

Abstract
FOXA1 and FOXA2, members of the forkhead transcription factor family, are critical for epithelial differentiation in many endoderm-derived organs, including the pancreas. However, their role in tumor progression is largely unknown. Here, we identified FOXA1 and FOXA2 as important antagonists of the epithelial-to-mesenchymal transition (EMT) in pancreatic ductal adenocarcinoma (PDA) through their positive regulation of E-cadherin and maintenance of the epithelial phenotype. In human PDA samples, FOXA1/2 are expressed in all epithelium from normal to well-differentiated cancer cells, but are lost in undifferentiated cancer cells. In PDA cell lines, FOXA1/2 expression is consistently suppressed in experimental EMT models and RNAi silencing of FOXA1/2 alone is sufficient to induce EMT. Conversely, ectopic FOXA1/2 expression can potently neutralize several EMT-related E-cadherin repressive mechanisms. Finally, ectopic FOXA2 expression could reactivate E-cadherin expression in a PDA cell line with extensive promoter hypermethylation. In fact, demethylation-mediated reactivation of E-cadherin expression in these cells required concurrent re-activation of endogenous FOXA2 expression. We conclude that suppression of FOXA1/2 expression is both necessary and sufficient for EMT during PDA malignant progression.
Keywords: E-cadherin, EMT, FOXA1, FOXA2, pancreatic adenocarcinoma
Introduction
The epithelial-to-mesenchymal transition (EMT) is a physiological process, originally described in embryonic development, in which cells lose epithelial characteristics and gain mesenchymal properties. EMT is accompanied by loss of cell-to-cell contact and increased cell motility. It is also implicated in late stage tumor progression as a prelude to cancer metastasis (1, 2). As a gate-keeper of the epithelial phenotype (2), E-cadherin regulation has been extensively studied during EMT, with most research focusing on its transcriptional repression by E12/47, TWIST (3) and members of the Snail (4, 5) and ZEB (6) protein families. In contrast, little attention has been paid to the involvement of E-cadherin transcriptional activators in EMT, since repressive factors are thought to be dominant.
FOXA proteins belong to subclass A of the Forkhead box containing transcription factor family (Fox proteins). FOXA1 and FOXA2 share 92% homology within their forkhead DNA-binding domains (7). Genetic ablation of Foxa1 or Foxa2 in mice causes post-natal or embryonic lethality, respectively(8–11). Individual or compound deletions of Foxa1 and Foxa2 demonstrate that both Foxa1 and Foxa2 are essential for terminal differentiation and maturation of many endoderm-derived cells, including α-cells in the endocrine pancreas (12), and liver (13), lung alveolar (14) and prostate luminal ductal epithelia (15). These findings suggest that Foxa1/2 proteins are critical for both early embryonic development and late or end stage epithelial differentiation.
Little is known about the role of FOXA1/2 in cancer even though their expression is observed in many human cancers including prostate (16), breast (17, 18), lung and esophagus (19). The involvement of FOXA proteins in pancreatic ductal adenocarcinoma (PDA) has yet been described. Foxa1 and Foxa2 are important transcription factors for endocrine pancreas development and function (7), and are expressed in exocrine pancreas where their function is unclear (20, 21).
Pancreatic ductal adenocarcinoma (PDA) has a 5-year survival rate of less than 4%, a statistic largely attributable to its aggressive invasive and metastasic behavior. We found that both FOXA1 and FOXA2 were expressed in most stages of PDA progression, but were lost in undifferentiated cancer cells. In PDA cell lines, FOXA1/2 expression was suppressed in experimental EMT models and forced inhibition of FOXA1/2 factors was sufficient to induce EMT. As activators of E-cadherin transcription, FOXA1/2 over-expression could overcome several repressive signals of E-cadherin expression, including Snail1 over-expression, TGF-β treatment and E-cadherin promoter hypermethylation. Interestingly, the FOXA2 gene itself was suppressed by DNA methylation and this repression was critical for methylation-mediated silencing of E-cadherin gene expression. Taken together, our study demonstrates that loss of FOXA1/2 is a critical event in EMT during pancreatic cancer progression.
Materials and methods
Human Tissue and Immunohistochemistry
Sections of archival formalin-fixed, paraffin-embedded human PDA and pancreatitis samples were obtained from the Tissue Core, Vanderbilt University Medical Center. Primary antibodies were used for immunohistochemical analysis: anti-FOXA1 (SC-6553, Santa Cruz Biotechnology Inc.); anti-FOXA2 (Seven Hills Bioreagents). Hemotoxylin was used at 1:10 dilution as counterstain to avoid interference with nuclear FOXA1/2 staining. Imagines were captured on an Olympus U-DO3 microscope.
Cell culture and treatments
Human PDA cell lines were purchased from American Type Culture Collection and maintained at 37°C in 5% CO2 in ATCC recommended media, supplemented with 10% fetal bovine serum (FBS) and 0.5μg/ml gentamicin. Phase contrast images were captured on a Zeiss Axiovert 200M microscope. Immunofluorescence images were captured on a Zeiss LSM-510 Meta confocal microscope. In indicated experiments, cells were treated with 5ng/ml TGFβ, 20ng/ml HGF (R&D system), 5′-Aza-deoxycytidine (Sigma-Aldrich), or their corresponding vehicle controls. See supplemental materials for antibodies, primers and other methods.
