

Abstract
Quiescin-sulfhydryl oxidase (QSOX) flavoenzymes catalyze the direct, facile, insertion of disulfide bonds into reduced unfolded proteins with the reduction of oxygen to hydrogen peroxide. To date, only QSOXs from vertebrates have been characterized enzymatically. These metazoan sulfhydryl oxidases have 4 recognizable domains: a redox-active thioredoxin (Trx) domain containing the first of three CxxC motifs (CI-CII), a second Trx domain with no obvious redox-active disulfide, a helix-rich domain, and then an Erv/ALR domain. This last domain contains the FAD moiety, a proximal CIII-CIV disulfide and a third CxxC of unknown function (CV-CVI). Plant and protist QSOXs lack the second Trx domain, but otherwise appear to contain the same complement of redox centers. This work presents the first characterization of a single-Trx QSOX. Trypanosoma brucei QSOX was expressed in Escherichia coli using a synthetic gene and found to be a stable, monomeric, FAD-containing protein. Although evidently lacking an entire domain, TbQSOX shows catalytic activity and substrate specificity similar to the vertebrate QSOXs examined previously. Unfolded reduced proteins are more than 200-fold more effective substrates on a per-thiol basis than glutathione, and some 10-fold better than the parasite bis-glutathione analog, trypanothione. These data are consistent with a role for the protist QSOX in oxidative protein folding. Site-directed mutagenesis of each of the 6 cysteine residues (to serines) show that the CxxC motif in the single Trx domain is crucial for efficient catalysis of the oxidation of both reduced RNase and the model substrate dithiothreitol. As expected, the proximal disulfide CIII-CIV, which interacts with the flavin, is catalytically crucial. However, as observed with human QSOX1, the third CxxC motif shows no obvious catalytic role during the in vitro oxidation of reduced RNase or dithiothreitol. Pre-steady state kinetics demonstrates that turnover in TbQSOX is limited by an internal redox step leading to 2-electron reduction of the FAD cofactor. In sum, the single-Trx domain QSOX studied here shows a striking similarity in enzymatic behavior to its double-Trx metazoan counterparts.
The Quiescin sulfhydryl oxidase (QSOX) family of enzymes catalyze the net generation of disulfide bonds with reduction of oxygen to hydrogen peroxide (1–4):
Reduced unfolded proteins are excellent substrates of these multi-domain flavin-dependent oxidases suggesting that they contribute to the net generation of disulfide bonds during oxidative protein folding. QSOXs are found in the genomes of almost all eukaryotes, with the notable exception of the fungi (2–4). However, all of our current insights into the substrate specificity and the mechanism of these oxidases have come from study of metazoan QSOXs: first from murine seminal vesicles (5, 6), then from egg white (7, 8), bovine milk (9) and, most recently, from a human recombinant enzyme expressed in Escherichia coli (10).
The domain organization of a metazoan QSOX is shown in Figure 1A. These enzymes have two N-terminal thioredoxin domains (Trx1 and Trx2) joined to a C-terminal Erv/ALR domain by a helix-rich region (HRR) (1, 3, 4, 10, 11). A transmembrane helix is found at the C-terminus of most QSOX sequences, although alternately-spliced transcripts lacking this feature have been identified (1, 4). Collectively, the Trx1 and Trx2 domains are similar to a and b domains of the protein disulfide isomerase (PDI) (12–15): with only the first containing a redox-active CxxC motif. Following the second thioredoxin domain in metazoan QSOXs is the HRR domain: an apparently unique feature of these oxidases. The Erv/ALR domain was first described as a yeast growth factor, essential for respiration and viability (Erv1) (16), and later found to be a small stand-alone flavin-linked sulfhydryl oxidase (17). Crystal structures of yeast Erv2 (18), rat augmenter of liver regeneration (ALR) (19) and other Erv/ALR family members (18, 20–22) show that the flavin ring is bound at the mouth of a compact 4-helix bundle. Additionally, the Erv/ALR domain houses a second redox center: a proximal CxxC disulfide that relays reducing equivalents from dithiol substrates, or from distal dithiol motifs, to the isoalloxazine ring. While the roles of thioredoxin and proximal disulfides in QSOX catalysis are established (10), there is a third CxxC motif of unknown function that is also conserved in all QSOX sequences available to date (1, 10). It lies at the C-terminus of the Erv/ALR domain and is not catalytically essential for the in vitro oxidation of DTT or reduced RNase (rRNase) by the recombinant human QSOX1 (10). It might play a role in the regulation of catalytic activity (10).
FIGURE 1.

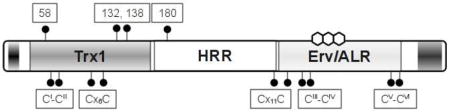
Domain organization of human and trypanosomal QSOX proteins and amino acid sequence of TbQSOX. Panels A and B show the domain organization of human and trypanosomal QSOX proteins respectively. CxxC motifs are shown as red bars; labeled as previously (10) and using the modified nomenclature implemented in this work. The flavin ring system of FAD is shown above the Erv/ALR label. Panel C represents the amino acid sequence (NCBI Acc. No. XP_845306), and predicted secondary structure of TbQSOX. The predicted signal sequence is underlined. Trx, HRR and Erv/ALR domains are highlighted in blue, orange and yellow respectively. A predicted transmembrane region is highlighted in pink. CxxC motifs are highlighted in red and underlined in boldface. The remaining cysteine residues are shown against a grey background.
Plant and protist QSOXs lack all, or parts, of the second thioredoxin domain found in their metazoan counterparts (Figure 1B) (1, 3, 4). Since all our current insights into the QSOX family have come from oxidases with two thioredoxin domains, we wanted to investigate a representative example of a structurally simpler QSOX. We chose the enzyme from Trypanosoma brucei, a protozoan pathogen (family Trypanosomatidae, order Kinetoplastida) that causes African sleeping sickness. In its predominant blood-stream phase, the parasite counters the host immune response with the help of a monolayer of variable surface glycoproteins (VSG) anchored to the outer leaflet of the plasma membrane by a glycosylphosphatidylinositol moiety (23). Although this coating is highly immunogenic, at each division cycle a proportion of the daughter cells change their VSG coat and thus evade detection by the human immune system. These highly variable, VSGs do however contain multiple conserved disulfide bonds (24–28) and hence the ability of the parasite to generate these structural disulfides would be expected to be important for pathogenicity.
Trypanosoma are known to have an unusual redox biology, in which a bis-glutathione derivative, trypanothione, plays a major role (29, 30). However, we know very little about the ways these parasites catalyze the net generation of disulfide bridges during oxidative protein folding. Three flavin-linked sulfhydryl oxidases can be identified from the T. brucei genome. One is homologous to yeast Ero1p and its metazoan counterparts; these flavoproteins are resident in the endoplasmic reticulum and are thought to play significant roles in disulfide bond generation (31–35). A second, TbALR, is the counterpart of yeast Erv1p and human ALR and hence would be expected to support oxidative protein folding in the mitochondrial intermembrane space (36–39). The third is a QSOX sequence of the type discussed above with three, not four, recognizable domains.
As part of our effort to understand catalytic strategies in the QSOX family of proteins, we report the first enzymatic characterization of a protist QSOX. The protein was expressed using a synthetic gene, and its behavior was analyzed by a range of static methods, together with steady-state and rapid-reaction studies. These approaches reveal striking commonalities of behavior between these evolutionarily distant and structurally diverse representatives of the QSOX family.
EXPERIMENTAL PROCEDURES
Materials
Chemical reagents were obtained as described previously (40). Primers for sub-cloning of all constructs and for mutagenesis were from Integrated DNA Technologies. Protein and DNA molecular weight standards, and all the enzymes used for sub-cloning, were from Promega. All bacterial culture media components were from Fisher Scientific.
TbQSOX Constructs
A synthetic gene encoding TbQSOX protein (NCBI Accession Number: XP_845306) codon-optimized for expression in E. coli was designed using Gene Designer software (DNA 2.0 www.dna20.com) and was obtained from DNA 2.0 in a pJ201 vector (see Supporting Information, Figure S1). All protein expression constructs were then obtained by sub-cloning the desired region into a pET-28a(+) vector (Novagen) using appropriate PCR primers (Supporting Information, Table S1). DNA sequences of all the inserts were confirmed by sequencing to ensure in-frame ligation and the absence of unwanted mutations. Specific cysteine mutations were introduced by using QuikChange site-directed mutagenesis kit (Stratagene) with the primers shown in Table S1. All mutant constructs were also sequenced to verify mutations.
