

Abstract
Objective
Periostin is dramatically upregulated in rat carotid arteries after balloon injury. The objective of the present study was to understand mechanisms underlying periostin upregulation in balloon injured rat carotid arteries and in cultured vascular smooth muscle cells (VSMCs).
Methods and Results
Periostin protein was strongly expressed at 3d (in the medial SMCs) and 7d (in the neointima) after injury. It was also abundantly expressed in the neointima in the late phase (at 14d and 28d) after injury. Periostin upregulation was mediated through PI3-kinase-dependent signaling pathway. In vivo, wortmannin, a PI-3-kinase inhibitor, inhibited balloon injury-induced Akt phosphorylation and periostin mRNA expression. In vitro, periostin mRNA expression in cultured VSMCs was stimulated by growth factors (TGF-β1, FGFs, PDGF-BB, and angiotensin II). This stimulatory effect was inhibited by the PI3-kinase inhibitor LY294002. Further, periostin protein was mostly located in the cytoplasma of VSMCs in culture and abundantly secreted into the culture medium (CM) after stimulation with FGF-2, which significantly promoted VSMC migration in vitro. Immunodepletion of periostin from the VSMC-CM or blockade of periostin function with an anti-periostin antibody significantly reduced VSMC migration.
Conclusions
Upregulation of periostin expression in rat carotid arteries following balloon injury and in cultured VSMCs after stimulation by growth factors is mediated through PI-3 kinase-dependent signaling pathway. Periostin protein secreted by VSMCs plays a significant role in regulating VSMC migration in vitro.
Keywords: periostin, PI-3 kinase, smooth muscle, migration
Introduction
Periostin (originally termed OSF2 for osteoblast-specific factor 2) is a novel secreted adhesion molecule (~90 kDa) and putative soluble extracellular matrix (ECM) protein (1, 2). It has a nuclear localization signal at the carboxy-terminus and a secretion signal peptide at the amino terminus, suggesting that it can localize to the nucleus and be secreted (1). Periostin is expressed in several normal adult tissues, preferentially in bone and to a lesser extent in lung, kidney and heart valves, but is not found in normal adult blood vessels (3-5). Multiple reports show that periostin is overexpressed in several types of human tumors and acts as a cell adhesion molecule through binding to cell surface integrins. For example, periostin promotes ανβ3 and ανβ5 integrin-dependent cell adhesion and motility in human epithelial ovarian carcinoma (6). It has also been shown to prevent stress-induced tumor cell apoptosis and to promote angiogenesis in colon cancers through the Akt/PKB signaling- and ανβ3 integrin-dependent mechanisms (7). The periostin-mediated induction of angiogenesis is mediated in part through upregulation of vascular endothelial growth factor receptor 2 expression via integrin ανβ3-focal adhesion kinase-mediated signaling pathways (8).
Abnormal expression of periostin has been reported in several cardiovascular pathologic conditions, such as infarcted rat myocardium and hypertrophied mouse hearts (9). In a rat gene transfer model, overexpression of the periostin gene leads to cardiac dilation and dysfunction, and inhibition of periostin gene expression using an antisense strategy results in a significant increase in survival rate accompanied by an improvement in LV function (10).
Periostin is strongly upregulated in balloon injured rat carotid arteries (11). It has been implicated in cell differentiation (in A404 cells) and migration (in C3H10T1/2 cells, a mouse fibroblast cell line) (11). To date, mechanisms underlying periostin upregulation and its possible biological functions in the vasculature remain unknown. The objective of the present study was to investigate signaling mechanisms by which periostin was upregulated in rat carotid arteries following balloon injury and in cultured VSMCs after stimulation by growth factors. In addition, we investigated effects of periostin on VSMC migration in vitro.
Methods
Rat Carotid Balloon Injury Model
Male Sprague-Dawley rats were obtained from Charles River Breeding Laboratories (Wilmington, MA) at 10 weeks of age, fed standard pellet diet and given water ad libitum. All animals used in this study were cared for and handled according to the National Institutes of health Guide for the Care and Use of Laboratory Animals. Balloon injury of the left common carotid artery of the rat was performed under anesthesia with ketamine (80 mg/kg) and xylazine (5 mg/kg) essentially as previously described (12). The right carotid artery was not damaged and served as a control. Animals were killed by an overdose of pentobarbital (210 mg/kg IP) at indicated times after balloon injury.
Cell Isolation and Culture
Rat aortic SMCs (RASMCs) were isolated from the thoracic aortas of Sprague-Dawley rats and cultured as previously described (13, 14). Cells were cultured in complete medium containing Dulbecco’s modified Eagle’s medium (DMEM, GIBCO BRL) supplemented with 10% (v/v) fetal bovine serum (FBS), 4 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Experiments were performed with cells from passages 4 through 9.