Plasmids, siRNA,shRNA and generation of stable cells
Expression vectors for mouse Foxa1 and rat FoxA2 were gifts of Dr. Robert Matusik (Vanderbilt University, Nashville, TN). Expression vector of Snail1 (pCDNA3.1/GS-Snail1) was purchased from Invitrogen. FOXA1-specific or FOXA2-specific siRNA (ON-TARGETplus, Dharmacon, Table S2 for sequences) was transfected using TransIT-TKO (Mirus). A scrambled siRNA sequence (BLOCK-iT Alexa Fluor fluorescent, Invitrogen) was employed as control siRNA and an indicator of transfection efficiency. Lentiviral-based shRNA constructs (pLKO.1-puro) targeting either FOXA1 or FOXA2 gene were purchased from Sigma (see Table S2 for sequences). Control pLKO.1 vector was a gift from Dr. Holly Colognato (Dept. of Pharmacology, Stony Brook University). Freshly prepared viruses were used to infect BxPC3 cells, selected and maintained with 0.8 μg/ml puromycin. The coding sequence of rat Foxa2 was sub-cloned into the pMIG retrovirus vector with an IRES-GFP. Viruses containing either Foxa2-IRES-GFP or control IRES-GPF were packaged in amphi-Phoenix cells and GFP+ PANC-1 cells were enriched by FACS sorting.
Promoter constructs and Luciferase reporter assay
A 921 bp human E-cadherin promoter region was sub-cloned into pGL3Basic (Promega). Three FOXA binding elements were mutated according to a previous report (22). Promoter activity was measured by the Dual Luciferase Kit (Promega). See Supplemental Material for details.
Migration assay
Cells were seeded onto Transwell membrane inserts (8 μm, Corning) in serum-free medium. 10% FBS containing media was added to the lower chamber. After 6 hr incubation at 37°C, cells that migrated to the lower surface of the membrane were fixed in 4% paraformaldehyde and stained with 0.5% crystal violet. When siRNA-mediated FOXA1/2 silencing was employed, cells were assessed 48 hours after transfection. For each membrane, 5 random fields were counted at ×10 magnification. The mean was calculated and data are presented as mean±SEM from three independent experiments performed in triplicate.
Statistics
Data are presented as mean±SEM. Student’s t test was used in two-group comparisons. p<0.05 was considered statistically significant. Expression of FOXA1/2 in PDA was analyzed by Chi-squared test.
Results
Expression of FOXA1/2 is lost in undifferentiated PDA cells
The critical function of FOXA1/2 in pancreas development and in several types of cancer led us to examine their expression in human PDA using immunohistochemistry. 25 human PDA and 5 chronic pancreatitis samples contained multiple epithelial pathologies, including metaplasia, pancreatic intraepithelial neoplasia (PanIN), differentiated invasive PDA and undifferentiated invasive PDA. Results are summarized in Table S1, divided by pathology, as found in each sample.
In nearby normal tissue, FOXA1 and FOXA2 were expressed in all islet cell types and at lower levels in acinar and ductal cells, primarily localized to the nucleus (Fig. 1A), a pattern consistent with that found in mouse pancreas (20, 21). Among the 25 PDA specimens, 24 contained invasive cancer and 12 contained various stages of PanIN. FOXA1 and FOXA2 localized primarily to the nucleus and were expressed in invasive adenocarcinoma cells in 79% (19/24) and 75% (18/24) of PDA samples (Fig. 1B), respectively, though strong cytoplasmic staining was observed occasionally for FOXA1/2 (3/24) in poorly differentiated cancers. Both FOXA1 and FOXA2 were detected in 83% (10/12) of PanINs at all stages (Fig. 1A). FOXA1 and FOXA2 were also expressed in all identifiable ductal metaplasia associated with PDA (25/25), and in 5/5 chronic pancreatitis cases examined (Fig. 1A).
Figure 1. FOXA1 and FOXA2 expression in human PDA samples.
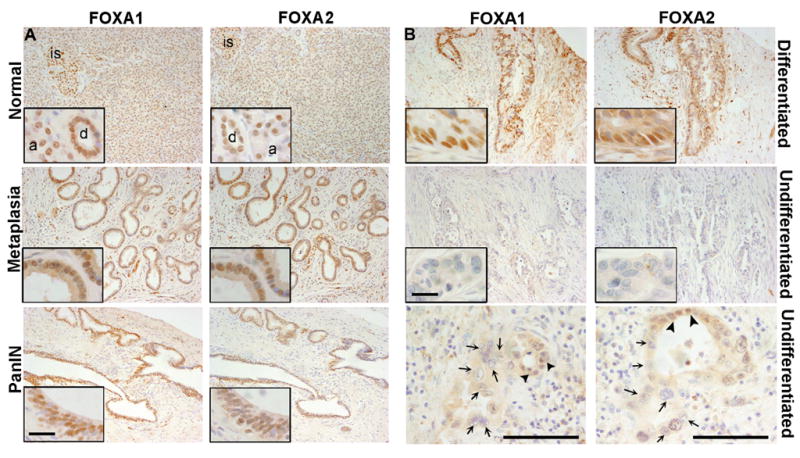
Human PDA samples were assessed for FOXA1 and FOXA2 by immunohistochemistry (IHC) shown in (A) nearby normal epithelia, metaplasia and PanIN lesions, and (B) well-differentiated or undifferentiated PDA. Note strong nuclear staining for FOXA1/2 in all the stages except in undifferentiated PDA. Undifferentiated cancer cells with large atypical nuclei (arrows) were consistently negative for both FOXA1 and FOXA2, in contrast to the positive staining of nearby moderately differentiated cancer cells (arrow heads). Scale bars = 100 μm in the main pictures and 50 μm for the insets. Representative images are shown from 24 human PDA specimens. is: islet; a: acini; d: duct.