Expression of TbQSOX Constructs and Mutants
Full length TbQSOX constructs started at either amino acid residues S20 or G32 and ended at G485. Initial attempts at expression and purification from E. coli BL21 Star (DE3) (Invitrogen) cells induced at either 15 °C or 37 °C were unsuccessful. However, a full length construct starting at residue S20 was expressed in a soluble form in E. coli Origami2 (DE3) (Novagen) cells. Thereafter, all TbQSOX constructs were expressed in this strain. Starter cultures were grown overnight at 37 °C in LB broth containing 15 μg/mL kanamycin and 12.5 μg/mL tetracycline. A 5 mL volume of starter culture was used to inoculate 500 mL of Terrific Broth containing the same levels of antibiotics and the media were incubated at 37 °C until an absorbance of 0.8–1.0 at 600 nm was reached. Protein expression was induced with 0.5 mM IPTG at 15 °C for 24 h. Cells were then harvested by centrifugation at 5000 g at 4 °C and stored at −20 °C.
Purification of TbQSOX Constructs and Mutants
Cell pellets from 2 L of culture were resuspended in 20 mL of 50 mM potassium phosphate buffer, pH 7.5 containing 300 mM NaCl, 100 μM FAD and two tablets of protease inhibitor cocktail for His-tagged proteins (Complete, Mini, EDTA free; Roche). Cells were lysed by two passages through a French Press (at 10,000 psi) followed by brief sonication to shear DNA. The resulting lysate was centrifuged at 17,000 g for 30 min at 4 °C. The supernatant was carefully removed and added to Pro Bond Ni-NTA resin (Invitrogen) previously equilibrated in 50 mM phosphate buffer, pH 7.5 containing 300 mM NaCl. The suspension was rocked at 4 °C for 3 h and poured into an empty column. Flow-through was collected for further analysis and the column was washed with four column volumes of 50 mM phosphate buffer, pH 7.5 containing 300 mM NaCl followed by four column volumes of 50 mM phosphate buffer, pH 6.0. Protein bound to the resin was eluted using a gradient of imidazole in 50 mM phosphate buffer, pH 6.0 and collected in 1.5 mL aliquots. Fractions containing TbQSOX enzyme were pooled and further purified using hydrophobic interaction chromatography. Here, ammonium sulfate was added to 40 % saturation at 4 °C and the solution clarified by centrifugation at 17,000 g for 30 min. The supernatant was then loaded onto a butyl-Sepharose column (6 × 2 cm) previously equilibrated with 50 mM phosphate buffer, pH 7.5 containing 40 % saturated ammonium sulfate and 1 mM EDTA. The column was washed with three column volumes of 50 mM phosphate buffer, pH 7.5, containing 40 % saturated ammonium sulfate and 1 mM EDTA. TbQSOX was eluted with a decreasing gradient of ammonium sulfate. Fractions (5 mL) were pooled and concentrated using Amicon Ultra centrifugal filter devices (Millipore).
UV/Vis Spectroscopy, Determination of Extinction Coefficients and Thiol Titers
All absorption spectra were collected on an Agilent 8453 UV/Vis spectrophotometer and the data were analyzed using the ChemStation software (Agilent Technologies). Extinction coefficients for the bound flavin of wild type TbQSOX and the shorter HRR-Erv construct (12,250 and 12,300 M−1 cm−1, respectively) were determined as described earlier (10). Thiol titer of full length TbQSOX was determined by following the reaction of the native and SDS-denatured enzyme with 0.5 mM DTNB (10).
rRNase Assays
RNase was reduced for 2 h at 37 °C in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA, a 10-fold excess of DTT over total thiols, and 6 M GdnHCl. The mixture (0.6 mL) was applied to a PD10 column (Amersham Biosciences) equilibrated with 10 mM sodium acetate, pH 4.0 giving a baseline separation between rRNase and excess DTT. Thiol titer confirmed complete reduction, and the resulting protein was stored under anaerobic conditions at 4 °C (10). rRNase assays were performed as described earlier (10). Briefly, 50 nM TbQSOX was added to a 300 μL reaction containing varying amounts of rRNase in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA at 25 °C. Aliquots (20 μL) of the reaction mixture were transferred into 0.5 mM DTNB in phosphate buffer, pH 7.5, and thiol titer was monitored at 412 nm. Control reactions were run under identical conditions in the absence of TbQSOX.
Reduced Riboflavin Binding Protein Assays
Chicken riboflavin binding protein (a gift from Dr. Harold B. White III, University of Delaware) was reduced for 3 h at 37 °C in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA, a 10-fold excess of DTT over total thiols, and 6 M GdnHCl. Reduced protein was purified using a PD10 column equilibrated at 4 °C with 10 mM sodium acetate buffer at pH 5.0 containing 1 mM EDTA and 1 M GdnHCl. The thiol content of reduced RfBP was measured using DTNB to confirm complete reduction and rRfBP was stored under anaerobic conditions at 4 °C. rRfBP assays were performed essentially as above, by adding 125 nM TbQSOX to a 300 μL reaction containing varying amounts of reduced protein in phosphate buffer containing 1 mM EDTA at pH 7.5 at 25 °C. Thiol titers were measured using DTNB to allow calculation of rate of thiol oxidation and of enzymatic turnover numbers.
Trypanothione Assays
A solution of 14 mg/mL oxidized trypanothione (Bachem) in 50 mM phosphate buffer at pH 7.5 containing 1 mM EDTA was incubated with an equal amount of immobilized TCEP disulfide reducing gel (Pierce) for 90 min. Reduced trypanothione was separated from the matrix by centrifugation and its thiol titer was measured by treatment with DTNB. Trypanothione assays were performed at 25 °C in a 150 μL reaction volume containing 2.5, 5 or 10 mM reduced trypanothione in phosphate buffer, pH 7.5, containing 1 mM EDTA. Reactions were started by the addition of 1 μM TbQSOX and followed by thiol titers as above. Control incubations lacked enzyme.
Oxygen Electrode Assays
Catalytic activities of all TbQSOX constructs were determined using DTT and GSH by using a Clark-type oxygen electrode as described previously (7). DTT and GSH were prepared in 50 mM phosphate buffer, containing 1 mM EDTA. Solutions were readjusted to pH 7.5 as necessary and standardized using DTNB before use.
pH dependence of turnover with DTT and rRNase
The following 50 mM buffers containing 1 mM EDTA were used: sodium acetate, potassium phosphate, Tris-HCl and CAPS, with either 5 mM DTT or 30 μM rRNase. Assays were initiated by the addition of 50 nM TbQSOX, using the oxygen electrode for DTT and discontinuous thiol titer measurements for rRNase.
Dithionite Titrations
Dithionite titrations were conducted under anaerobic conditions as before (7).
Stopped-flow Spectrophotometry
Data were collected using an SF-61 DX2 double mixing stopped-flow system (Hi-Tech Instruments) at 25 °C in either diode-array or monochromator modes and analyzed by their KinetAsyst 3 software. For experiments under aerobic conditions, TbQSOX and DTT were prepared in air-saturated 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA at 25 °C. rRNase was prepared as described earlier. For experiments under anaerobic conditions, wild type or mutant TbQSOX proteins were added to tonometers containing phosphate buffer, pH 7.5, with 1 mM EDTA, 5 mM glucose, 5 nM glucose oxidase and 1 nM catalase. Separate tonometers contained DTT in the same buffer. The tonometers were made thoroughly anaerobic by rocking for 20 min using alternate cycles of vacuum and flushing with nitrogen. For experiments to determine reactivity towards oxygen, the CIS TbQSOX mutant dissolved in phosphate buffer was made anaerobic as before, except that 1 nM glucose oxidase was used. The mutant enzyme was then reduced by the addition of 2-electron equivalents from a standardized solution of dithionite.