RNA Extraction and Northern Blot Analysis
For in vivo studies, total RNA was extracted from pooled injured left or uninjured right carotid arteries, with use of TRIzol Reagent (Invitrogen) treated with DNase I to remove genomic DNA and then purified with an RNA purification kit (Invitrogen). Each group contained 5 to 8 rats. For in vitro studies, total RNA was extracted from RASMCs that were grown to subconfluence (~95%) in complete medium, serum deprived for 24 hours, and then treated with defined reagents for varying times or concentrations (see figure legends for details). Northern blot analysis was carried out using 32P-labeled cDNA probes for rat periostin (PN) and osteopontin (OPN) as previously described (14,15). A 32P-labeled 18S rRNA oligonucleotide (5’-ACGGTATCTGATCGTCTTCGAACC-3’) was used as the control probe to normalize data. Quantification of autoradiographic signals was performed by densitometry. Results were expressed as the ratios of specific mRNA to 18S rRNA.
Protein Extraction and Western Blot Analysis
For in vivo studies, total proteins were extracted from pooled injured or naïve uninjured carotid arteries. Each group contained 5 rats. Briefly, tissues samples were ground to a fine powder under liquid nitrogen using a tissue homogenizer, washed twice in ice-cold PBS/1 mM Na3VO4, and then lysed in RIPA-II buffer (500 mM NaCl, 50 mM Tris/HCl, pH 7.4, 0.1% SDS, 1% NP-40, 0.5% Na-DOC, 0.05% NaN3, 1 mM Na3VO4) plus Complete Protease Inhibitor cocktail (Roche) for 1 hour on ice. Protein concentration was measured using a BCA Protein Assay kit (Pierce, Rockford, IL). Equal amounts (30 μg/lane) of protein lysates were run out on a 10% SDS-PAGE and transferred to nitrocellulose membrane (Amersham). Nonspecific binding was blocked for 1.5 hour with 5% nonfat dry milk in TBS-T buffer (10 mmol/L Tris [pH 7.5], 100 mmol/L NaCl, 0.1% Tween 20).
For periostin protein assay, the blots were incubated with rabbit anti-periostin polyclonal antibody (1:800 dilution) (provided by Drs. Meiru Dai and Lan Bo Chen, Harvard Medical School) and anti-α-tubulin antibody (1:3000 dilution, oncogene) to demonstrate equal protein loading. For Akt phosphorylation analysis, the blots were incubated for 1 hour with anti-phospho-Akt (Ser473) (1: 500 dilution) or anti-total-Akt antibody (1: 500 dilution) (both from Cell Signaling Technology). An identical blot was incubated with nonimmune normal IgG as a control. Blots were washed 3 times with TBS-T buffer between incubations. All incubations were carried out with agitation at room temperature in TBS-T containing 2.5% nonfat dry milk. Immune complexes were detected by Phototop-horseradish peroxidase Western detection kit (Cell Signaling Technology).
For in vitro studies, western blot assay was carried out to measure secreted periostin protein that released into RASMC-conditioned medium. The medium was conditioned by cultured RASMCs under defined conditions (see figure legends for details) and concentrated approximately 5-fold using Ultra Concentrator (50 kDa cut-off, Amicon). The concentrated medium (1.0 mL) was incubated (16 hours, 4°C) with 10 μg of rabbit anti-periostin polyclonal antibody. Immune complexes were precipitated with (1.5 hours, 4°C) with 100 μl protein A-Sepharose and fractionated by 10% SDS-PAGE for western blot assay of periostin protein, as described above.
Immunohistochemistry
At the indicated times after balloon injury, animals were perfusion-fixed with 4% phosphate-buffered paraformaldehyde. Each time point contained five rats. Both injured left and uninjured right carotid arteries were harvested. Serial 5-μm paraffin-embedded arterial sections were prepared for immunostaining of periostin using the avidin-biotin-peroxidase method (Vector Labs). Endogenous peroxidase activity was blocked with 0.45% hydrogen peroxide in methanol for 30 minutes. After blocking with normal goat serum, sections were incubated with rabbit anti-periostin polyclonal antibody at a concentration of 2 μg/mL overnight at °4C in humidified chamber. Sections were then incubated with biotinylated goat anti-rabbit secondary antibody (1:300) followed by avidin-biotin-peroxidase complex in a Vectastain Elite kit (both from Vector Labs). The reaction product was visualized with the chromogenic substrate DAB (Dako), which produces brown stain. The sections were counterstained with hematoxylin I (Richard-Allan Scientific). For negative controls, the sections were stained without the primary or secondary antibody or both, which did not produce brown stain.