Strikingly, very few poorly differentiated cancer cells maintained FOXA1 (26%, 5/19) or FOXA2 (11%, 2/18) expression in contrast to well-differentiated cancer cells in the same sections (Fig. 1B, Table 1). This was especially evident in isolated invasive anaplastic cancer cells, which commonly showed no expression of FOXA1 or FOXA2, in contrast to the nuclear expression of FOXA1 or FOXA2 in nearby moderately well-differentiated carcinomas (Fig. 1B). Thus, FOXA1/2 are expressed in normal pancreas epithelium and maintained in metaplastic epithelium, PanIN lesions and well-differentiated PDA, but are lost in poorly differentiated cancer cells.
Expression of FOXA1/2 factors in PDA cell lines and experimental EMT
Loss of FOXA1/2 expression in poorly differentiated PDA provoked the question whether these factors play a role in the epithelial-to-mesenchymal transition (EMT). To begin to address this question, a cohort of pancreatic cancer cell lines were examined for the expression of FOXA1/2 and EMT related genes including E-cadherin, vimentin, Snail1and Slug by quantitative RT-PCR (Fig. S1) and western blot analysis (Fig. 2A). FOXA1 and FOXA2 expression was found in every cell line with the exception of MiaPaCa-2 cells, an E-cadherin-negative, vimentin positive line (Fig. 2A). Even though E-cadherin transcriptional repressors, such as Snail1, have been characterized as dominant mediators of the undifferentiated status in a variety of cancer cell lines, including MiaPaCa-2 (4, 23), PDA lines characterized as moderately-differentiated, CFPAC-1 and PANC-1 (24), expressed levels of Snail1 or Slug comparable to that in MiaPaCa-2 cells (Fig. 2A & S1).
Figure 2. FOXA1 and FOXA2 are suppressed during EMT in vitro.

(A) Expression of E-cadherin, vimentin, Snail1, FOXA1 and FOXA2 were analyzed by western blot analysis in human PDA cell lines (A). (B–D) PANC-1 or HPAFII cells were treated for 48 hours with 5ng/ml TGFβ or 20ng/ml HGF, respectively, to induce EMT. (B) Immunofluorescence staining confirmed repression of E-cadherin (Red) and activation of vimentin (Green). DAPI is shown in blue. Scale bar = 100 nm. (C,D) Expression of EMT related genes was determined by western blot analysis (C) and qRT-PCR (D). Actin was used as a loading control in western blots. A representative western blot is shown from three independent experiments. GAPDH served as the normalization control for qRT-PCR. Data are presented as mean±SEM from three independent experiments. *: p<0.05, **: p<0.01.
To test if FOXA1/2 factors are involved in EMT, we employed two experimental models of EMT. PANC-1 and HPAFII are well-documented PDA cell lines that undergo EMT upon TGFβ (25, 26) and HGF stimulation (27), respectively. EMT was induced in both cell lines within 48 hours of the respective treatments, illustrated by loss of E-cadherin and gain of vimentin expression (Fig. 2B), though an increase of vimentin protein was undetectable in HPAFII cells. FOXA1 and FOXA2 mRNA and protein were consistently suppressed in both model systems (Fig. 2C&D), suggesting that downregulation of FOXA1/2 factors is part of the EMT program, in coordination with upregulation of E-cadherin transcriptional repressors, such as Snail1 and Slug (Fig. 2C&D).
Loss of FOXA1/2 induces EMT
We next tested whether direct inhibition of FOXA1/2 alone could induce EMT. Two well-differentiated PDA cell lines, HPAFII (Fig. 3) and BxPC3 (Fig. S2A&B), were transiently transfected with synthetic siRNAs against either FOXA1 or FOXA2. As FOXA1/2 were inhibited to 50–60% of control levels, E-cadherin expression was diminished at both mRNA and protein levels (Fig. 3A&B). Knockdown of FOXA1 and FOXA2 together further reduced E-cadherin expression by 80%, indicating that the total quantity of FOXA proteins is critical for optimal E-cadherin expression. Consistent with a previous report indicating that FOXA2 is essential for FOXA1 expression (28), FOXA1 was consistently inhibited when FOXA2 was silenced, possibly explaining, why FOXA2 silencing generally had a stronger effect than FOXA1 silencing (Fig. 3A&B). Localization of E-cadherin was unchanged, remaining at cell-cell junctions (Fig. 3C). Transcripts for ZO-1, another epithelial marker, were also inhibited as FOXA1/2 factors were suppressed (Fig. 3B), supporting a loss of epithelial character. However, neither the mesenchymal markers vimentin and fibronectin nor E-cadherin-repressing Snail transcription factors were significantly upregulated (Fig. 3B), indicating that transient FOXA1/2 inhibition could initiate the loss of epithelial traits, but was not sufficient to induce the mesenchymal phenotype.
Figure 3. Transient inhibition of FOXA1/2 leads to loss of epithelial markers.