Sequence Analyses
The signalP server was used to predict signal sequence in TbQSOX (41). The transmembrane helix and the topology of the protein were predicted by the TMHMM server (41). Secondary structure predictions for all proteins were obtained by submitting sequences to PSIPRED (42). Protein sequence alignments were obtained using ClustalW (43).
RESULTS AND DISCUSSION
TbQSOX Gene and Protein
The single T. brucei QSOX gene is located on chromosome 6 between a cluster of snoRNA genes and a diacylglycerol kinase gene. TbQSOX is 1581 nucleotides long and encodes a 526-amino acid protein starting with an N-terminal signal sequence with a predicted cleavage site after residue 22 (Figure 1C). The full length sequence corresponds to a type I membrane protein with a single transmembrane helix (residues 491-513) and a short cytoplasmic C-terminal tail. In higher organisms, alternate splicing generates soluble QSOX forms lacking the membrane span (2, 4, 6). Whether the parasite generates correspondingly truncated QSOX forms is not yet known. As mentioned earlier, the domain structure of TbQSOX is distinctly different from its metazoan orthologs. Analysis of predicted secondary structure (Figure 1C) shows the presence of an alternating arrangement of strands and helices typical of a thioredoxin fold. This feature is supplemented by an N-terminal additional βα motif similar to that of an a domain of PDI. Following the HRR domain, encompassing residues 168-304 and dominated by alpha-helices, is the Erv/ALR domain, another helix-rich domain which serves to bind the FAD moiety and which houses the proximal disulfide that is critical for communication with the Trx1 domain in metazoan QSOXs.
The cysteine residues of TbQSOX
Table 1 lists the cysteine residues, and their domain context, in this single-Trx QSOX. Three CxxC motifs are conserved in all of the available QSOX sequences; from the smallest free-living eukaryote (Ostreococcus tauri) to humans (1). For simplicity, we refer to these cysteine pairs as CI-CII, CIII-CIV, and CV-CVI, together with their numbering in the context of the full-length protein (Table 1). CI-CII cysteine residues comprise the CxxC motif located at the N-terminus of the α1 helix of the thioredoxin domain. This motif is highly conserved among proteins with thioredoxin domains; it has been shown to be critical for the catalytic activity of, among other proteins, human QSOX1 (10) and PDI (12, 14, 44). This sequence in QSOXs from mammals and birds is typically CGHC, and is immediately preceded by a tryptophan residue (1). However, the corresponding sequence in TbQSOX is CGAC (Figure 1C), preceded by a glycine rather than an aromatic residue. In metazoan QSOXs, this disulfide receives reducing equivalents from dithiol substrates and then transmits them to CIII-CIV of the Erv/ALR flavin-binding domain (10, 11). Site directed mutagenesis with the human QSOX1 confirms that CIII-CIV is catalytically essential because it needs to communicate first with CI-CII in the Trx domain and then with the adjacent flavin moiety (10). The third CxxC motif, CV-CVI, is also invariant; however mutation of either, or both, of the CV-CVI cysteine residues did not result in a major change in activity towards small molecule or protein substrates in QSOX1 (10). The role of this conserved disulfide is currently cryptic.
TABLE 1.
The cysteine residues in TbQSOX
 | ||
|---|---|---|
| Cysteine Residue | Motif | Corresponding cysteines in related proteins |
| Trx domain | ||
| 58 | n/a | not conserved |
| 69, 72 | CI-CII | C70 – C73 (CxxCtrx) of human QSOX1b; C61 – C64 of yeast Pdi1p |
| 105, 112 | Cx6C | C101 – C110 (Cx8C) of human QSOX1b; C90 – C97 (Cx6C) of yeast Pdi1p |
| 132, 138 | Cx5C | not conserved |
| HRR domain | ||
| 180 | n/a | not conserved |
| Erv/ALR domain | ||
| 305, 317 | Cx11C | Unique to QSOX family; C393 – C405 (Cx11C) of human QSOX1b |
| 350, 353 | CIII-CIV | C449 – C452 (CxxCprox) of human QSOX1b; C121 – C124 of yeast Erv2p |
| 405, 408 | CV-CVI | C509 – C512 (CxxCtrm) of human QSOX1b; C176 – C178 of yeast Erv2p |
In addition to the three CxxC disulfides mentioned above, a further 8 cysteines, likely in disulfide linkage (see later), are present in full length TbQSOX. A Cx6C motif (C105-C112) is placed comparably to the Cx8C motif present in human QSOX1 protein (10), to the Cx6C motif present in yeast Pdi1p (13), and to the Cx6-8C motifs from several other members of PDI family. In yeast Pdi1p, these residues form a disulfide (13) and may be involved in regulation of catalytic function (45). The Cx11C motif (C305-C317) lies at the beginning of the Erv/ALR domains. TbQSOX contains an additional 4 non-conserved cysteine residues (58, 132, 138 and 180) of uncertain role. This report will address mutations of only the three CxxC motifs that have been previously suggested to play catalytic roles in QSOX family members.
Sub-cloning, Expression and Purification of TbQSOX
The TbQSOX cDNA sequence (NCBI Accession Number XM_840213) was edited to reflect codon preferences in E. coli and synthesized as described in Experimental Procedures. We initially attempted to express TbQSOX with an N-terminal His-tag by starting the QSOX sequence at residue G32. This residue lies in the middle of a region with no predicted secondary structure, between the end of the signal sequence and the beginning of a short beta strand that starts the PDI-like Trx domain (Figure 1C). This construct failed to express (see Experimental Procedures). However, the inclusion of an additional 12 residues at the N-terminus of the oxidase sequence yielded a stable, soluble protein in ample amounts for the work described in this study (see below). The codon-optimized gene sequence and the amino acid sequence of the wild-type protein, used throughout this work are included in Supporting Information (Figures S1 and S2).
The number of anticipated catalytic and structural disulfide bonds in TbQSOX suggested that expression in E. coli BL21 Star (DE3) cells might be problematic because this strain does not favor the cytosolic expression of disulfide-containing proteins. Indeed, expression trials using this strain led to the accumulation of QSOX in inclusion bodies (not shown). However, we could obtain soluble and active protein using a gor/trx mutant strain (E. coli Origami2 (DE3) ) grown until mid log phase at 37 °C and then induced with 0.5 mM IPTG at 15 °C for 24 h. Induction at higher temperatures led to progressively more QSOX appearing in the insoluble fraction (not shown). TbQSOX was then purified utilizing the N-terminal His-tag followed by an additional purification step using a butyl-Sepharose column to remove a contaminating flavoprotein (identified as alkyl hydroperoxide reductase; AhpF, data not shown). Typically we obtained ~7 mg of pure TbQSOX per liter of culture for wild type and cysteine mutants. The resulting protein was stable for at least 6 months at 4 °C in 50 mM potassium phosphate buffer containing 1 mM EDTA at pH 7.5. TbQSOX could also be stored for at least one year at −20 °C without significant loss of enzyme activity. The purified protein showed a typical unresolved flavin absorbance in the visible region with a maximum at 456 nm and an experimentally-determined molar extinction coefficient of 12,250 M−1cm−1 (Figure 2; see Experimental Procedures). No free thiols were detected when protein denatured with SDS was incubated with DTNB (see Experimental Procedures) suggesting that all 14 cysteine residues in TbQSOX comprise 7 disulfide bonds. SDS-PAGE, performed under reducing and non-reducing conditions, showed a single band at the expected molecular weight of 54 kDa (Figure 2, inset). Analytical ultracentrifugation experiments gave a solution molecular weight of 54.3 kDa (Supporting Information Figure S3).
FIGURE 2.

UV/Vis spectrum of TbQSOX. The spectrum was recorded in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA. The inset shows SDS-PAGE of TbQSOX under reducing (Red) and non-reducing (NR) conditions together with protein molecular weight standards in kDa.