Immunofluoresence staining was performed to determine localization of periostin protein in RASMCs essentially as described (16). Briefly, cells were cultured in chamber slides (Fisher Scientific) under defined conditions (see Figure legends for details). Periostin protein was detected by anti-periostin Ab and biotinylated secondary Ab and visualized by streptavidin conjugated-Alexa Fluor 488 (Molecular Probes) using fluorescence microscopy (Olympus BX51 microscope).
Migration Assay
Haptotaxis migration assays were performed using Transwell migration chambers (Costar Corp) essentially as previously described (13). For preparation of VSMC-conditioned medium, quiescent RASMCs (~95% confluence) were maintained in serum free medium for 12 hours in the absence or presence of 10 ng/mL FGF-2. After washing (PBS) 3 times, cells were incubated in fresh serum free medium for 24 hours, and then the conditioned medium was harvested. Upper-chamber membranes were coated (4°C, 48 hours) with the conditioned medium or the medium containing varying concentrations of recombinant rat vitronectin (Sigma). The lower chambers contained migration buffer (DMEM/0.1% BSA) only. A suspension (200 μL) of RASMCs (3×104 cells in migration buffer) alone or along with indicated concentrations of anti-periostin antibody or isotype-match irrelevant rabbit anti-HA antibody (Zymed) was added to the upper chamber. Migration was allowed to proceed for 6 hours at 37°C in a humidified incubator, and cells that migrated to the bottom of the filter were fixed with methanol and stained with hematoxylin. Migration was quantitated by cell counts of 4 random high-power (×100) fields in each well. Each assay was performed in quadruplicate.
Haptotaxis assay also included coating of the upper-chamber membranes with the periostin-deleted VSMC-conditioned medium. Depletion of periostin from VSMC-conditioned medium was achieved by immunoadsorption technique (13). Briefly, 10 μg of anti-periostin antibody or rabbit anti-HA antibody was incubated (4°C; 16 hours) with 50 μL protein A-Sepharose (Santa Cruz). After washing (ice pre-cooled PBS) 2 times, immuno-complexed beads were incubated (4°C; 1 hour) with 1.0 mL VSMC-conditioned medium. Clarified supernatants were used to coat upper-chamber membranes for haptotaxis migration assays, as described above.
Statistical Analysis
Each experiment was repeated 2-3 times. Data are expressed as mean±SEM. Statistical analysis was performed with one-way ANOVA or Student’s t test, as appropriate. Values of P<0.05 were considered significant.
Results
Carotid Balloon Injury Induces Periostin Expression via PI3-Kinase Pathway
Expression of periostin mRNA dramatically increased in injured left carotid arteries at 3 and 7 days after balloon injury, with a peak at 3 days, but was minimal in uninjured right carotid arteries (Figure 1A). Similarly, periostin protein was absent in normal uninjured carotid arteries but increased to significant high levels as seen in the 7-day injured left carotid arteries (Figure 1B).
Figure 1.

A. Northern blot analysis of periostin (PN) mRNA expression in the injured left carotid arteries at 3 and 7 days after balloon injury. Uninjured right carotid arteries were used as a control. Each lane was loaded with 10 μg of total RNA extracted from pooled three carotid arteries. B. Western blot analysis of periostin protein in the injured left carotid arteries at 7 days after balloon injury. Naive uninjured carotid arteries were used as a control. Each lane was loaded with 30 μg of total protein lysates extracted from pooled five carotid arteries.
Activation of the PI3-kinase signaling was evaluated by phosphorylation of Akt, which was negligible in normal blood vessels but was markedly induced in the 3-day injured carotid arteries (Figure 2A). The involvement of the PI3-kinase signaling in regulation of periostin expression in the vasculature was examined by using the PI3 kinase inhibitor wortmannin. Although the LY294002 compound is a more potent and highly selective PI3-kinase inhibitor, it appears not to be suitable for in vivo studies as it is very insoluble and thus unable to achieve an effective concentration in the artery in vivo according to previous reports (17, 18). Wortmannin (dissolved in 2%DMSO/PBS) was given by intravenous injection at 60 and 5 minutes before balloon injury followed by daily injections, at a dose of 10 μg per rat. This dosing regimen has been showed to effectively inhibit the activation of Akt, but not ERK1/2 kinases in rat carotid arteries after balloon injury (17, 18). The wortmannin treatment of rats inhibited the Akt phosphorylation (Figure 2A) and the periostin mRNA upregulation (Figure 2B) in the 3-day injured carotid arteries.
Figure 2.