HPAFII cells were transiently transfected with siRNA against either FOXA1 or FOXA2, or a combination of both. (A) Expression of E-cadherin and FOXA1/2 was examined 48 hours after siRNA transfection. Quantitation of the representative blot is shown in the bar graph (right). (B) Expression of FOXA1/2, E-cadherin, ZO-1, vimentin, fibronectin, Snail1 and Slug was examined by qRT-PCR 48 hours after siRNA transfection. (C) Representative immunofluorescence staining of E-cadherin 60 hours after transfection is shown. Scale bar = 100 nm. Representative data are shown in (A) and (C) from three independent experiments. Mean±SEM are shown for (B) from at least three independent experiments. *: p<0.05; **: p<0.01
To study the long term effects of FOXA1 or FOXA2 silencing, stable lines were established from BxPC3 cells that expressed short hairpin RNAs (shRNAs) against either FOXA1 or FOXA2. BxPC3 is a well-differentiated PDA cell line with abundant expression of E-cadherin and FOXA1/2 (Fig. 2A). In agreement with transient knockdown experiments, inhibition of either FOXA1 or FOXA2 alone impaired endogenous E-cadherin expression, with simultaneous knockdown of both FOXA1 and FOXA2 being more effective (Fig. 4A). In contrast to transient knockdowns, transcripts for vimentin, fibronectin and Snail1 were significantly elevated (Fig. 4B). Morphologically, cells lacking FOXA1 or FOXA2 showed an increase in cell-spreading, exhibiting a spindle shape and reduced cell-cell contact (Fig. 4C) compared to control cells. Functionally, cell migration was enhanced 4–6 fold when either FOXA1 or FOXA2 was silenced and by ~10-fold when both FOXA1/2 factors were silenced together, as assessed by Transwell migration assay (Fig. 4D). Cell proliferation was also moderately inhibited in FOXA1 or FOXA2 deficient BxPC3 cells (Fig. S2). Collectively, these findings indicate that FOXA1/2 factors are essential in maintaining the epithelial phenotype, with their long term loss being sufficient to induce a secondary mesenchymal transition.
Figure 4. Persistent inhibition of FOXA1/2 induces EMT in PDA cells.

BxPC3 cells with stably integrated shRNA constructs targeting either FOXA1 (shFOXA1) or FOXA2 (shFOXA2) were established by puromycin selection. Two independent shRNA constructs (designated A and B) were used for each gene. Cells with stable transfection of an empty pLKO.1 vector served as control. To analyze combined effects of simultaneous FOXA1 and FOXA2 knockdown, siRNA targeting either FOXA2 or FOXA1 were transiently introduced into stable shFOXA1 or shFOXA2 BxPC3 cells, labeled shA1+siA2 or shA2+siA1, respectively. (A) Expression of FOXA1/2 and E-cadherin was examined by western blot analysis in FOXA1/2 knockdown cells. When siRNAs were used, lysates were collected 48 h after siRNA transfection. Data shown are representative of three independent experiments. (B) Silencing of FOXA1/2 resulted in elevated mesenchymal markers analyzed by qRT-PCR. Mean and SEM are shown from three independent experiments. (C) Phase-constrast microscopy of control, shFOXA1, and shFOXA2 BxPC3 cells. Note cells have increased cell-spreading and a fibroblast-like shape in shFOXA1 and shFOXA2 BxPC3 cells. Scale bar =100 μm in the main pictures and 50 μm in insets. (D) Silencing of FOXA1/2 results in increased cell migration in Transwell migration assays. Cells that migrated from serum-free to serum-containing medium within 6 hours were stained with crystal violet (micrographs) and counted (bar graphs). Quantitation is of 5 random fields from each membrane, and averaged from triplicates of three independent experiments. Error bars represent SEM. *: p<0.05; **: p<0.01.
FOXA2 can overcome suppressive signals to activate the E-cadherin promoter
To further understand the relationship between FOXA1/2 factors and epithelial differentiation in PDA cells, we examined direct regulation of the E-cadherin promoter by these transcription factors. In both MiaPaCa-2 (data not shown) and PANC-1 cells (Fig. 5A), FOXA2 overexpression activated an E-cadherin promoter/luciferase reporter in a dose-dependent (Fig. 5A) and FOXA binding site dependent (Fig. S3A&B) manner. Even though FOXA1 appeared to contribute to endogenous E-cadherin expression (Figs. 3&4), its overexpression did not activate the E-cadherin promoter/reporter in either cell line. Nevertheless, chromatin immunoprecipitation assays showed that both endogenous FOXA1 and FOXA2 proteins interact with DNA regions that include proximal consensus FOXA binding sites (Fig. S3C) within the E-cadherin promoter.
Figure 5. FOXA1/2 proteins are potent activators of E-cadherin in PDA cells.