Catalytic Activity of TbQSOX
The divergent redox biology and the evolutionary distance between T. brucei and metazoan QSOXs justify an examination of thiol substrate specificity for the parasite oxidase. As for all the other QSOXs investigated (5, 7, 9, 10), DTT proves a useful model substrate of TbQSOX. Here, TbQSOX shows a pH dependence with a maximal activity at neutral pH values (with ascending and descending limbs fit to pK values of 5.7 and 8.9 respectively; Figure 3). This pH dependence, and the optimal pH value of about 7, is similar to those of the egg white oxidase (7). We utilized 50 mM potassium phosphate buffer, pH 7.5, containing 1 mM EDTA for most of the experiments described herein. Compared to DTT, glutathione is a very poor substrate of TbQSOX with an approximately 2000-fold lower kcat/Km value (reflecting a much larger Km term for the monothiol; Table 2). While glutathione is present in trypanosomes, the dithiol analog trypanothione, [N1,N8–bis(glutathionyl)spermidine], substitutes for many of its intracellular functions (29, 46). While trypanothione shows a roughly 20-fold increase in catalytic efficiency over glutathione, this dithiol is approximately 10-fold poorer as a substrate than two reduced proteins selected for study (Table 2).
FIGURE 3.

pH dependence for the oxidation of DTT by TbQSOX. The ascending and descending limbs for the oxidation of 5 mM DTT were fit to pK values of 5.7 and 8.9, respectively, setting upper and lower limits of 2608 s−1 and 0 s−1.
Table 2.
Comparison of Steady State Parameters for the Oxidation of Thiol Substrates between TbQSOX and Three Metazoan QSOXsa
| TbQSOX | GgQSOX1bb | BtQSOX1bc | HsQSOX1bc | |||
|---|---|---|---|---|---|---|
| kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | kcat/Km (M−1 s−1) | kcat/Km (M−1 s−1) | kcat/Km (M−1 s−1) | |
| DTT | 45 | 0.086 | 5.2 × 105 | 2.3 × 105 | 3.7 × 105 | 2.0 × 105 |
| GSH | -d | > 50 | 2.7 × 102d | 2.3 × 103 | 5.9 × 103 | 2.0 × 103 |
| TSH | 16 | 3.23 | 4.9 × 103 | n.d.e | n.d. | n.d. |
| rRNase A | 22 | 0.36 | 6.0 × 104 | 1.8 × 105 | 3.7 × 105 | 2.2 × 105 |
| rRfBP | 14 | 0.30 | 4.5 × 104 | 1.6 × 105 | n.d. | n.d. |
Turnover numbers are expressed in terms of thiols oxidized, not disulfides generated per s. For comparison between substrates with different numbers of thiol groups, Km values are expressed on a per thiol basis. The oxidation of trypanothione, rRNase and rRfBP was evaluated using discontinuous thiol titer assays with DTNB. Measurement of the turnover with DTT and GSH utilized the oxygen electrode (see Experimental Procedures).
Recalculated in terms of monothiols using data from avian QSOX enzymes (7).
Rates of GSH oxidation were essentially linear up to 40 mM: the catalytic efficiency is expressed as the apparent second-order rate constant for 10 mM GSH.
n.d.: not done.
Both reduced proteins, RNase and RfBP, are substantially unfolded in the absence of denaturant. Reduction of the disulfide bonds in RNase generates a largely unstructured protein (47) in which reaction of all 8 thiols with 2 mM of the polar reagent DTNB is complete in less than 200 ms (40). Similarly, about 13 of the 18 thiol groups of rRfBP react in 0.5 s under the same conditions (40). The catalytic efficiencies of TbQSOX in oxidizing rRNase are some 3- to 6-fold lower than for the avian, milk and human enzymes (9, 10, 48). An estimate of the pH dependence for the oxidation of rRNase shows ascending and descending limbs with pK values of 6.8 and 9.8 respectively (Figure S4). For rRfBP, the trypanosomal enzyme shows a catalytic efficiency that is 2-fold lower than the avian enzyme (48). These differences between protist and metazoan enzymes are largely a reflection of the Km term. In spite of these rather minor decreases in catalytic efficiency between parasite and metazoan QSOXs, the trypanosomal enzyme remains a facile oxidant of reduced proteins. Hence, TbQSOX may generate some fraction of secreted protein disulfides in trypanosomes. Finally, TbQSOX can cooperate with reduced PDI in the successful oxidative protein folding of RfBP ((49) data not shown).
Roles of CxxC Motifs in TbQSOX Catalysis
Recent work with the recombinant human QSOX1 showed that, while CI-CII and the CIII-CIV disulfides were crucial for the efficient oxidation of both DTT and rRNase thiols, the conserved CV-CVI disulfide was non-essential in vitro (10). These studies, combined with earlier results from the avian QSOX (11, 50), led to a revised model for catalysis in which the thioredoxin CI-CII motif receives reducing equivalents from a protein substrate and then interacts with CIII-CIV of the Erv/ALR domain within the same subunit of the oxidase (10). We were thus interested in comparing the behavior of the single-Trx QSOX with these prior results. In particular, we wished to examine whether the CV-CVI disulfide was catalytically important in this smaller parasite enzyme.
When CI (C69) or CII (C72) was mutated to serine, catalytic activity of TbQSOX towards 5 mM DTT decreased to ~5 % of wild type protein (Figure 4). This decrease in catalytic activity was even more drastic when rRNase was used as a substrate; now only ~0.5 % of wild type activity was observed (Figure 4). These values likely represent the ability of DTT, in the absence of a functional thioredoxin domain, to directly interact with the Erv/ALR oxidase domain. Accordingly, we prepared a truncated protein (HRR-Erv) which lacked the entire Trx domain (the sequence of this construct is shown in Supporting Information Figure S2). HRR-Erv binds FAD with essentially the same visible spectrum as that for full-length protein (Supporting Information Figure S5). The turnover number of the HRR-Erv truncated protein using 5 mM DTT was comparable to the full length protein containing either a CIS or a CIIS mutation (150, 114, and 112 thiols oxidized per min, respectively).
FIGURE 4.

Comparison of the turnover numbers of wild type TbQSOX and its CxxC mutants towards DTT and rRNase. Turnover numbers were measured at 25 °C in phosphate buffer, pH 7.5, using 5 mM DTT and 200 μM rRNase thiols (filled and open columns, respectively). Data were normalized with the wild-type protein set at 100 %.
Replacement of either cysteine of the proximal disulfide (CIII or CIV) with serine essentially abolished activity both towards DTT and rRNase (Figure 4). The placement of this proximal disulfide within the Erv/ALR domain of TbQSOX is likely to be very similar to that found in the other Erv/ALR family members (18, 19, 21). CIII is the interchange residue with a sulfur atom largely exposed to solvent and poised to accept an incoming mixed disulfide bond from the surface-accessible CI cysteinyl thiolate of the TbQSOX Trx domain. Following resolution of this mixed disulfide between Trx and Erv/ALR domains, the now-reduced CIII-CIV motif can then transfer a pair of electrons to the flavin probably via the intermediacy of a C4a adduct (1, 3, 51, 52). Clearly, both CIII and CIV residues would then be expected to be catalytically critical in TbQSOX. The visible spectra of the two mutant proteins are noticeably different. The CIIIS mutant leaves CIV unpaired and it forms a thiolate to oxidized flavin charge-transfer complex (showing a wedge-shaped long-wavelength band; Figure S6). In contrast, the corresponding CIVS mutant protein shows a typical oxidized flavin spectrum but it is blue shifted by 6 nm compared to the wild type protein (Figure S6). This behavior likely reflects greater access of solvent to the flavin when the proximal disulfide is reduced, and/or an increase in polarity when the hydroxyl group of serine replaces the S atom of CIV.
Finally, mutations of the terminal CxxC disulfide were prepared. The visible spectrum of both CVS and CVIS mutants were comparable to that of the wild-type protein (Figure S6). In terms of catalysis, neither mutant showed significant loss of activity towards DTT or rRNase (Figure 4). In aggregate, these studies show that the first two CxxC redox centers (CI-CII and CIII-CIV) in TbQSOX are crucial for the efficient oxidation of rRNase. The third conserved CxxC motif (CV-CVI) may play an important role in vivo, but it is not revealed in these in vitro experiments.