A. Western blot analysis of Akt phosphorylation in the injured left carotid arteries at 3 days after balloon injury. Naïve uninjured carotid arteries were used as a control. Protein lysates (~30 μg/lane) were immunoblotted with anti-phospho-Akt or anti-total-Akt antibody. Lane 1: uninjured arteries; lanes 2: 3-day injured arteries; lane 3:3-day injured arteries treated with vehicle (V, 2% DMSO/PBS); lane 4: 3-day injured arteries treated with wortmannin (WM). B. Northern blot analysis of periostin (PN) mRNA expression in uninjured (control), injured and untreated, injured and WM-treated carotid arteries at 3 days after balloon injury. Each lane was loaded with 10 μg of total RNA extracted from pooled three carotid arteries.
Growth Factors Induce Vascular Smooth Muscle Cell Expression of Periostin via PI3-Kinase Pathway In Vitro
Expression of periostin mRNA was readily detectable in quiescent RASMCs in vitro (~95% confluence) and robustly stimulated by multiple growth factors (all from Sigma). Transforming growth factor-β1 (TGF-β1) has been showed to stimulate periostin mRNA expression in bone cells (2, 3). Similarly, it stimulated periostin mRNA in cultured RASMCs in a dose-dependent manner (Figure 3A). In contrast to the previous report (11), expression of periostin mRNA was upregulated by multiple growth factors. FGF-2 was showed to stimulate periostin mRNA expression in a time-dependent manner (Figure 3B). In addition, other growth factors, such as FGF-1, PDGF-BB and angiotensin II, also robustly stimulated periostin mRNA expression (Figure 3C).
Figure 3.


Northern blot analysis of periostin (PN) mRNA expression in RASMCs in vitro. Cells were grown to subconfluence (~95%) in 100-mm dishes, deprived of serum for 24 hours, and then treated under defined experimental conditions. Each lane was loaded with 10 μg of total RNA. A. Cells were treated with the varying concentrations of TGF-β1 for 12 hours. B. Cells were treated with 10 ng/mL FGF-2 for various times. C. Cells were treated with 10 ng/mL of FGF-1, or PDGF-BB, or 10-7mol/L Ang II for 12 hours. Data are mean ± SEM. *P<0.001 vs control.
Further, the involvement of PI3-kinase signaling in regulation of periostin mRNA expression was examined in RASMCs in vitro. The PI3-kinase inhibitor LY294002 pretreatment (45 minutes) almost completely inhibited the expression of periostin mRNA induced by TGF-β1 (Figure 3A), FGF-1 (Figure 4A) and angiotensin II (Figure 4B). To test the specificity of the inhibitory effects of LY294002 on periostin expression, the same membrane was re-hybridized with the osteopontin (OPN) cDNA probe. Intriguingly, the LY294002 pretreatment did not block OPN expression induced by TGF-β1 (Figure 3A) and FGF-1 (Figure 4A). In addition, other signaling kinase inhibitors such as PD98059 (for p42/p44 MAP kinase) and SB203580 (for p38 MAP kinase) exhibited a partial inhibition of periostin mRNA expression induced by FGF-1 and angiotensin II (Figure 4C and 4D).
Figure 4.


Effects of signaling kinase inhibitors on periostin (PN) mRNA expression in RASMCs in vitro. Cells were prepared as described in Figure 3, and pretreated (45 min) with indicated kinase inhibitors followed by treatment with 10 ng/mL FGF-1 or 10-7mol/L angiotensin II (Ang II) for 12 hours. Each lane was loaded with 10 μg of total RNA. Note that LY294002 (10 μmol/L) blocks PN mRNA expression induced by FGF-1 (A) or Ang II (B), but it does not block osteopontin (OPN) mRNA expression induced by FGF-1 when the same membrane was re-hybridized with the OPN cDNA probe. The p42/p44 MAP kinase inhibitor PD98059 (25 μmol/L) (C) and the p38 MAP kinase inhibitor SB20580 (2.5 μmol/L) (D) results in partial inhibition of PN mRNA expression induced by FGF-1 and AngII.
Spatiotemporal Expression of Periostin Following Carotid Balloon Injury
The spatial and temporal expression pattern of periostin protein in rat carotid arteries after balloon injury were showed in Figure 5. Consistent with the protein assay by Western blotting (Figure 1B), immunohistochemistry showed that periostin protein (brown stain) was robustly expressed in the developing neointimal lesions in the 7-day injured left carotid arteries (panel a), but was minimal in uninjured right carotid arteries (panel d). Periostin was also abundantly expressed in the neointimal lesions in the late phase (at 14d and 28d) after injury (panels b and c, respectively). In addition, abundant levels of periostin expression were observed in the medial SMCs at 3 days after carotid balloon injury (data not shown).