(A) FOXA2 activates E-cadherin promoter/reporter. Schematic of the E-cadherin promoter fragment from −921 bp to +47 bp relative to the transcription start site is depicted with three FOXA binding sites. PANC-1 cells were co-transfected with an expression vector encoding EGFP, Foxa1 or Foxa2 with a wild type −921 bp E-cadherin promoter/reporter. Fold induction is calculated relative co-transfection with control EGFP-vector. (B) FOXA2 overcomes suppression of E-cadherin promoter mediated by Snail1 or TGFβ. In PANC-1 cells, pGS-Snail1 (2 μg) was cotransfected with an EGFP or FOXA2 expression vector (2 μg). 24 hours after transfection, cells were treated with 5 ng/ml TGFβ, as indicated. Promoter activity was determined by dual luciferase assay 48 hours after transfection. Luciferase assays were performed in triplicate and data were summarized from at least three independent experiments. *: p<0.05; **: p<0.01. ##: p<0.01 in comparison to control. (C) Ectopic expression of FOXA2 enhances endogenous E-cadherin expression. Retrovirus based FOXA2-IRES-GFP or IRES-GFP (control) construct was used to infect PANC-1 cells. GFP+ cells were enriched by FACS. Expression of E-cadherin, vimentin, Snail1 and FOXA2 was examined by western blot (upper panel) and qRT-PCR (lower panel) from GFP+ PANC-1 cells. (D) Ectopic expression of FOXA1/2 induces E-cadherin transcription in MiaPaCa-2 cells. MiaPaCa-2 cells were transiently transfected with an expression vector encoding EGFP, Foxa1 or Foxa2. E-cadherin mRNA was examined by semi-quantitative RT-PCR (upper panel) and by qRT-PCR along with Snail1 and Slug (lower panel) 48 hours after transfection. Representative results from three independent experiments are shown for (C,D). Mean±SEM are shown for qRT-PCR from at least three independent experiments. *: p<0.05; **: p<0.01.
Given that FOXA2 can activate the E-cadherin promoter/reporter in Snail1-expressing PANC-1 and MiaPaCa2 cells (Fig. 5A), we asked whether FOXA2 is a dominant activating factor, capable of overcoming E-cadherin suppressing signals. Using the TGFβ1-responsive PANC-1 cells, we showed that both ectopic Snail1 overexpression and TGFβ1 treatment were able to repress wild-type E-cadherin promoter/reporter activity compared to control (Fig. 5B), as expected. However, neither Snail1 nor TGFβ1 prevented FOXA2 activation of the E-cadherin promoter (Fig. 5B). In fact, FOXA2 activated the promoter to a level comparable to when FOXA2 was expressed alone. These results indicate that downregulation of FOXA2 is important in order for Snail1 or TGFβ to effectively repress the E-cadherin promoter.
FOXA1/2 are potent activators of endogenous E-cadherin
To test whether FOXA2 can activate the endogenous E-cadherin gene in the presence of other repressive mechanisms, a retrovirus based FOXA2-IRES-EGFP construct or an IRES-EGFP construct (as control) was stably expressed in PANC-1 cells. GFP+ cells were identified and enriched by FACS. Compared to control GFP+ cells, E-cadherin expression was strongly elevated in FOXA2-GFP+ cells, while vimentin expression was suppressed (Fig. 5C), consistent with FOXA2 promoting epithelial character, despite Snail1 being highly expressed in parental PANC-1 cells (Fig. 2B) and remaining unchanged in FOXA2 overxpressing cells (Fig. 5C).
We then tested whether FOXA1/2 overexpression can re-activate endogenous E-cadherin in E-cadherin negative MiaPaCa-2 cells. MiaPaCa-2 cells express Snail1 (4) and have an E-cadherin promoter reported to be silenced by hypermethylation (29, 30), and an undetectable FOXA1/2 expression (Fig. 2). Given these multiple modes of E-cadherin repression, it was surprising that transient expression of either FOXA1 or FOXA2 elevated E-cadherin transcript levels, which were undetectable in parental and vector control cells, to an easily detectable level (Fig. 5D), a quantitative increase of over 200-fold. Neither Snail nor Slug expression was repressed in these experiments (Fig. 5D). While both promoter/reporter experiments (Fig. 5B) and ectopic FOXA2 overexpression in PANC-1 cells (Fig. 5C) demonstrated that FOXA1/2 could counteract Snail1-mediated repression, it was unexpected that it could overcome promoter hypermethylation. It should be noted that E-cadherin protein remained undetectable by western blot with transient FOXA1 or FOXA2 expression (data not shown). It is possible that even with the dramatic elevation induced by FOXA1/2 factors, E-cadherin transcripts remains too low (~1% of BxPC3 levels, when adjusted for transfection efficiency) to lead to high levels of E-cadherin protein.
FOXA2 induction is responsible for demethylation-mediated E-cadherin re-activation
Hypermethylation of the E-cadherin promoter is thought to represent a silenced E-cadherin locus because it introduces an insurmountable structural impediment to transcription. Since overexpression of FOXA1/2 proteins in MiaPaCa-2 cells, which contains mostly methylated E-cadherin promoter (30) (Fig. S4A), reactivated E-cadherin expression, demonstrating that the promoter in these cells is amenable to transactivation. With this surprising result, we questioned whether promoter hypermethylation was demonstrably involved in E-cadherin silencing in MiaPaCa2 cells. To test this, we reversed DNA methylation by treating cells with 5′-Aza-deoxycytidine (5′-Aza-dC) (31) (Fig. S4A), and quantitated E-cadherin expression by qRT-PCR. Indeed, E-cadherin transcripts were elevated by >20-fold after treatment (Fig. 6A), consistent with silencing by hypermethylation. However, we found that FOXA2 transcripts were simultaneously elevated by ~10-fold after treatment (Fig. 6A), indicating that the FOXA2 gene is also suppressed by methylation. A corresponding enhancement of E-cadherin and FOXA2 protein expression was found by FACS analysis (Fig. 6B). In contrast, no change was observed for FOXA1 (Fig. 6A, S4B). Taken together, these results suggested the possibility that restoration of E-cadherin expression after demethylation may be indirect, through reactivation of FOXA2 expression.