Dithionite Titrations of TbQSOX
In preparation for the rapid reaction experiments to be presented later, we wished to assess how many redox centers could be revealed during dithionite titrations of the wild type enzyme. Sodium dithionite solutions were standardized by anaerobic titrations against solutions of free FAD before being used as redox titrants of the oxidase (7, 53). The dithionite ion, S2O42−, serves as an overall 2-electron reductant of the flavin, although reduction may proceed via the intermediacy of the SO2− radical anion (54). In addition, disulfides can be reduced indirectly by dithionite (requiring an additional 2-electron equivalents per disulfide) if these redox centers can communicate with the flavin ring. Figure 5 shows that complete reduction of the flavin in TbQSOX occurs with concomitant reduction of a redox-active disulfide. The apparent stoichiometry of 4.6 electron equivalents per QSOX monomer, slightly more than the expected 4.0 electron equivalents, might reflect kinetic sluggishness towards the end of the titration. The approximate linearity of the absorbance decrease at 456 nm with reductant added (inset Figure 5) shows that both the flavin and the redox-active disulfide are almost equipotential. However, when dithionite titrations are repeated with the CIS mutant, removing any potential involvement of the Trx redox-active disulfide (Figure 1B), full reduction of the flavin now requires only 2-electron equivalents (open circles, inset Figure 5). These data suggest that the second redox center is CI-CII and that the proximal CIII-CIV disulfide is considerably more reducing than either the flavin or the Trx disulfide. Removal of the entire Trx domain, as in the HRR-Erv construct, again results in a stoichiometry of 2-electron equivalents for complete reduction of the flavin prosthetic group (open squares, inset Figure 5).
FIGURE 5.

Dithionite titrations of wild type, CIS and HRR-Erv forms of TbQSOX. WT TbQSOX in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA, was made anaerobic, and titrated at 25 °C with a standardized solution of sodium dithionite yielding curves 1–7. The inset plots the reduction of the oxidase as a function of electron equivalents added per flavin (■) together with comparable titrations with CIS (○) and HRR-Erv (□) proteins.
Consistent with these results using TbQSOX, the proximal disulfide in a number of other Erv/ALR/QSOX family members is found to be significantly more reducing than the flavin prosthetic group. In Erv2p, reducing equivalents from PDI enter via a CxC shuttle (distal) disulfide, and are then passed to the proximal disulfide, to the adjacent flavin prosthetic group, and finally to molecular oxygen (18, 55). The 4-electron equivalents needed to completely bleach the flavin in Erv2p are distributed, more or less evenly, between a distal disulfide and the isoalloxazine thereby leaving the proximal disulfide largely oxidized (55). Secondly, the cytokine-like form of augmenter of liver regeneration, ALR, has both flavin and proximal disulfide redox centers. Here, 2-electron reduction of the flavin occurs before significant reduction of the proximal disulfide again reflecting their relative redox potentials (56). Finally, partial proteolysis of avian egg white QSOX generates a fragment analogous to the HRR-Erv fragment of TbQSOX mentioned above (11). The subsequent removal of the redox-active thioredoxin domain in the avian QSOX causes the stoichiometry required for flavin reduction to revert from 4- to 2-electron equivalents (11). In all these examples, the proximal (CIII-CIV) disulfide is more reducing than either the isoalloxazine ring or any additional shuttle disulfides that communicate reducing equivalents from distal loci to the flavin binding domain.
Monitoring Turnover with DTT and rRNase in the Stopped-Flow Spectrophotometer
Aerobic solutions of TbQSOX and saturating concentrations of DTT were mixed in the stopped flow instrument and the evolution of spectral changes was followed using a diode-array spectrophotometer (Figure 6). The inset plots the corresponding changes at 456 and 560 nm. Here, the flavin is almost completely oxidized in the steady state showing that the reoxidation of reduced flavin species by molecular oxygen is considerably faster than the rate-determining steps in the reductive half-reaction (see later). The small (3 %) decline in 456 nm absorbance in the steady state is accompanied by a correspondingly rapid formation of a weak long-wavelength band consistent with a thiolate to oxidized flavin charge transfer complex (monitored here at 560 nm and observed, under static conditions, in the spectrum of the CIIIS mutant, Figure S6). The steady-state persists for about 0.6 s before reduced flavin begins to accumulate as the dissolved oxygen is depleted. While reduction of the flavin is largely complete by 0.9 s (inset Figure 6), further smaller decreases in absorbance continue over several seconds. These minor secondary changes are too slow to be catalytically significant; they are also encountered during the reduction of TbQSOX by DTT under anaerobic conditions (see later).
FIGURE 6.

Enzyme monitored turnover of DTT by TbQSOX. The oxidase was mixed with DTT to give final concentrations of 18.4 μM TbQSOX and 2.75 mM DTT in 50 mM phosphate buffer, pH 7.5, 25 °C containing 240 μM dissolved oxygen. Only selected spectra between 5 ms to 3 s are shown for clarity. The inset shows absorbance at 456 nm and 560 nm plotted as a function of time.
The area under the curve for the absorbance changes at 560 nm (inset Figure 6) allows calculation of the turnover values as a function of remaining oxygen concentration (Figure S7) and shows a Km for oxygen of 10.5 μM, with a limiting turnover of 43 s−1, in good agreement with the data obtained from the oxygen electrode (Table 2). A comparable progression of changes was seen during the aerobic turnover of rRNase, with the rapid generation of the charge-transfer band and the eventual reduction of the flavin (Figure S8).
Reductive Half-reaction Using DTT, Kinetic Evidence for the Importance of a Functional Thioredoxin Domain
Figure 7 shows that anaerobic reduction of TbQSOX by 0.5 mM DTT rapidly generates small levels of a charge-transfer species which decays more slowly to yield reduced flavin (inset). The rate constant for the appearance of the long wavelength intermediate is shown in Figure 7B (solid squares; with a limiting rate constant of 280 s−1 and an apparent Kd for DTT of 0.66 mM). This phase is more than 10-fold faster than overall turnover (23 disulfides/s, 45 thiols/s; Table 2). In contrast, the concentration dependence and the limiting rate constant of the disappearance of 560 nm absorbance closely parallels the main reduction phase (Figure 7B, solid diamonds; 0.31 mM Kd and 18.8 s−1). Comparable values are observed for the major phase of flavin reduction at 456 nm (accounting for ~85 % of the total bleaching observed with a Kd of 0.28 mM DTT and a limiting rate of 17.9 s−1; Figure 7B, open circles). The average between the two values for this slower phase (18.4 s−1) is in good agreement with the 22.5 s−1 expected for overall turnover (disulfides generated) under these conditions. About 15 % of the total bleaching of the flavin observed at 456 nm in Figure 7A requires 50 s for completion and can be fit to two exponentials with rate constants of approximately 0.8 and 0.1 s−1 (not shown). Both these slower phases are irrelevant for aerobic turnover conditions: they likely entail eventual reduction of all three redox centers in TbQSOX (see later).
FIGURE 7.

Anaerobic reduction of TbQSOX with DTT. Anaerobic solutions of TbQSOX and DTT (in 50 mM phosphate buffer, pH 7.5, containing 1 mM EDTA, 5 mM glucose, 5 nM glucose oxidase and 1 nM catalase) were mixed in a stopped-flow spectrophotometer at 25 °C to give final concentrations of 8 μM and 0.5 mM respectively. Panel A shows selected spectra from the diode-array detector and the inset follows the absorbance at 560 nm using a monochromator. Panel B plots observed rate constants as a function of the concentration of DTT. Appearance of the long wavelength species at 560 nm shows a limiting rate constant of 280 s−1 with an apparent Kd of 0.66 mM (■). Disappearance of the long wavelength species (◆) and the reduction of flavin (○) show limiting rate constants of 18.8 s−1 and 17.9 s−1 respectively, both with an apparent Kd of 0.31 mM.
Anaerobic stopped flow experiments were then repeated using the CIS mutant of TbQSOX so that the CxxC motif of the Trx domain could no longer participate in reduction of the enzyme by DTT. The consequences of lack of a functional thioredoxin domain are profound. Full reduction of the flavin at 456 nm can be fit to a single exponential and occurs without the formation of a significant charge transfer complex (Supporting Information, Figure S9). Unlike the wild-type protein, these rate constants show a linear dependence up to 20 mM DTT (Figure S10). At 5 mM DTT, the resulting rate constant of 1.2 s−1 is in good agreement with the overall turnover number determined in the oxygen electrode (1.25 s−1 and Figure 4). Removing the Trx domain entirely (as in the HRR-Erv construct) yields a similar turnover number (0.93 s−1).