Figure 5.
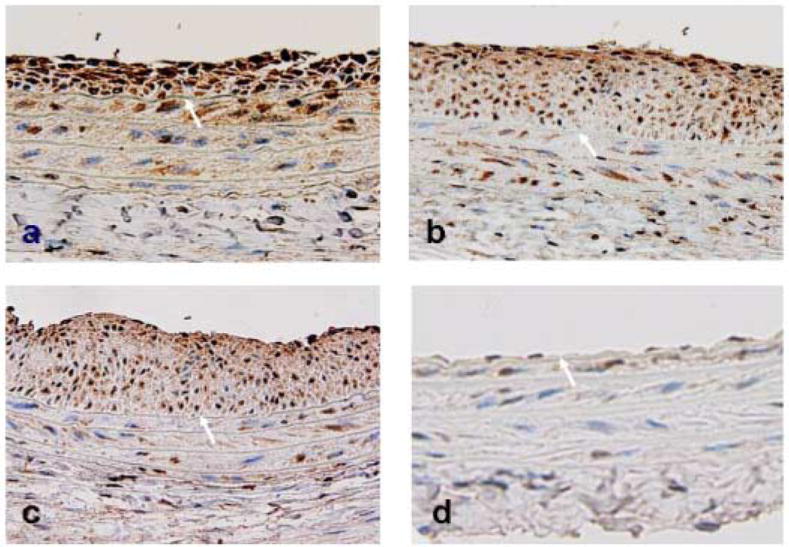
Immunohistochemistry illustrating the time-course expression of periostin protein in the injured left carotid arteries at 7 days (panel a), 14 days (panel b), and 28 days (panel c) after balloon injury, and in the uninjured contralateral right carotid arteries (panel d). Arrows indicate internal elastic lamina. Positive periostin staining is shown in a brown color. Magnification × 400.
In order to better characterize the subcellular localization of periostin protein in VSMCs, immunofluoresence staining was performed using rat aortic SMCs cultured in chamber slides. Periostin protein was localized robustly in the cytoplasma and to a much lesser extent in the nuclei in the cultured aortic SMCs (Figure 6A).
Figure 6.

A. Immunofluoresence staining illustrating the subcellular localization of periostin protein in VSMCs in culture. Cells were grown in chamber slides until ~70% confluence, deprived of serum for 24 hours, and then incubated (for 24 hours) in fresh medium as follows: serum free medium alone (panel a), plus 10% FBS (panel b), or plus 10 ng/ml FGF-2 (panels c and d). Positive periostin staining is shown in a green color. Cells in panel d was stained without primary antibody and served as a negative control. Note that periostin was mostly localized in the cytoplasma and to a much lesser extent in the nuclei. B. Western blot analysis of periostin protein that secreted into the conditioned-medium from RASMCs in culture. Subconfluent (95%) quiescent RASMCs were incubated in the absence (lane 1) or presence of 10 ng/mL FGF-2 (lane 2), or pretreated (45 min) with 10 μmol/L Y294002 followed by FGF-2 treatment (lane 3) for 12 hours. Then, cells were maintained in fresh serum-free medium for additional 24 hours and medium was harvested as conditioned-medium. Western analysis was carried out after concentration (5-fold) and immunoprecipitation of the conditioned-medium.
Periostin Secretion and Migratory Effect on VSMC Migration in Vitro
Periostin is a secreted protein in bone and tumor cells, but its secretion in VSMCs has not been reported. Periostin protein was readily detectable in serum-free medium conditioned by quiescent RASMCs as measured by Western blot (Figure 6B, lane 1). FGF-2 treatment of the quiescent RASMCs markedly increased secretion levels of periostin protein (Figure 6B, lane 2), and this increase was inhibited by the PI3-kinase inhibitor LY294002 pretreatment (Figure 6B, lane 3).
Subsequent experiments investigated whether the VSMC-secreted periostin protein enhances cell migration, by use of two approaches: (1) antibody blockade of periostin function, which was achieved by incubation of RASMCs that were placed in the upper-chambers alone or along with anti-periostin antibody or rabbit anti-HA antibody (Iso-Ab) during the 6-hour migration assay period, and (2) depletion of periostin protein from VSMC-conditioned medium by immunoadsorption technique (14). Under basal conditions, the medium conditioned by quiescent RASMCs stimulated RASMC haptotaxis migration (~130 cells/field). The FGF-2 treatment of RASMCs further enhanced RASMC haptotaxis migration by approximately 80%. The antibody blockade of periostin function and immunodepletion of periostin protein significantly reduced the FGF-2-enhanced RASMC haptotaxis migration by 65 % (Figure 7A) and 63% (Figure 7B) (both P<0.01), respectively. Moreover, recombinant rat vitronectin dose-dependently stimulated VSMC migration, but this stimulation was not blocked by anti-periostin Ab (Figure 7C), indicating the specificity of the anti-periostin antibody for blocking the periostin-induced VSMC migration.