Figure 6. FOXA2 is required for demethylation-mediated E-cadherin re-activation.

(A, B) 5′-Aza-dC treatment induces E-cadherin and FOXA2 expression. MiaPaCa-2 cells were treated with 5′-Aza-dC for 5 days at indicated doses. Expression of E-cadherin, FOXA1 and FOXA2 were examined by qRT-PCR (A) and flow cytometry (B). (C, D) 5′-Aza-dC treatment induced E-cadherin expression is dependent on FOXA2 elevation. MiaPaCa-2 cells were treated with 1 μM 5′-Aza-dC for 5 days. On the 3rd day, two different synthetic siRNAs (#7 and #8) against FOXA2 were independently transfected into 5′-Aza-dC treated or untreated MiaPaCa-2 cells. Expression of E-cadherin and FOXA2 were examined by qRT-PCR (C) and flow cytometric analysis (D). For qRT-PCR analysis, mean±SEM from three independent experiments are shown. For FACS analysis, representative data from at least three independent experiments are shown. Only four data lines are shown for simplicity (representative complete data set is presented in Fig S4). Bar graph summarizes at least three independent experiments. *: p<0.05; **: p<0.01.
To test this possibility, synthetic siRNAs targeting FOXA2 were transfected into MiaPaCa-2 cells with or without 5′-Aza-dC treatment. When FOXA2 expression was inhibited by either of the two FOXA2 siRNAs, re-expression of E-cadherin mRNA was also inhibited by ~80%, compared to control siRNA (Fig. 6C). This inhibition was confirmed at the protein level by FACS analysis (Fig. 6D). Thus, we conclude that, in MiaPaCa-2 cells, an indirect methylation-dependent regulatory mechanism controls E-cadherin expression through methylation-mediated suppression of FOXA2.
Discussion
Metastasis causes >90% of cancer deaths. The epithelial-to-mesenchymal transition is considered a prerequisite to metastasis for most carcinomas, allowing cancer cells to disassociate from the primary tumor and enhancing cell motility. Early observations showing that gain-of-function signals, such as oncogene activation and growth factor responses, are sufficient to induce EMT has led to the extensive study and identification of several EMT-promoting pathways, while relatively little attention has been paid to EMT-suppressing pathways. In this study, we demonstrate for the first time that inhibition of FOXA1/2 transcription factors is an integral part of the EMT program. Suppression of endogenous FOXA1/2 is sufficient to induce EMT and sustained FOXA1/2 expression can inhibit EMT induced by a variety of pathways.
Based on our findings from in vivo human PDA specimens and in vitro studies in PDA cell lines, we propose a model where FOXA1/2 factors are constitutively expressed in normal pancreatic epithelia, PanINs and well-differentiated PDA where they maintain optimal E-cadherin levels, thus suppressing EMT. As the cancer progresses, FOXA1/2 factors are suppressed by EMT inducing signals (eg. TGFβ, DNA methylation), leading to downregulation of E-cadherin and possibly other epithelial markers. This model is consistent with previous observations of FOXA1/2 in breast cancer (17, 18, 22) and their functions during development (7). Concurrent with FOXA1/2 repression in PDA cells, E-cadherin repressors, such as Snail, are activated, further contributing to E-cadherin silencing and EMT induction. This model strongly suggests that the balance between FOXA factors and Snail repressors is critical. This idea is bolstered by our promoter/reporter studies in PANC-1 cells (Fig. 5B), which express both endogenous FOXA1/2 and Snail1 proteins, yet are still susceptible to both Snail1 repression and FOXA2 activation of the E-cadherin promoter.
Similarly, observed antagonism between FOX factors themselves may require their relative expression to be appropriately balanced as well. For instance, FOXC2 was shown to induce EMT and suppress E-cadherin expression (32), which could be achieved by antagonizing FOXA transactivation. Also, FOXA1 interferes with FOXA2 mediated transactivation as a result of its inferior transactivation activity (28). While no activation of the E-cadherin promoter/reporter was observed with FOXA1 overexpression, co-expression of FOXA1 with FOXA2 dampens the degree of transactivation observed with FOXA2 overexpression alone by about 30–40% (data not shown), supporting the possibility of antagonism. However, since knockdown of endogenous FOXA1 in FOXA2 expressing lines consistently reduces E-cadherin expression (Figs 3&4), we conclude that FOXA1 plays an overall positive role in endogenous E-cadherin gene expression, possibly by interacting with distal regulatory elements.
Another important mechanism implicated in E-cadherin regulation is DNA methylation-mediated gene silencing, conferred by establishment of repressive chromatin complexes composed of methyl CpG binding proteins and histone deacetylases that mediate chromatin condensation (33). Nevertheless, methylated promoters can still be activated by several transcriptional activators (34, 35), that compete with repressive chromatin complexes. Moreover, FOXA1 has been demonstrated as a “pioneer factor” of chromatin remodeling that can decondense compacted chromatin through its C-terminal domain to facilitate further transcription (36, 37). We hypothesize that abundantly expressed FOXA proteins can activate a methylated E-cadherin promoter (Fig. 5D) due to their dual functions as potent trans-activators and chromatin remodeling factors.