Oxidative Half-reaction
As mentioned previously, dithionite reduction of the wild type TbQSOX leads to the reduction of both flavin and CI-CII redox centers (Figure 5). In contrast, the CIS mutant can be reduced essentially exclusively at the flavin center (Figure 5) allowing its reaction with molecular oxygen to be followed without the associated spectral changes involved in bringing the second pair of reducing equivalents from the Trx domain to the flavin. At a final concentration of 120 μM oxygen, reoxidation is very rapid and proceeds without detectable intermediates (Figure S11). Assuming that the TbQSOX reaction with oxygen is bimolecular, as has been observed for avian QSOX (50) and for most flavin dependent oxidases (57), the second order rate constant would be approximately 2.5 × 106 M−1s−1. For comparison, a value of 9 × 105 M−1s−1 was reported for the oxygen reactivity of the reduced flavin in avian QSOX at 4 °C (50).
A Model for Turnover
Figure 8 presents a simplified depiction of catalysis with the model substrate DTT. Turnover is initiated by the reduction of CI-CII disulfide by DTT. Reducing equivalents from CI-CII are transferred to CIII-CIV via a mixed disulfide between CI and CIII (form C), allowing CIV to form a thiolate to oxidized flavin charge-transfer species (shown by a dotted line from CIV to FAD, forms C and D). FAD is reduced by the transfer of electrons from CIII-CIV and then reoxidized by molecular oxygen to complete the catalytic cycle. In some cases, the molecular basis for the kinetic transients observed in this work is not certain. For example, while it is clear that the weak long-wavelength band that appears rapidly in the steady state represents a charge-transfer complex between the thiolate form of CIV and the flavin (see earlier, Figure 8), the relative contributions of forms C and D to this absorbance feature are unclear. The appearance of the C/D charge-transfer species occurs with a limiting rate of ~ 280 s−1 and is half-saturated at 0.66 mM DTT. What is the significance of this DTT concentration dependence? In an early kinetic study of avian QSOX we interpreted a comparable saturation of the apparent rate constant in terms of a binding site for DTT (50). This prior work was performed before the realization that these QSOX enzymes contain at least three redox centers, and that input of reducing equivalents occurred primarily through the CI-CII disulfide of a thioredoxin domain. While a binding site for reducing substrates (e.g. DTT or reduced proteins) is certainly plausible, there is currently no structural or kinetic evidence for such a site in any member of the QSOX family. For simplicity, we have here assumed that the reduction of the enzyme by DTT occurs by a simple bimolecular encounter with the CI-CII disulfide of the Trx domain. Assuming that the conversion of A to C/D reflects two irreversible steps, k1 and k2 (Figure 8), and analyzing for the formation of C/D as in Strickland et al. (58), predicts a value for this bimolecular reduction step of 4.6 × 105 M−1s−1.
FIGURE 8.

Simplified Model for Turnover in TbQSOX. Reduction of the CI-CII disulfide by DTT (k1) yields form B. A thiolate to oxidized flavin charge-transfer species (represented by the curved dotted line: involving either, or both, of forms C and D) then forms with a limiting rate of 280 s−1 (k2). The charge-transfer absorbance decays (k3) with a limiting rate of 18 s−1 yielding reduced flavin (form E). The extent to which the interdomain redox reaction between forms C and D contribute to this rate liming step is unknown. The anticipated C4a adduct in the reduction of the flavin is omitted from the scheme because it has not been observed experimentally. The second substrate oxygen participates in k4 by converting forms E to A. For simplicity, the scheme omits the possibility that DTT could reduce the CI-CII disulfide in forms D and E to generate 4-electron reduced TbQSOX prior to the oxidative half reaction (k4). A contribution of 4-electron reduced oxidase to overall turnover would be favored by high DTT concentrations and low oxygen tensions.
Form D is likely to react via a C4a thiol-flavin adduct (not shown, (51, 59–61)) yielding form E. Currently it is unknown whether the rate limiting step of 18 s−1 reflects a slow interconversion of forms C and D (followed by a rapid reaction generating reduced flavin), or whether it actually represents the direct accumulation of form E from D. There is a further uncertainty in Figure 8: whether TbQSOX turnover is necessarily restricted to cycling between 0 and 2-electron reduced states. Indeed, this seems unlikely. Since DTT reacts rapidly with the CI-CII disulfide in the Trx domain of form A, it might re-react with the disulfide in form E (and/or form D) before the reoxidation event (k4). For illustration, using 240 μM oxygen and 5 mM DTT, form E would be predicted to undergo a second reduction by DTT in preference to reoxidation by step k4 (with pseudo first-order rate constants of 1500 vs. 600 s−1 respectively). Partition in favor of 4-electron reduction of TbQSOX would be favored by low oxygen tensions. In contrast, low thiol substrate concentrations would favor the simple 0- to 2-electron cycling scheme shown in Figure 8. Further work is aimed at a more detailed examination of the pre-steady state behavior of TbQSOX and the possible roles of 4-electron reduced forms of the oxidase in turnover.
CONCLUSIONS
This characterization of a single-Trx QSOX shows strong similarities with the double-Trx enzymes investigated to date. Lack of the second Trx domain evidently does not result in major changes in the catalytic specificity of the parasite QSOX. While TbQSOX is between 3- to 6-fold less efficient at the oxidation of the rRNase and rRfBP than the metazoan enzymes, reduced unfolded proteins are its best known substrates. The average kcat/Km values for the metazoan enzymes for DTT are about one-half of those for TbQSOX. Both single- and double-Trx QSOXs oxidize the monothiol GSH very poorly. The dithiol trypanothione shows a kcat/Km value per thiol that is at least 10-fold lower than that for rRNase and rRfBP.
The similarities between single- and double-Trx QSOXs extend to pre-steady state kinetic behavior. Both enzymes rapidly form a 2-electron reduced charge transfer complex with dithiol substrates, and in both cases this species decays to yield reduced flavin in a process that strongly limits overall turnover. Reoxidation of both enzymes by molecular oxygen does not limit turnover; illustrated by the almost exclusively oxidized flavin component found in the steady-state during enzyme-monitored aerobic turnover experiments (Figure 6, (50)). Finally, both enzymes show a low Km for oxygen (10.5 μM and 4.6 μM for T. brucei and the avian QSOX (50) respectively, when the reductant is DTT).
The mutagenesis studies reported in this work complement, and extend, those performed recently with the human enzyme (10). Two particular aspects of the behavior of TbQSOX are of note. First, stopped-flow studies of the recombinant parasite enzyme reinforce how important the CI-CII motif is for overall turnover. The observation that complete removal of the Trx domain by truncation has no more serious catalytic impact than a CI to serine mutation in full-length TbQSOX suggests that the thioredoxin domain does not serve additional effector roles under the conditions of our experiments. A second issue is that one could envisage that the simpler domain structure of a protist QSOX would result in an overt catalytic role for the CV-CVI disulfide: a motif conserved in all QSOX family members. However, we see no significant impact in TbQSOX: mutations at this locus behave like the CV-CVI mutations of the human recombinant QSOX enzyme.
The pathways of oxidative protein folding in Trypanosoma remain to be uncovered. In addition to QSOX, the parasite has a typical Ero1 protein and several PDI-like proteins. Neither parasite flavin-dependent sulfhydryl oxidases has an obvious ER retention sequence, but it is possible that one, or both, could be retained in the ER lumen by binding to a PDI homolog (as has been suggested for human Ero1-Lalpha and -Lbeta (62)). In terms of TbQSOX, we show that GSH and its dithiol analog, trypanothione, are relatively poor substrates. Thus TbQSOX might introduce disulfide bonds into client proteins in the secretory system of Trypanosoma while minimizing collateral perturbation of the small molecule thiol redox pool and the consequent added oxidative stress that this may engender (3, 49). Alternatively, TbQSOX may function post-ER (1, 4): in the Golgi apparatus (as has been suggested for the long form of human QSOX1 (63)), on the plasma membrane, or following secretion from the cell. Since trypanosomes seem to contain single paralogs of QSOX and Ero1, it will be interesting to examine their relative roles in oxidative protein folding. More generally, oxidative protein folding pathways in protists might represent an additional target for antimicrobial therapies including the arsenicals (40) that have been used for over a century to combat trypanosomiases.