Figure 7.

Effects of periostin protein secretion on VSMC hapotaxis migration. A. Antibody blockade of periostin reduces VSMC haptotaxis migration. Blockade was achieved by incubation of VSMCs in migration buffer alone (non-Ab) or along with 10 ug/mL of anti-periostin (anti-PN) or rabbit anti-HA antibody (Iso-Ab) in the upper-chamber. Upper-chamber membranes were pre-coated with conditioned medium of quiescent VSMCs that were maintained in the absence (bar 1) or presence of FGF-2 (bars 2, 3, 4). Results are expressed as percentage of the migration observed in the control group (bar 1). *P<0.001 vs bar 1. #P<0.01 vs bar 2 and bar 3.
B. Depletion of secreted periostin protein from VSMC-conditioned medium reduces VSMC haptotaxis migration. Depletion was achieved by immunoadsorption using beads alone (non-Ab) or beads immuno-complexed with indicated concentrations of anti-PN or rabbit anti-HA antibody (Iso-Ab). Upper-chamber membranes were pre-coated with periostin-depleted conditioned-medium obtained in the absence (bar 1) or presence (bars 2 to 5) of FGF-2 treatment. Results are expressed as percentage of the migration observed in the control group (bar 1). *P<0.001 vs bar 1. #P<0.01 vs bar 2 and bar 3.
C. Anti-periostin antibody (anti-PN) does not block vitronectin-induced VSMC haptotaxis migration. Blockade was achieved by incubation of VSMCs in migration buffer containing 10 ug/ml of anti-PN in the upper chambers. Up-chamber membranes were pre-coated with migration buffer alone (control) or containing varying concentrations of vitronectin, or with the conditioned-medium of the FGF-2-treated VSMCs (FGF2-CM).
Discussion
The present study for the first time demonstrated that upregulation of periostin expression in the vasculature, in the response to either endoluminal arterial injury in vivo or stimulation by growth factors in vitro, is mediated through the PI-3 kinase-dependent signaling pathway. This study further demonstrated that periostin protein was mostly located in the cytoplasma of VSMCs in culture and was abundantly secreted into the culture medium, which significantly promoted VSMC migration in vitro.
Periostin mRNA is among the most strongly upregulated transcripts in rat carotid arteries after balloon injury. Using in situ hybridization (ISH), the previous study showed that periostin mRNA expression was localized to smooth muscle cells of the neointima and to the adventitia in rat carotid arteries after balloon injury (11). Using immunohistochemistry, the present study further revealed the spatial and temporal expression pattern of periostin protein after rat carotid balloon injury (Figure 5). In consistent with the protein assay by the Western blotting (Figure 1B), periostin protein was expressed robustly in the developing neointimal lesions in the 7-day injured carotid arteries. The neointimal cells were showed to express abundant levels of the protein out to 4 weeks (Figure 5). In contrast to the mRNA expression determined by ISH (11), periostin protein was sparse in the adventitia, indicating that mRNA transcripts do not necessarily indicate pattern and amount of resulting protein. Moreover, the present study demonstrated that periostin protein was mostly localized in the cytoplasma and to a much lesser extent in the nuclei of RASMCs in vitro (Figure 6A), and was abundantly secreted in the culture medium (Figure 6B).
So far, few studies have addressed the expression and regulation of periostin in the vasculature. Lindner et al (11) reported that expression of periostin in human and rat aortic SMCs in vitro was not regulated by cytokines such as fibroblast growth factor-2 (FGF-2). In contrast, Li et al (15) reported that expression of periostin mRNA in rat pulmonary arterial SMCs in vitro was upregulated by FGF-1 and angiotensin II, in a dose- and time- dependent manner. The present study clearly demonstrated that expression of periostin mRNA in rat aortic SMCs in vitro was robustly stimulated, in a dose- and time-dependent manner, by multiple growth factors, such as TGF-β1, FGF-1, FGF-2, PDGF-BB, and angiotensin II (Figure 3). These growth factors are believed to act as important mediators of the vascular response to injury. The discrepancy between different studies may be explained by distinct experimental conditions in which cells were prepared and treated. In the present study and the Li’s report (15), vascular SMCs were made quiescent prior to stimulation by growth factors whereas in the Lindner’s report (11) cells were stimulated by growth factors in the presence of 10% fetal bovine serum. Non-quiescent cells, particularly in the presence of serum, likely contain high levels of endogenous growth factors such as FGF-2, which could limit the ability of cells to respond to exogenous growth factors (11). This speculation was also supported by our data that showed abundant levels of periostin expressed in RASMCs in vitro in the presence of 10% fetal bovine serum (Figure 6A).