Furthermore, we found that the FOXA2 gene, but not the FOXA1 gene, was regulated by methylation in an undifferentiated PDA cell line. Whether this is by direct methylation of the FOXA2 promoter, by methylation of upstream regulators or both, is unknown. Two large CpG islands are predicted with heavily clustered CpG sites within a 2100 bp 5′ regulatory region of the FOXA2 gene, using CpG island prediction softwares (cpgislands and cpgplot), making FOXA2 a candidate for silencing by direct hypermethylation. Given recent reports that methylation of E-cadherin promoter in less than 2% of PDA patient samples (30, 38), we propose that loss of E-cadherin expression may be strongly influenced by methylation-mediated silencing of the FOXA2 gene, independent of direct methylation of the E-cadherin gene itself.
Besides regulation of FOXA1/2 by EMT-inducing paracrine signals or DNA methylation, FOXA1/2 proteins frequently localized to the cytoplasm of undifferentiated cancer cells, suggesting an alternative mechanism of inhibiting their transcription activity. In an insulinoma cell line, cytoplasmic translocation of FOXA proteins was induced by AKT phosphorylation of the Fkhd domain (39), in a manner analogous to FOXO (40). However, such translocation of FOXA1 or FOXA2 was not found in vivo in response to PI3K/AKT activation (7). In several PDA cell lines, we observed FOXA1 and FOXA2 in both the nucleus and cytoplasm, but neither activation of AKT by growth factors nor pharmacological inhibition of PI3K or AKT altered this distribution (data not shown). Nevertheless, we speculate that signaling pathways controlling nuclear-to-cytoplasmic shuttling of FOXA1/2 are likely to be important regulators of EMT in PDA.
In summary, we have shown a consistent inhibition of FOXA1 and FOXA2 during EMT both in vivo and in vitro by diverse signals. Suppression of endogenous FOXA1/2 alone is sufficient to induce EMT in PDA cell lines and is required for E-cadherin silencing previously attributed directly to promoter hypermethylation. Together these data lead us to conclude that loss of FOXA1/2 is an essential step in the EMT program during pancreatic cancer progression.
Supplementary Material
Acknowledgments
We thank Drs. Robert Matusik (Vanderbilt University) and Holly Colognato (Stony Brook University) for plasmid gifts; Dr. Klaus Kaestner (University of Pennsylvania) for helpful advice; Dr. Jian Cao, Antoine Dufour and Dr. Xing Chang (Yale University) for technical advice and Dun Li for technical assistance.
The project was supported by National Cancer Institute Grant Number R01CA100126 and the Knapp Chair for Pancreatic Cancer Research to HCC and by Grant Number P50CA095103 for the Vanderbilt University Medical Center GI Spore Tissue Core.
Abbreviations
- BrdU
Bromo-Deoxyuridine
- EMT
Epithelial-to-Mesenchymal Transition
- E-cadherin
Epithelial-cadherin
- FOXA transcription factors
Forkhead box transcription factors, subclass A
- HGF
Hepatocyte Growth Factor
- PanIN
Pancreatic Intraepithelial Neoplasia
- PDA
Pancreatic Ductal Adenocarcinoma
- 5′-Aza-dC
5′-Aza-deoxycytidine
- TGFβ
transforming Growth Factor β
Footnotes
Competing Interest: No conflicts of interest exist
References
- 1.Bailey JM, Singh PK, Hollingsworth MA. Cancer metastasis facilitated by developmental pathways: Sonic hedgehog, Notch, and bone morphogenic proteins. J Cell Biochem. 2007;102:829–39. doi: 10.1002/jcb.21509. [DOI] [PubMed] [Google Scholar]
- 2.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Cano A, Perez-Moreno MA, Rodrigo I, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 5.Nieto MA, Sargent MG, Wilkinson DG, Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264:835–9. doi: 10.1126/science.7513443. [DOI] [PubMed] [Google Scholar]
- 6.Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–78. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 7.Friedman JR, Kaestner KH. The Foxa family of transcription factors in development and metabolism. Cell Mol Life Sci. 2006;63:2317–28. doi: 10.1007/s00018-006-6095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shih DQ, Navas MA, Kuwajima S, Duncan SA, Stoffel M. Impaired glucose homeostasis and neonatal mortality in hepatocyte nuclear factor 3α-deficient mice. Proc Natl Acad Sci U S A. 1999;96:10152–7. doi: 10.1073/pnas.96.18.10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaestner KH, Katz J, Liu Y, Drucker DJ, Schutz G. Inactivation of the winged helix transcription factor HNF3α affects glucose homeostasis and islet glucagon gene expression in vivo. Genes Dev. 1999;13:495–504. doi: 10.1101/gad.13.4.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinstein DC, Ruiz i Altaba A, Chen WS, et al. The winged-helix transcription factor HNF-3β is required for notochord development in the mouse embryo. Cell. 1994;78:575–88. doi: 10.1016/0092-8674(94)90523-1. [DOI] [PubMed] [Google Scholar]
- 11.Ang SL, Rossant J. HNF-3β is essential for node and notochord formation in mouse development. Cell. 1994;78:561–74. doi: 10.1016/0092-8674(94)90522-3. [DOI] [PubMed] [Google Scholar]
