMed] [Google Scholar]
- 2.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–174. [DOI] [PubMed] [Google Scholar]
- 3.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 4.Myers RH, Schaefer EJ, Wilson PW, et al. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology 1996;46:673–677. [DOI] [PubMed] [Google Scholar]
- 5.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 6.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 2003;160:636–645. [DOI] [PubMed] [Google Scholar]
- 7.Gould TD, Gottesman II. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav 2006;5:113–119. [DOI] [PubMed] [Google Scholar]
- 8.Glahn DC, Thompson PM, Blangero J. Neuroimaging endophenotypes: strategies for finding genes influencing brain structure and function. Hum Brain Mapp 2007;28:488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rockman MV, Wray GA. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol 2002;19:1991–2004. [DOI] [PubMed] [Google Scholar]
- 10.Monks SA, Leonardson A, Zhu H, et al. Genetic inheritance of gene expression in human cell lines. Am J Hum Genet 2004;75:1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon AL, Liang L, Moffatt MF, et al. A genome-wide association study of global gene expression. Nat Genet 2007;39:1202–1207. [DOI] [PubMed] [Google Scholar]
- 12.Stranger BE, Forrest MS, Clark AG, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet 2005;1:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT. Mapping determinants of human gene expression by regional and genome-wide association. Nature 2005;437:1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrasquillo MM, Zou F, Pankratz VS, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet 2009;41:192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Z, Hu J, Li X, Jiang Y, Zhou W, Lu D. Expression analyses of 27 DNA repair genes in astrocytoma by TaqMan low-density array. Neurosci Lett 2006;409:112–117. [DOI] [PubMed] [Google Scholar]
- 16.Steg A, Wang W, Blanquicett C, et al. Multiple gene expression analyses in paraffin-embedded tissues by TaqMan low-density array: application to hedgehog and Wnt pathway analysis in ovarian endometrioid adenocarcinoma. J Mol Diagn 2006;8:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 19.Storey JD. The Positive False Discovery Rate: a Bayesian interpretation and the q-value. Ann Stat 2003;31:2013–2035. [Google Scholar]
- 20.Storey JD, Tibshirani R. Statistical significance for genome-wide experiments. Proc Natl Acad Sci 2003;100 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storey JD. A direct approach to false discovery rates. J R Stat Soc B 2002;64:479–498. [Google Scholar]
- 22.Vitart V, Rudan I, Hayward C, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet 2008;40:437–442. [DOI] [PubMed] [Google Scholar]
- 23.Campbell H, Carothers AD, Rudan I, et al. Effects of genome-wide heterozygosity on a range of biomedically relevant human quantitative traits. Hum Mol Genet 2007;16:233–241. [DOI] [PubMed] [Google Scholar]
- 24.Zemunik T, Boban M, Lauc G, et al. Genome-wide association study of biochemical traits in Korcula Island, Croatia. Croat Med J 2009;50:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grupe A, Abraham R, Li Y, et al. Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum Mol Genet 2007;16:865–873. [DOI] [PubMed] [Google Scholar]
- 26.Reiman EM, Webster JA, Myers AJ, et al. GAB2 alleles modify Alzheimer's risk in APOE epsilon4 carriers. Neuron 2007;54:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Wetten S, Li L, et al. Candidate single-nucleotide polymorphisms from a genome-wide association study of Alzheimer disease. Arch Neurol 2008;65:45–53. [DOI] [PubMed] [Google Scholar]
- 28.Bertram L, Lange C, Mullin K, et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet Epub 2008. [DOI] [PMC free article] [PubMed]
- 29.Beecham GW, Martin ER, Li YJ, et al. Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 2009;84:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet Epub 2009. [DOI] [PubMed]
- 31.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet Epub 2009. [DOI] [PMC free article] [PubMed]
- 32.Myers AJ, Gibbs JR, Webster JA, et al. A survey of genetic human cortical gene expression. Nat Genet 2007;39:1494–1499. [DOI] [PubMed] [Google Scholar]
- 33.Webster JA, Gibbs JR, Clarke J, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet 2009;84:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 2007;39:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurochkin IV, Goto S. Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett 1994;345:33–37. [DOI] [PubMed] [Google Scholar]
- 36.Qiu WQ, Walsh DM, Ye Z, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem 1998;273:32730–32738. [DOI] [PubMed] [Google Scholar]
- 37.Vekrellis K, Ye Z, Qiu WQ, et al. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci 2000;20:1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myers A, Holmans P, Marshall H, et al. Susceptibility locus for Alzheimer's disease on chromosome 10. Science 2000;290:2304–2305. [DOI] [PubMed] [Google Scholar]
- 39.Bertram L, Blacker D, Mullin K, et al. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science 2000;290:2302–2303. [DOI] [PubMed] [Google Scholar]
- 40.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 2007;39:17–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.