Supplementary Material
Acknowledgments
We thank NIH GM26643 for financial support, Drs. Deborah Fass and Luise Krauth-Siegel for helpful discussions, and anonymous reviewers for their insight.
Abbreviations
- ALR
augmenter of liver regeneration
- DTNB
5,5′-dithiobis(2-nitrobenzoate)
- DTT
dithiothreitol
- ERV
a protein essential for respiration and viability in yeast
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GdnHCl
guanidine hydrochloride
- HRR
helix-rich region
- IPTG
isopropyl-β-D-thiogalactopyranoside
- QSOX
Quiescin-sulfhydryl oxidase
- RfBP
riboflavin binding protein
- RNase
ribonuclease A
- SDS
sodium dodecyl sulfate
- TCEP
triscarboxyethylphosphine
- Trx
thioredoxin domain
- VSG
variable surface glycoproteins
- WT
wild-type
Footnotes
This work was supported in part by National Institutes of Health Grant (GM26643). The content of this work is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
SUPPORTING INFORMATION AVAILABLE
Table S1 lists the sequences of primers used in this study. Figures S1 – S11 show the DNA and protein sequences of the constructs used, analysis of analytical ultracentrifugation data, UV/Vis spectra of TbQSOX constructs and mutants, and data from steady state and pre-steady state kinetic measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Heckler EJ, Rancy PC, Kodali VK, Thorpe C. Generating disulfides with the Quiescin-sulfhydryl oxidases. Biochim Biophys Acta. 2008;1783:567–577. doi: 10.1016/j.bbamcr.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thorpe C, Hoober K, Raje S, Glynn N, Burnside J, Turi G, Coppock D. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch Biochem Biophys. 2002;405:1–12. doi: 10.1016/s0003-9861(02)00337-5. [DOI] [PubMed] [Google Scholar]
- 3.Thorpe C, Coppock DL. Generating disulfides in multicellular organisms: Emerging roles for a new flavoprotein family. J Biol Chem. 2007;282:13929–13933. doi: 10.1074/jbc.R600037200. [DOI] [PubMed] [Google Scholar]
- 4.Coppock DL, Thorpe C. Multidomain flavin-dependent sulfhydryl oxidases. Antioxid Redox Signal. 2006;8:300–311. doi: 10.1089/ars.2006.8.300. [DOI] [PubMed] [Google Scholar]
- 5.Ostrowski MC, Kistler WS. Properties of a flavoprotein sulfhydryl oxidase from rat seminal vesicle secretion. Biochemistry. 1980;19:2639–2645. doi: 10.1021/bi00553a016. [DOI] [PubMed] [Google Scholar]
- 6.Benayoun B, Esnard-Feve A, Castella S, Courty Y, Esnard F. Rat seminal vesicle FAD-dependent sulfhydryl oxidase. Biochemical characterization and molecular cloning of a member of the new sulfhydryl oxidase/quiescin Q6 gene family. J Biol Chem. 2001;276:13830–13837. doi: 10.1074/jbc.M010933200. [DOI] [PubMed] [Google Scholar]
- 7.Hoober KL, Joneja B, White HB, III, Thorpe C. A Sulfhydryl Oxidase from Chicken Egg White. J Biol Chem. 1996;271:30510–30516. doi: 10.1074/jbc.271.48.30510. [DOI] [PubMed] [Google Scholar]
- 8.Hoober KL, Glynn NM, Burnside J, Coppock DL, Thorpe C. Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J Biol Chem. 1999;274:31759–31762. doi: 10.1074/jbc.274.45.31759. [DOI] [PubMed] [Google Scholar]
- 9.Jaje J, Wolcott HN, Fadugba O, Cripps D, Yang AJ, Mather IH, Thorpe C. A flavin-dependent sulfhydryl oxidase in bovine milk. Biochemistry. 2007;46:13031–13040. doi: 10.1021/bi7016975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heckler EJ, Alon A, Fass D, Thorpe C. Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry. 2008;47:4955–4963. doi: 10.1021/bi702522q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raje S, Thorpe C. Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxin domain in disulfide bond formation. Biochemistry. 2003;42:4560–4568. doi: 10.1021/bi030003z. [DOI] [PubMed] [Google Scholar]
- 12.Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci. 2006;31:455–464. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell. 2006;124:61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 14.Hatahet F, Ruddock LW. Protein Disulfide Isomerase: A Critical Evaluation of Its Function in Disulfide Bond Formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 15.Atkinson HJ, Babbitt PC. An atlas of the thioredoxin fold class reveals the complexity of function-enabling adaptations. PLoS Comput Biol. 2009;5:e1000541. doi: 10.1371/journal.pcbi.1000541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lisowsky T. ERV1 is involved in the cell-division cycle and the maintenance of mitochondrial genomes in Saccharomyces cerevisiae. Curr Genet. 1994;26:15–20. doi: 10.1007/BF00326299. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Hofhaus G, Lisowsky T. Erv1p from Saccharomyces cerevisiae is a FAD-linked sulfhydryl oxidase. FEBS Lett. 2000;477(1–2):62–66. doi: 10.1016/s0014-5793(00)01767-1. [DOI] [PubMed] [Google Scholar]
- 18.Gross E, Sevier CS, Vala A, Kaiser CA, Fass D. A new FAD-binding fold and intersubunit disulfide shuttle in the thiol oxidase Erv2p. Nat Struct Biol. 2002;9:61–67. doi: 10.1038/nsb740. [DOI] [PubMed] [Google Scholar]
- 19.Wu CK, Dailey TA, Dailey HA, Wang BC, Rose JP. The crystal structure of augmenter of liver regeneration: A mammalian FAD-dependent sulfhydryl oxidase. Protein Sci. 2003;12:1109–1118. doi: 10.1110/ps.0238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vitu E, Bentzur M, Lisowsky T, Kaiser CA, Fass D. Gain of function in an ERV/ALR sulfhydryl oxidase by molecular engineering of the shuttle disulfide. J Mol Biol. 2006;362:89–101. doi: 10.1016/j.jmb.2006.06.070. [DOI] [PubMed] [Google Scholar]
- 21.Fass D. The Erv family of sulfhydryl oxidases. Biochim Biophys Acta. 2008;1783:557–566. doi: 10.1016/j.bbamcr.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Hakim M, Fass D. Dimer interface migration in a viral sulfhydryl oxidase. J Mol Biol. 2009;391:758–768. doi: 10.1016/j.jmb.2009.06.070. [DOI] [PubMed] [Google Scholar]
- 23.Barry D, Carrington M. Antigenic Variation. In: Maudlin I, Holmes P, Miles M, editors. The Trypanosomiases. International CABI Publishing; UK: 2004. pp. 25–38. [Google Scholar]
- 24.Chattopadhyay A, Jones NG, Nietlispach D, Nielsen PR, Voorheis HP, Mott HR, Carrington M. Structure of the C-terminal domain from Trypanosoma brucei variant surface glycoprotein MITat1.2. J Biol Chem. 2005;280:7228–7235. doi: 10.1074/jbc.M410787200. [DOI] [PubMed] [Google Scholar]
- 25.Carrington M, Miller N, Blum M, Roditi I, Wiley D, Turner M. Variant specific glycoprotein of Trypanosoma brucei consists of two domains each having an independently conserved pattern of cysteine residues. J Mol Biol. 1991;221:823–835. doi: 10.1016/0022-2836(91)80178-w. [DOI] [PubMed] [Google Scholar]
- 26.Freymann D, Down J, Carrington M, Roditi I, Turner M, Wiley D. 2.9 A resolution structure of the N-terminal domain of a variant surface glycoprotein from Trypanosoma brucei. J Mol Biol. 1990;216:141–160. doi: 10.1016/S0022-2836(05)80066-X. [DOI] [PubMed] [Google Scholar]
- 27.Blum ML, Down JA, Gurnett AM, Carrington M, Turner MJ, Wiley DC. A structural motif in the variant surface glycoproteins of Trypanosoma brucei. Nature. 1993;362:603–609. doi: 10.1038/362603a0. [DOI] [PubMed] [Google Scholar]
- 28.Jones NG, Nietlispach D, Sharma R, Burke DF, Eyres I, Mues M, Mott HR, Carrington M. Structure of a glycosylphosphatidylinositol-anchored domain from a trypanosome variant surface glycoprotein. J Biol Chem. 2008;283:3584–3593. doi: 10.1074/jbc.M706207200. [DOI] [PubMed] [Google Scholar]