The PI3-kinase pathway is important for survival, replication, and migration of mammalian cells, including VSMCs (19-21). This pathway has been showed to be rapidly activated by growth factors such as PDGF and angiotensin II in vitro (20,21) and by mechanical vascular injury in vivo (17,18). Inhibition of the PI3-kinase/Akt signaling pathway has been showed to limit the neointima formation in rat vascular injury models. Administration of the PI3-kinase inhibitor wortmannin reduced the Akt phosphorylation, with a concomitant reduction in medial SMC proliferation in the 2-day balloon injured rat carotid arteries (17). Further, administration of wortmannin to stented rats resulted in a significant reduction in neointima formation at 3 weeks after stent implantation (18). The present study tested the hypothesis that the activation of PI3-kinae signaling mediates periostin expression in rat carotid arteries after balloon injury. Similar to the previous reports (17,18), our data showed high levels of phosphorylation of Akt, a downstream target of the PI3-kinase, in the 3-day-injured carotid arteries (Figure 2A). Further, our data demonstrated that in vivo treatment of rats with the PI-3-kinase inhibitor wortmannin inhibited both the phosphorylation of Akt and periostin mRNA expression in the 3-day balloon injured carotid arteries (Figure 2A and 2B). These findings suggest that the balloon injury-induced periostin expression is mediated through the PI3-kinase/Akt dependent signaling pathway. To further confirm these findings in vivo, we investigated whether the PI3 kinase signaling regulates periostin expression in RASMCs in vitro. Our data demonstrated that periostin mRNA expression in RASMCs in vitro was stimulated by multiple growth factors, such as TGF-β1, FGFs, PDGF-BB, and angiotensin II (Figure 3). This stimulatory effect was inhibited by the PI3-kinase inhibitor LY294002 (Figure 4A and 4B). Taken together, our in vivo and in vitro data suggest that the PI3-kinase pathway appears to act as a common pathway for regulation of periostin expression in the vasculature in response to a variety of stimuli. In addition, as growth factor-mediated gene regulation occurred in most molecules, the periostin gene regulation also involves a variety of parallel or intermediate signaling pathways. Indeed, other signaling kinase inhibitors such as PD98059 (for p42/p44 MAP kinase) and SB203580 (for p38 MAP kinase) resulted in partial inhibition of periostin mRNA expression induced by FGF-1 and angiotensin II (Figure 4C and 4D).
To date, the biological functions of periostin upregulation in the vasculature is unknown. Periostin protein is a secreted protein in bone and tumor cells and may act as a ligand for ανβ3 and ανβ5 integrins (6-8), both integrins are regulators of adhesion and migration in a variety of cell types. The secretion and potential importance of periostin directly derived from vascular SMCs have not been reported. The present study demonstrated that abundant levels of periostin protein were secreted into culture medium conditioned by RASMCs in vitro in the presence of FGF-2 (Figure 6B). Further, our data showed that the VSMC-secreted periostin protein played a significant role in directing VSMC migration in vitro (Figure 7). Intriguingly, we have recently observed that periostin expression is markedly upregulated in hyperlipidemic and atherosclerosis-prone apoE-deficient mouse models of carotid wire denudation injury and innominate artery atherosclerosis (unpublished data). In the future study, we propose to define the functional importance of periostin upregulation in atherosclerotic vascular diseases and potential mechanisms involved, by using our unique periostin knockout mice and perioatin/apoE double knockout mice, together with other approaches, such as small RNA interference.