- 12.Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic α-cells. Developmental Biology. 2005;278:484–95. doi: 10.1016/j.ydbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 13.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–7. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 14.Wan H, Kaestner KH, Ang SL, et al. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development. 2004;131:953–64. doi: 10.1242/dev.00966. [DOI] [PubMed] [Google Scholar]
- 15.Gao N, Ishii K, Mirosevich J, et al. Forkhead box A1 regulates prostate ductal morphogenesis and promotes epithelial cell maturation. Development. 2005;132:3431–43. doi: 10.1242/dev.01917. [DOI] [PubMed] [Google Scholar]
- 16.Janni M, Nan G, Aparna G, Scott BS, Richard J, Robert JM. Expression and role of Foxa proteins in prostate cancer. The Prostate. 2006;66:1013–28. doi: 10.1002/pros.20299. [DOI] [PubMed] [Google Scholar]
- 17.Thorat MA, Marchio C, Morimiya A, et al. Forkhead box A1 expression in breast cancer is associated with luminal subtype and good prognosis. J Clin Pathol. 2008;61:327–32. doi: 10.1136/jcp.2007.052431. [DOI] [PubMed] [Google Scholar]
- 18.Wolf I, Bose S, Williamson EA, Miller CW, Karlan BY, Koeffler HP. FOXA1: Growth inhibitor and a favorable prognostic factor in human breast cancer. Int J Cancer. 2007;120:1013–22. doi: 10.1002/ijc.22389. [DOI] [PubMed] [Google Scholar]
- 19.Lin L, Miller CT, Contreras JI, et al. The Hepatocyte Nuclear Factor 3 α Gene, HNF3 α (FOXA1), on Chromosome Band 14q13 Is Amplified and Overexpressed in Esophageal and Lung Adenocarcinomas. Cancer Res. 2002;62:5273–9. [PubMed] [Google Scholar]
- 20.Wu KL, Gannon M, Peshavaria M, et al. Hepatocyte nuclear factor 3 β is involved in pancreatic beta-cell- specific transcription of the pdx-1 gene. Mol Cell Biol. 1997;17:6002–13. doi: 10.1128/mcb.17.10.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Besnard V, Wert SE, Hull WM, Whitsett JA. Immunohistochemical localization of Foxa1 and Foxa2 in mouse embryos and adult tissues. Gene Expression Patterns. 2004;5:193–208. doi: 10.1016/j.modgep.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24:8277–90. doi: 10.1038/sj.onc.1208991. [DOI] [PubMed] [Google Scholar]
- 23.Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 24.Sipos B, Moser S, Kalthoff H, Torok V, Lohr M, Kloppel G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch. 2003;442:444–52. doi: 10.1007/s00428-003-0784-4. [DOI] [PubMed] [Google Scholar]
- 25.Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras Signaling in the Induction of Snail by Transforming Growth Factor-β. J Biol Chem. 2009;284:245–53. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- 26.Ellenrieder V, Hendler SF, Boeck W, et al. Transforming Growth Factor β1 Treatment Leads to an Epithelial-Mesenchymal Transdifferentiation of Pancreatic Cancer Cells Requiring Extracellular Signal-regulated Kinase 2 Activation. Cancer Res. 2001;61:4222–8. [PubMed] [Google Scholar]
- 27.Hirota M, Egami H, Corra S, et al. Production of scatter factor-like activity by a nitrosamine-induced pancreatic cancer cell line. Carcinogenesis. 1993;14:259–64. doi: 10.1093/carcin/14.2.259. [DOI] [PubMed] [Google Scholar]
- 28.Duncan SA, Navas MA, Dufort D, Rossant J, Stoffel M. Regulation of a Transcription Factor Network Required for Differentiation and Metabolism. Science. 1998;281:692–5. doi: 10.1126/science.281.5377.692. [DOI] [PubMed] [Google Scholar]
- 29.Ueki T, Toyota M, Sohn T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–9. [PubMed] [Google Scholar]
- 30.Winter JM, Ting AH, Vilardell F, et al. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin Cancer Res. 2008;14:412–8. doi: 10.1158/1078-0432.CCR-07-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombaerts M, van Wezel T, Philippo K, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer. 2006;94:661–71. doi: 10.1038/sj.bjc.6602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mani SA, Yang J, Brooks M, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–74. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–26. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 34.Niesen MI, Osborne AR, Yang H, et al. Activation of a methylated promoter mediated by a sequence-specific DNA-binding protein, RFX. J Biol Chem. 2005;280:38914–22. doi: 10.1074/jbc.M504633200. [DOI] [PubMed] [Google Scholar]
- 35.Curradi M, Izzo A, Badaracco G, Landsberger N. Molecular Mechanisms of Gene Silencing Mediated by DNA Methylation. Mol Cell Biol. 2002;22:3157–73. doi: 10.1128/MCB.22.9.3157-3173.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–89. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 37.Carroll JS, Liu XS, Brodsky AS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Peng DF, Kanai Y, Sawada M, et al. DNA methylation of multiple tumor-related genes in association with overexpression of DNA methyltransferase 1 (DNMT1) during multistage carcinogenesis of the pancreas. Carcinogenesis. 2006;27:1160–8. doi: 10.1093/carcin/bgi361. [DOI] [PubMed] [Google Scholar]
- 39.Wolfrum C, Besser D, Luca E, Stoffel M. Insulin regulates the activity of forkhead transcription factor Hnf-3β/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc Natl Acad Sci U S A. 2003;100:11624–9. doi: 10.1073/pnas.1931483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunet A, Bonni A, Zigmond MJ, et al. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.