- 29.Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science. 1985;227:1485–1487. doi: 10.1126/science.3883489. [DOI] [PubMed] [Google Scholar]
- 30.Krauth-Siegel LR, Comini MA, Schlecker T. The trypanothione system. Subcell Biochem. 2007;44:231–251. doi: 10.1007/978-1-4020-6051-9_11. [DOI] [PubMed] [Google Scholar]
- 31.Sevier CS, Kaiser CA. Disulfide transfer between two conserved cysteine pairs imparts selectivity to protein oxidation by Ero1. Mol Biol Cell. 2006;17:2256–2266. doi: 10.1091/mbc.E05-05-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mezghrani A, Fassio A, Benham A, Simmen T, Braakman I, Sitia R. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 2001;20:6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 2000;290:1571–1574. doi: 10.1126/science.290.5496.1571. [DOI] [PubMed] [Google Scholar]
- 34.Frand AR, Kaiser CA. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol Cell. 1999;4:469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]
- 35.Baker KM, Chakravarthi S, Langton KP, Sheppard AM, Lu H, Bulleid NJ. Low reduction potential of Ero1alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 2008;27:2988–2997. doi: 10.1038/emboj.2008.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daithankar VN, Farrell SR, Thorpe C. Augmenter of liver regeneration: substrate specificity of a flavin-dependent oxidoreductase from the mitochondrial intermembrane space. Biochemistry. 2009;48:4828–4837. doi: 10.1021/bi900347v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deponte M, Hell K. Disulfide bond formation in the intermembrane space of mitochondria. J Biochem. 2009;146:599–608. doi: 10.1093/jb/mvp133. [DOI] [PubMed] [Google Scholar]
- 38.Bihlmaier K, Mesecke N, Terziyska N, Bien M, Hell K, Herrmann JM. The disulfide relay system of mitochondria is connected to the respiratory chain. J Cell Biol. 2007;179:389–395. doi: 10.1083/jcb.200707123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tokatlidis K. A disulfide relay system in mitochondria. Cell. 2005;121:965–967. doi: 10.1016/j.cell.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 40.Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C. Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry. 2009;48:424–432. doi: 10.1021/bi801988x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 42.Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilkinson B, Gilbert HF. Protein disulfide isomerase. BBA-Proteins Proteom. 2004;1699:35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 45.Wilkinson B, Xiao R, Gilbert HF. A structural disulfide of yeast protein-disulfide isomerase destabilizes the active site disulfide of the N-terminal thioredoxin domain. J Biol Chem. 2005;280:11483–11487. doi: 10.1074/jbc.M414203200. [DOI] [PubMed] [Google Scholar]
- 46.Krauth-Siegel RL, Comini MA. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta. 2008;1780:1236–1248. doi: 10.1016/j.bbagen.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 47.Navon A, Ittah V, Landsman P, Scheraga HA, Haas E. Distributions of intramolecular distances in the reduced and denatured states of bovine pancreatic ribonuclease A. Folding initiation structures in the C-terminal portions of the reduced protein. Biochemistry. 2001;40:105–118. doi: 10.1021/bi001946o. [DOI] [PubMed] [Google Scholar]
- 48.Hoober KL, Sheasley SS, Gilbert HF, Thorpe C. Sulfhydryl oxidase from egg white: a facile catalyst for disulfide bond formation in proteins and peptides. J Biol Chem. 1999;274:22147–22150. doi: 10.1074/jbc.274.32.22147. [DOI] [PubMed] [Google Scholar]
- 49.Rancy PC, Thorpe C. Oxidative Protein Folding in vitro: a Study of the Cooperation between Quiescin-sulfhydryl Oxidase and Protein Disulfide Isomerase. Biochemistry. 2008;47:12047–12056. doi: 10.1021/bi801604x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoober KL, Thorpe C. Egg white sulfhydryl oxidase: Kinetic mechanism of the catalysis of disulfide bond formation. Biochemistry. 1999;38:3211–3217. doi: 10.1021/bi9820816. [DOI] [PubMed] [Google Scholar]
- 51.Thorpe C, Williams CH. Spectral evidence for a flavin adduct in a monoalkylated derivative of pig heart lipoamide dehydrogenase. J Biol Chem. 1976;251:7726–7728. [PubMed] [Google Scholar]
- 52.Dmitrenko O, Thorpe C. A computational analysis of the interaction between flavin and thiol(ate) groups. Implications for flavoenzyme catalysis. J Sulfur Chem. 2008;29:415–C421. [Google Scholar]
- 53.Williams CH, Arscott LD, Matthews RG, Thorpe C, Wilkinson KD. Methodology employed for anaerobic spectrophotometric titrations and for computer-assisted data analysis. Methods Enzymol. 1979:185–198. doi: 10.1016/0076-6879(79)62217-6. [DOI] [PubMed] [Google Scholar]
- 54.Mayhew SG. Redox Potential of Dithionite and So2- from Equilibrium Reactions with Flavodoxins, Methyl Viologen and Hydrogen Plus Hydrogenase. European Journal of Biochemistry. 1978;85:535–547. doi: 10.1111/j.1432-1033.1978.tb12269.x. [DOI] [PubMed] [Google Scholar]
- 55.Wang W, Winther JR, Thorpe C. Erv2p: characterization of the redox behavior of a yeast sulfhydryl oxidase. Biochemistry. 2007;46:3246–3254. doi: 10.1021/bi602499t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farrell SR, Thorpe C. Augmenter of liver regeneration: a flavin dependent sulfhydryl oxidase with cytochrome C reductase activity. Biochemistry. 2005;44:1532–1541. doi: 10.1021/bi0479555. [DOI] [PubMed] [Google Scholar]
- 57.Mattevi A. To be or not to be an oxidase: challenging the oxygen reactivity of flavoenzymes. Trends Biochem Sci. 2006;31:276–283. doi: 10.1016/j.tibs.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 58.Strickland S, Palmer G, Massey V. Determination of dissociation constants and specific rate constants of enzyme-substrate (or protein-ligand) interactions from rapid reaction kinetic data. J Biol Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- 59.Williams CH., Jr . Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase-A family of flavoenzyme transhydrogenases. In: Müller F, editor. Chemistry and Biochemistry of Flavoenzymes. CRC Press; 1992. pp. 121–211. [Google Scholar]
- 60.Miller SM, Massey V, Ballou D, Williams CH, Jr, Distefano MD, Moore MJ, Walsh CT. Use of a site-directed triple mutant to trap intermediates: demonstration that the flavin C(4a)-thiol adduct and reduced flavin are kinetically competent intermediates in mercuric ion reductase. Biochemistry. 1990;29:2831–2841. doi: 10.1021/bi00463a028. [DOI] [PubMed] [Google Scholar]
- 61.O’Donnell ME, Williams CH., Jr Reconstitution of Escherichia coli thioredoxin reductase with 1-deazaFAD. Evidence for 1-deazaFAD C-4a adduct formation linked to the ionization of an active site base. J Biol Chem. 1984;259:2243–2251. [PubMed] [Google Scholar]
- 62.Otsu M, Bertoli G, Fagioli C, Guerini-Rocco E, Nerini-Molteni S, Ruffato E, Sitia R. Dynamic retention of Ero1alpha and Ero1beta in the endoplasmic reticulum by interactions with PDI and ERp44. Antioxid Redox Signal. 2006;8:274–282. doi: 10.1089/ars.2006.8.274. [DOI] [PubMed] [Google Scholar]
- 63.Chakravarthi S, Jessop CE, Willer M, Stirling CJ, Bulleid NJ. Intracellular catalysis of disulphide bond formation by the human sulphydryl oxidase, QSOX1. Biochem J. 2007;404:403–411. doi: 10.1042/BJ20061510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.