Acknowledgments
This study was supported in part by a Cardiovascular Research Center Grant (G.H.L) from University of Virginia Health System and a Frommeyer Fellowship in Investigative Medicine (G.H.L) from University of Alabama at Birmingham and an American Heart Association Grant 0530166N (G.H.L), and by National Institutes of Health (NIH) Grants HL64614 (S.O), HL-062522 (C.A.M), and HL-66264 (I.J.S).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takeshita S, Kikuno R, Tezuka K, et al. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J. 1993;294(Pt 1):271–278. doi: 10.1042/bj2940271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horiuchi K, Amizuka N, Katsuura M, et al. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14(7):1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 3.Litvin J, Selim AH, Montgomery MO, et al. Expression and function of periostin-isoforms in bone. J Cell Biochem. 2004;92(5):1044–61. doi: 10.1002/jcb.20115. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki H, Lo KM, Chen LB, et al. Expression of Periostin, homologous with an insect cell adhesion molecule, as a prognostic marker in non-small cell lung cancers. Jpn J Cancer Res. 2001;92(8):869–73. doi: 10.1111/j.1349-7006.2001.tb01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruzynska-Frejtag A, Machnicki M, Rogers R, et al. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103(12):183–8. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 6.Gillan L, Matei D, Fishman DA, et al. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62(18):5358–64. [PubMed] [Google Scholar]
- 7.Bao S, Ouyang G, Bai X, et al. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell. 2004;5(4):329–339. doi: 10.1016/s1535-6108(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 8.Shao R, Bao S, Bai X, et al. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol. 2004;24(9):3992–4003. doi: 10.1128/MCB.24.9.3992-4003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang D, Oparil S, Feng JA, et al. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension. 2003;42(1):88–95. doi: 10.1161/01.HYP.0000074905.22908.A6. [DOI] [PubMed] [Google Scholar]
- 10.Katsuragi N, Morishita R, Nakamura N, et al. Periostin as a novel factor responsible for ventricular dilation. Circulation. 2004;110(13):1806–1813. doi: 10.1161/01.CIR.0000142607.33398.54. [DOI] [PubMed] [Google Scholar]
- 11.Lindner V, Wang Q, Conley BA, et al. Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler Thromb Vasc Biol. 2005;25(1):77–83. doi: 10.1161/01.ATV.0000149141.81230.c6. [DOI] [PubMed] [Google Scholar]
- 12.Levine RL, Chen SJ, Durand J, et al. Medroxyprogesterone attenuates estrogen-mediated inhibition of neointima formation after balloon injury of the rat carotid artery. Circulation. 1996;94:2221–2227. doi: 10.1161/01.cir.94.9.2221. [DOI] [PubMed] [Google Scholar]
- 13.Li G, Chen YF, Kelpke SS, et al. Estrogen attenuates integrin-beta(3)-dependent adventitial fibroblast migration after inhibition of osteopontin production in vascular smooth muscle cells. Circulation. 2000;101(25):2949–55. doi: 10.1161/01.cir.101.25.2949. [DOI] [PubMed] [Google Scholar]
- 14.Li G, Oparil S, Kelpke SS, et al. Fibroblast growth factor receptor-1 signaling induces osteopontin expression and vascular smooth muscle cell-dependent adventitial fibroblast migration in vitro. Circulation. 2002;106(7):854–9. doi: 10.1161/01.cir.0000024113.26985.cc. [DOI] [PubMed] [Google Scholar]
- 15.Li P, Oparil S, Feng W, et al. Hypoxia-responsive growth factors upregulate periostin and osteopontin expression via distinct pathways in rat pulmonary arterial smooth muscle cells. J Appl Physiol. 2004;97:1550–1558. doi: 10.1152/japplphysiol.01311.2003. [DOI] [PubMed] [Google Scholar]
- 16.Li GH, Sanders JM, Phan ET, et al. Arterial macrophages and regenerating endothelial cells express P-selectin in atherosclerosis-prone apolipoprotein-deficient mice. Am J Pathol. 2005 doi: 10.1016/S0002-9440(10)61237-0. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shigematsu K, Koyama H, Olson NE, et al. Phosphatidylinositol 3-kinase signaling is important for smooth muscle cell replication after arterial injury. Arterioscler Thromb Vasc Biol. 2000;20(11):2373–8. doi: 10.1161/01.atv.20.11.2373. [DOI] [PubMed] [Google Scholar]
- 18.Zhou RH, Lee TS, Tsou TC, et al. Stent implantation activates Akt in the vessel wall: role of mechanical stretch in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23(11):2015–20. doi: 10.1161/01.ATV.0000095161.06906.ED. [DOI] [PubMed] [Google Scholar]
- 19.Duan C, Bauchat JR, Hsieh T. Phosphatidylinositol 3-kinase is required for insulin-like growth factor-I-induced vascular smooth muscle cell proliferation and migration. Circ Res. 2000;86:15–23. doi: 10.1161/01.res.86.1.15. [DOI] [PubMed] [Google Scholar]
- 20.Dugourd C, Gervais M, Corvol P, et al. Akt is a major downstream target of PI3-kinase involved in angiotensin II-induced proliferation. Hypertension. 2003;41:882–890. doi: 10.1161/01.HYP.0000060821.62417.35. [DOI] [PubMed] [Google Scholar]
- 21.Goncharova EA, Ammit AJ, Irani C, et al. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L354–63. doi: 10.1152/ajplung.00010.2002. [DOI] [PubMed] [Google Scholar]